Ticagrelor holsten
Poland
Table of Contents
- 1. NAME OF THE MEDICINAL PRODUCT
- 2. QUALITATIVE AND QUANTITATIVE COMPOSITION
- 3. PHARMACEUTICAL FORM
- 4.2 Dosage and administration
- 4.3 Contraindications
- 4.4 Special warnings and precautions for use
- 4.5 Interactions with other medicinal products and other forms of interaction
- 4.6 Fertility, pregnancy and lactation
- 4.7 Effects on ability to drive and use machines
- 4.8 Undesirable effects
- 4.9 Overdose
- 5.2 Pharmacokinetic Properties
- 5.3 Preclinical safety data
- 6.2 Pharmaceutical incompatibilities
- 6.3 Shelf life
- 6.4 Special precautions during storage
- 6.5 Type and contents of the container
- 6.6 Special precautions for disposal
- 7. MARKETING AUTHORISATION HOLDER
- 8. MARKETING AUTHORISATION NUMBERS
- 9. DATE OF FIRST AUTHORISATION OR RENEWAL OF THE AUTHORISATION
- 10. DATE OF ADOPTION OR PARTIAL CHANGE OF THE TEXT
SUMMARY OF PRODUCT CHARACTERISTICS
1. NAME OF THE MEDICINAL PRODUCT
Ticagrelor Holsten, 60 mg, film-coated tablets
2. QUALITATIVE AND QUANTITATIVE COMPOSITION
Each coated tablet contains 60 mg of ticagrelor.
For a complete list of excipients, see section 6.1.
3. PHARMACEUTICAL FORM
Film-coated tablet.
Round (8.1 x 8.1 mm), biconvex, pink tablets marked with the number "60" on one side and smooth on the other side.
4. CLINICAL PARTICULARS
4.1 Therapeutic Indications
The medicinal product Ticagrelor Holsten, in combination with acetylsalicylic acid (ASA), is indicated for the prevention of cardiovascular events in adult patients:
- with acute coronary syndrome (ACS), or
- with a history of myocardial infarction (heart attack) and high risk of cardiovascular events (see sections 4.2 and 5.1).
4.2 Dosage and administration
Dosage
Patients taking the medicinal product Ticagrelor Holsten should also take a daily maintenance dose of low-dose acetylsalicylic acid (ASA) 75–150 mg, unless contraindicated for individual patients.
Acute coronary syndromes
Treatment with the medicinal product Ticagrelor Holsten should be initiated with a single 180 mg loading dose (2 tablets of 90 mg) and continued with a dose of 90 mg twice daily.
In patients with ACS, treatment duration with Ticagrelor Holsten 90 mg twice daily should be 12 months, unless there are clinical indications for earlier discontinuation (see section 5.1).
Discontinuation of ASA may be considered after 3 months in patients with ACS who have undergone percutaneous coronary intervention (PCI) and who are at increased risk of bleeding. In such cases, continued treatment with ticagrelor as the sole antiplatelet agent should be maintained for an additional 9 months (see section 4.4).
Prior myocardial infarction
The recommended dose of the medicinal product Ticagrelor Holsten is 60 mg twice daily when prolonged treatment is required in patients with a history of myocardial infarction at least one year previously and who are at high risk of cardiovascular events (see section 5.1). Treatment may be initiated without interruption as a continuation of initial 12-month treatment with Ticagrelor Holsten 90 mg or another adenosine diphosphate (ADP) receptor inhibitor in patients with ACS and high risk of cardiovascular events. Treatment may also be initiated up to 2 years after myocardial infarction or within one year of stopping prior ADP receptor inhibitor therapy. Data on the efficacy and safety of ticagrelor use beyond 3 years of long-term treatment are limited.
If a switch from another antiplatelet drug is required, the first dose of Ticagrelor Holsten should be administered 24 hours after the last dose of the previous antiplatelet drug.
Missed dose
Dosage errors should be avoided. If a dose of Ticagrelor Holsten is missed, the patient should take only one tablet (the next dose) according to the prescribed dosing schedule.
Special patient groups
Elderly patients
No dosage adjustment is required in elderly patients (see section 5.2).
Renal impairment
Dosage adjustment is not necessary in patients with renal impairment (see section 5.2).
Hepatic impairment
Studies on the use of ticagrelor in patients with severe hepatic impairment have not been conducted; therefore, its use is contraindicated in these patients (see section 4.3). Limited data are available on the use of the medicinal product in patients with moderate hepatic impairment. Dosage adjustment is not required; however, ticagrelor should be used with caution (see sections 4.4 and 5.2). No dosage adjustment is necessary in patients with mild hepatic impairment (see section 5.2).
Children and adolescents
The safety and efficacy of ticagrelor have not been established in children under 18 years of age.
The use of ticagrelor in children is not appropriate for the indication sickle cell anaemia (see sections 5.1 and 5.2).
Method of administration
Oral administration.
Ticagrelor Holsten may be administered with or without food.
For patients who have difficulty swallowing the tablet(s) whole, the tablets may be crushed into a fine powder, mixed with half a glass of water, and immediately consumed. The glass should then be rinsed with water (another half glass of water), and the rinse water should also be consumed.
The crushed tablet(s) mixed with water may also be administered via a nasogastric tube (CH8 or larger). It is important to flush the nasogastric tube with water (half a glass: minimum 50 ml up to a maximum of 125 ml) after administration of the mixture.
4.3 Contraindications
- Hypersensitivity to the active substance or to any of the excipients listed in section 6.1 (see section 4.8).
- Active pathological bleeding.
- History of intracranial haemorrhage (see section 4.8).
- Severe hepatic impairment (see sections 4.2, 4.4 and 5.2).
- Concomitant use of ticagrelor and strong inhibitors of the CYP3A4 enzyme (e.g. ketoconazole, clarithromycin, nefazodone, ritonavir and atazanavir), as this may lead to a significant increase in exposure to ticagrelor (see section 4.5).
4.4 Special warnings and precautions for use
Bleeding risk
In patients with an increased risk of bleeding, the benefit-risk ratio regarding prevention of cardiovascular events should be carefully considered (see sections 4.8 and 5.1). If there are clinical indications for using ticagrelor, it should be used with caution in the following patient groups:
- Patients with a tendency to bleed (e.g. due to recent trauma, surgical procedures, coagulation disorders, active or recent gastrointestinal bleeding) or those at increased risk of injury. Ticagrelor is contraindicated in patients with active pathological bleeding, a history of intracranial haemorrhage, or severe hepatic impairment (see section 4.3).
- Patients receiving concomitant medications that may increase bleeding risk (e.g. non-steroidal anti-inflammatory drugs (NSAIDs), oral anticoagulants, and/or fibrinolytic agents) administered within 24 hours prior to ticagrelor dosing.
In two randomised, controlled trials (TICO and TWILIGHT) involving patients with ACS who underwent PCI with a drug-eluting stent, discontinuation of ASA after 3 months of dual antiplatelet therapy with ticagrelor and ASA, followed by continuation of ticagrelor as the sole antiplatelet agent for 9 and 12 months, respectively, reduced the risk of bleeding without increasing the observed risk of major adverse cardiovascular events (MACE) compared to continuation of dual antiplatelet therapy. The decision to discontinue ASA after 3 months and continue ticagrelor as the sole antiplatelet agent for 9 months in patients at increased risk of bleeding should be based on a clinical assessment weighing bleeding risk against thrombotic risk (see section 4.2).
Platelet transfusion did not reverse the antiplatelet effect of ticagrelor in healthy volunteers and is unlikely to be clinically beneficial in patients with bleeding.
Since desmopressin co-administered with ticagrelor does not shorten the standard bleeding time, its clinical efficacy in treating bleeding episodes is questionable (see section 4.5).
Antifibrinolytic treatment (aminocaproic acid or tranexamic acid) and/or recombinant factor VIIa may enhance haemostasis. Ticagrelor may be reinitiated if the cause of bleeding has been identified and controlled.
Surgical procedures
Patients should be instructed to inform physicians and dentists about their use of ticagrelor prior to any planned surgical procedures and before starting any new medications.
In patients enrolled in the PLATO study who underwent coronary artery bypass grafting (CABG), more bleeding events occurred in the ticagrelor group compared to the clopidogrel group when the study drug was discontinued one day before surgery. However, when the drug was stopped two or more days before surgery, the number of severe bleeding events was similar between the two groups (see section 4.8). If a patient is scheduled for elective surgery and antiplatelet effect is undesirable, ticagrelor should be discontinued 5 days prior to the procedure (see section 5.1).
Patients with prior ischaemic stroke
Patients with ACS and a history of ischaemic stroke may be treated with ticagrelor for up to 12 months (PLATO study).
Patients with prior myocardial infarction and prior ischaemic stroke were not included in the PEGASUS study. Therefore, due to lack of data, treatment in these patients beyond one year is not recommended.
Hepatic impairment
Ticagrelor is contraindicated in patients with severe hepatic impairment (see sections 4.2 and 4.3). Experience with ticagrelor in patients with moderate hepatic impairment is limited; therefore, caution is recommended in these patients (see sections 4.2 and 5.2).
Patients at risk of bradyarrhythmia events
Holter ECG monitoring showed an increased frequency of mostly asymptomatic ventricular pauses during ticagrelor treatment compared to clopidogrel. Patients at increased risk of bradyarrhythmia events (e.g. patients without pacemakers who have sick sinus syndrome, second- or third-degree atrioventricular block, or those with syncope related to bradycardia) were excluded from the main trials assessing the safety and efficacy of ticagrelor. Therefore, due to limited clinical experience, ticagrelor should be used with caution in these patients (see section 5.1).
Additionally, caution is advised when ticagrelor is used concomitantly with medicinal products that may cause bradycardia. However, no evidence of clinically significant adverse effects was observed in the PLATO study with concomitant use of one or more bradycardia-inducing agents (i.e. 96% beta-blockers, 33% calcium channel blockers diltiazem and verapamil, and 4% digoxin) (see section 4.5).
In the Holter substudy of the PLATO trial, ventricular pauses >3 seconds were observed more frequently in ticagrelor-treated patients than in clopidogrel-treated patients during the acute phase of acute coronary syndrome (ACS). The increase in Holter-detected ventricular pauses during ticagrelor treatment was more pronounced in patients with chronic heart failure than in the overall ACS population, but not during the one-month ticagrelor observation period or compared to clopidogrel. No adverse clinical consequences (including syncope or pacemaker implantation) related to this disproportion were observed in this patient group (see section 5.1).
Since marketing authorisation, cases of bradyarrhythmias and AV blocks have been reported in patients receiving ticagrelor (see section 4.8), particularly in patients with ACS, where myocardial ischaemia and concomitant use of drugs that reduce heart rate or affect cardiac conduction may be contributing factors. Before adjusting treatment, the patient's clinical status and concomitant medications should be evaluated as potential causes.
Dyspnoea
Patients treated with ticagrelor have reported dyspnoea. Dyspnoea is usually mild to moderate and often resolves without discontinuing the drug. In patients with asthma/chronic obstructive pulmonary disease (COPD), there may be an increased absolute risk of dyspnoea during ticagrelor treatment. Ticagrelor should be used with caution in patients with a history of asthma and/or COPD. The mechanism of dyspnoea is not fully understood. If a patient reports new episodes of dyspnoea, prolonged duration, or worsening of symptoms during ticagrelor treatment, a full diagnostic evaluation should be performed and, if the patient tolerates the condition poorly, ticagrelor treatment should be discontinued. Further details are provided in section 4.8.
Central sleep apnoea
Since marketing authorisation, central sleep apnoea, including Cheyne-Stokes respiration, has been reported in patients receiving ticagrelor. If central sleep apnoea is suspected, further clinical evaluation should be considered.
Increased creatinine levels
Creatinine levels may increase during ticagrelor treatment. The mechanism of this phenomenon has not been established. Renal function should be monitored according to standard clinical practice. In patients with ACS, renal function should also be checked one month after initiating ticagrelor treatment, with particular attention to patients aged ≥75 years, those with moderate to severe renal impairment, and those receiving angiotensin receptor blockers (ARBs).
Increased uric acid levels
Hyperuricaemia may develop during ticagrelor treatment (see section 4.8). Caution is advised in patients with a history of hyperuricaemia or gouty arthritis.
As a precautionary measure, ticagrelor is not recommended in patients with urate nephropathy.
Thrombotic thrombocytopenic purpura (TTP)
Thrombotic thrombocytopenic purpura (TTP) has been very rarely reported during ticagrelor treatment. TTP is characterised by thrombocytopenia and microangiopathic haemolytic anaemia associated with neurological symptoms, renal dysfunction, or fever. TTP is a potentially life-threatening condition requiring prompt treatment, including plasmapheresis.
Interference with platelet function tests used to diagnose heparin-induced thrombocytopenia (HIT)
In the functional heparin-induced platelet activation (HIPA) test used to diagnose HIT, antibodies against the platelet factor 4/heparin complex in patient serum activate platelets from healthy donors in the presence of heparin.
False-negative results in platelet function tests (including HIPA) used to diagnose HIT have been reported in patients receiving ticagrelor. This is due to P2Y12 receptor inhibition on healthy donor platelets by ticagrelor present in patient serum/plasma. Information on concomitant ticagrelor treatment is required for correct interpretation of platelet function test results used in HIT diagnosis.
In patients who develop heparin-induced thrombocytopenia, the benefit-risk ratio of continuing ticagrelor treatment should be evaluated, considering both the prothrombotic state of HIT and the increased bleeding risk associated with concomitant anticoagulant and ticagrelor therapy.
Other
Based on the observed relationship in the PLATO study between the maintenance dose of acetylsalicylic acid and the relative efficacy of ticagrelor compared to clopidogrel, concomitant use of ticagrelor and high maintenance doses of acetylsalicylic acid (>300 mg) is not recommended (see section 5.1).
Premature discontinuation of treatment
Premature discontinuation of any antiplatelet treatment, including Ticagrelor Holsten, may increase the risk of cardiovascular death, myocardial infarction, or stroke due to the underlying disease. Therefore, premature discontinuation of treatment should be avoided.
Sodium
Ticagrelor Holsten contains less than 1 mmol sodium (23 mg) per dose, i.e., essentially "sodium-free".
4.5 Interactions with other medicinal products and other forms of interaction
Ticagrelor is primarily a substrate of the CYP3A4 isoenzyme and also a mild inhibitor of this enzyme.
Ticagrelor is also a substrate of P-glycoprotein (P-gp) and a weak inhibitor of P-gp, and may increase exposure
to P-gp substrates.
Effect of other medicinal products and substances on ticagrelor
Inhibitors of CYP3A4
- Strong CYP3A4 inhibitors – concomitant administration of ketoconazole with ticagrelor resulted in a 2.4-fold increase in C\textsubscript{max} and a 7.3-fold increase in AUC of ticagrelor. C\textsubscript{max} and AUC of the active metabolite were reduced by 89% and 56%, respectively. It is expected that other strong CYP3A4 inhibitors (clarithromycin, nefazodone, ritonavir, atazanavir) will produce a similar effect; therefore, co-administration of strong CYP3A4 inhibitors with ticagrelor is contraindicated (see section 4.3).
- Moderate CYP3A4 inhibitors – concomitant administration of diltiazem and ticagrelor increased C\textsubscript{max} of ticagrelor by 69% and AUC by 2.7-fold, while C\textsubscript{max} of the active metabolite decreased by 38%, with no effect on its AUC. Ticagrelor did not affect plasma concentrations of diltiazem. Other moderate CYP3A4 inhibitors (e.g. amprenavir, aprepitant, erythromycin, and fluconazole) may have similar effects and can also be used concomitantly with ticagrelor.
- A 2-fold increase in ticagrelor exposure was observed after daily consumption of large amounts of grapefruit juice (3 x 200 ml). This level of increased exposure to ticagrelor is not expected to be clinically significant in most patients.
Inducers of CYP3A
Concomitant administration of rifampicin and ticagrelor reduced C\textsubscript{max} and AUC of ticagrelor by 73% and 86%, respectively. C\textsubscript{max} of the active metabolite remained unchanged, while its AUC decreased by 46%. Other CYP3A inducers (e.g. phenytoin, carbamazepine, and phenobarbital) are expected to similarly reduce exposure to ticagrelor. Concomitant use of ticagrelor with strong CYP3A inducers may reduce ticagrelor concentrations and efficacy; therefore, such combination is not recommended.
Cylosporine (inhibitor of P-gp and CYP3A)
Concomitant administration of cyclosporine (600 mg) and ticagrelor increased C\textsubscript{max} of ticagrelor by 2.3-fold and AUC by 2.8-fold. In the presence of cyclosporine, AUC of the active metabolite of ticagrelor increased by 32%, while C\textsubscript{max} decreased by 15%.
There are no data on concomitant use of ticagrelor with other active substances that are strong inhibitors of P-glycoprotein (P-gp) and moderate inhibitors of CYP3A4 (e.g. verapamil, quinidine), which may increase exposure to ticagrelor. If such combination cannot be avoided, concomitant use requires caution.
Others
Clinical interaction studies have shown that concomitant administration of ticagrelor with heparin, enoxaparin, and acetylsalicylic acid (ASA), or desmopressin did not affect the pharmacokinetics of ticagrelor or its active metabolite, or ADP-induced platelet aggregation, compared to ticagrelor alone. If clinically indicated, medicinal products affecting haemostasis should be used cautiously in combination with ticagrelor.
In patients with ACS treated with morphine, delayed and reduced exposure to oral P2Y\textsubscript{12} inhibitors, including ticagrelor and its active metabolite (reduced exposure to ticagrelor by 35%), has been observed. This interaction may be related to reduced gastrointestinal motility and also applies to other opioids. The clinical significance of this is unknown, but data suggest a potential reduction in the efficacy of ticagrelor in patients receiving morphine concomitantly.
In patients with ACS in whom morphine administration cannot be withheld and rapid P2Y\textsubscript{12} inhibition is considered critically important, administration of an intravenous P2Y\textsubscript{12} inhibitor may be considered.
Effect of ticagrelor on other medicinal products
Medicinal products metabolized by CYP3A4
- Simvastatin – concomitant administration of ticagrelor with simvastatin increased C\textsubscript{max} of simvastatin by 81% and AUC by 56%, and increased C\textsubscript{max} of simvastatin acid by 64% and AUC by 52%, with individual cases of 2- or 3-fold increases. Concomitant use of ticagrelor with simvastatin at doses exceeding 40 mg daily could lead to adverse effects of simvastatin; therefore, the potential benefits of this combination should be carefully considered. No effect of simvastatin on plasma concentrations of ticagrelor was observed. Ticagrelor may have a similar effect on lovastatin use. Concomitant use of ticagrelor with simvastatin or lovastatin at doses exceeding 40 mg is not recommended.
- Atorvastatin – concomitant administration of atorvastatin and ticagrelor increases C\textsubscript{max} and AUC of atorvastatin acid by 23% and 36%, respectively. Similar increases in AUC and C\textsubscript{max} were observed for all metabolites of atorvastatin acid. This is considered not clinically significant.
- A similar effect on other statins metabolized by CYP3A4 cannot be excluded. In the PLATO study, however, patients were taking various statins, and in 93% of all patients enrolled in this study, there were no safety concerns related to statin use.
Ticagrelor is a moderate inhibitor of CYP3A4. Concomitant use of ticagrelor with CYP3A4 substrates having a narrow therapeutic index (e.g. cisapride and ergot alkaloids) is not recommended, as ticagrelor may increase exposure to these medicinal products.
P-gp substrates (including digoxin, cyclosporine)
Concomitant administration of ticagrelor increases C\textsubscript{max} and AUC of digoxin by 75% and 28%, respectively. Mean trough concentrations of digoxin increased by approximately 30% with concomitant use of ticagrelor, while individual peak concentrations increased up to 2-fold. The presence of digoxin does not affect C\textsubscript{max} and AUC of ticagrelor or its active metabolite. Therefore, appropriate clinical and/or laboratory monitoring is recommended during concomitant use of medicinal products with a narrow therapeutic index that are P-gp dependent, such as digoxin and ticagrelor.
Ticagrelor did not affect blood concentrations of cyclosporine. The effect of ticagrelor on other P-gp substrates has not been studied.
Medicinal products metabolized by CYP2C9
Concomitant administration of ticagrelor and tolbutamide did not alter plasma concentrations of either medicinal product, suggesting that ticagrelor is not an inhibitor of CYP2C9 and is unlikely to alter the metabolism of medicinal products such as warfarin or tolbutamide via CYP2C9.
Rosuvastatin
Ticagrelor may affect renal excretion of rosuvastatin, increasing the risk of rosuvastatin accumulation. Although the exact mechanism is unknown, in some cases, concomitant use of ticagrelor and rosuvastatin has led to worsening renal function, increased CPK (creatine phosphokinase) activity, and rhabdomyolysis.
Oral contraceptives
Concomitant administration of ticagrelor with levonorgestrel and ethinylestradiol increased exposure to ethinylestradiol by approximately 20%, but did not affect the pharmacokinetics of levonorgestrel. No clinically significant effect on the efficacy of oral contraceptives is expected when levonorgestrel and ethinylestradiol are used concomitantly with ticagrelor.
Medicinal products causing bradycardia
Due to observed, usually asymptomatic, ventricular pauses and bradycardia, caution should be exercised when using ticagrelor concomitantly with medicinal products that cause bradycardia (see section 4.4). However, in the PLATO study, no evidence of clinically significant adverse effects was observed with concomitant use of one or more medicinal products causing bradycardia (i.e. 96% beta-blockers, 33% calcium channel blockers: diltiazem and verapamil, and 4% digoxin).
Concomitant use with other medicinal products
In clinical studies, ticagrelor was administered concomitantly with ASA, proton pump inhibitors, statins, beta-blockers, angiotensin-converting enzyme (ACE) inhibitors, and angiotensin receptor blockers over long periods due to the need for treatment of concomitant conditions, as well as with heparin, low molecular weight heparin, and intravenous GpIIb/IIIa inhibitors for short durations (see section 5.1). No clinically significant interactions were observed with these medicinal products.
Concomitant administration of ticagrelor with heparin, enoxaparin, or desmopressin did not affect activated partial thromboplastin time (aPTT), activated clotting time (ACT), or anti-factor Xa activity. However, due to potential pharmacodynamic interactions, caution should be exercised when using ticagrelor concomitantly with medicinal products affecting haemostasis.
Due to observed skin bleeding events with selective serotonin reuptake inhibitors (SSRIs) (e.g. paroxetine, sertraline, and citalopram), caution should be exercised when using SSRIs concomitantly with ticagrelor, as this may increase the risk of bleeding.
4.6 Fertility, pregnancy and lactation
Women of childbearing potential
Women of childbearing potential should use appropriate contraceptive measures to prevent pregnancy during treatment with ticagrelor.
Pregnancy
There are no or limited data on the use of ticagrelor during pregnancy. Animal studies have shown harmful effects on fertility (see section 5.3). Ticagrelor is not recommended during pregnancy.
Breast-feeding
Available pharmacodynamic and toxicological data from animal studies have shown that ticagrelor and its active metabolites are excreted into milk (see section 5.3). A risk to newborns/infants cannot be excluded. A decision should be made whether to discontinue breast-feeding or to discontinue/abandon ticagrelor therapy, taking into account the benefits of breast-feeding for the child and the benefits of therapy for the woman.
Fertility
In animals, ticagrelor had no effect on fertility in males or females (see section 5.3).
4.7 Effects on ability to drive and use machines
Ticagrelor has no or negligible influence on the ability to drive vehicles and operate machinery.
Dizziness and confusion have been reported in patients treated with ticagrelor. Therefore, patients
experiencing these symptoms should exercise caution when driving or operating machinery.
4.8 Undesirable effects
Summary of the safety profile
The safety profile of ticagrelor was evaluated based on results from two large phase 3 studies (PLATO and PEGASUS), involving over 39,000 patients (see section 5.1).
In the PLATO study, a higher frequency of treatment discontinuation due to adverse events was observed in patients receiving ticagrelor compared to those receiving clopidogrel (7.4% vs. 5.4%).
In the PEGASUS study, a higher frequency of treatment discontinuation due to adverse events was observed in patients receiving ticagrelor compared to those treated with ASA monotherapy (16.1% in the group treated with ticagrelor 60 mg in combination with ASA versus 8.5% in the group receiving ASA monotherapy). The most commonly reported adverse reactions in patients treated with ticagrelor were bleeding and dyspnoea (see section 4.4).
Tabulated list of adverse reactions
The adverse reactions listed below have been identified from clinical trials or reported following the marketing of ticagrelor (Table 1).
Adverse reactions are listed by MedDRA system organ class (SOC). Within each SOC group, adverse reactions are ranked by frequency.
Frequency categories are defined as follows: very common (≥1/10), common (≥1/100 to <1/10), uncommon (≥1/1,000 to <1/100), rare (≥1/10,000 to <1/1,000), very rare (<1/10,000), and not known (frequency cannot be estimated from available data).
Table 1 – Adverse reactions listed by frequency and system organ class (SOC)
| System organ classes | Very common | Common | Uncommon | Frequency not known |
| Benign, malignant and unspecified neoplasms (including cysts and polyps) | Bleeding from tumora | |||
| Blood and lymphatic system disorders | Blood disorders, haemorrhageb | Thrombotic thrombocytopeniac | ||
| Immune system disorders | Hypersensitivity, including angioedemac | |||
| Metabolism and nutrition disorders | Hyperuricemiad | Gout/Gouty arthritis | ||
| Psychiatric disorders | Disorientation | |||
| Nervous system disorders | Dizziness, syncope, headache | Intracranial haemorrhage | ||
| Eye disorders | Ocular haemorrhagee | |||
| Ear and labyrinth disorders | Vertigo of vestibular origin | Ear haemorrhage | ||
| Cardiac disorders | Bradyarrhythmia, AV blockc | |||
| System organ classes | Very common | Common | Uncommon | Frequency not known |
| Vascular disorders | Hypotension | |||
| Respiratory, thoracic and mediastinal disorders | Dyspnoea | Bleeding from respiratory tractf | ||
| Gastrointestinal disorders | Gastrointestinal haemorrhageg, diarrhoea, nausea, dyspepsia, constipation | Retroperitoneal haemorrhage | ||
| Skin and subcutaneous tissue disorders | Subcutaneous or dermal bleeding, rash, pruritus | |||
| Musculoskeletal and connective tissue disorders | Haemorrhage into musclesi | |||
| Renal and urinary disorders | Bleeding from urinary tractj | |||
| Reproductive system and breast disorders | Bleeding from reproductive tractk | |||
| Investigations | Increased blood creatinine concentrationd | |||
| Injury, poisoning and procedural complications | Post-procedural haemorrhage, traumatic bleedingl |
Description of selected adverse reactions
Bleeding
Results of the PLATO study regarding bleeding
The overall results on the frequency of bleeding in the PLATO study are presented in Table 2.
Table 2 – Analysis of all bleeding events, values estimated using the Kaplan-Meier method after 12 months (PLATO)
| Ticagrelor 90 mg twice daily N=9235 | Clopidogrel N=9186 | p-value* | |
| Major bleeding, PLATO | 11.6 | 11.2 | 0.4336 |
| Major fatal/life-threatening bleeding, PLATO | 5.8 | 5.8 | 0.6988 |
| Major non-CABG-related bleeding, PLATO | 4.5 | 3.8 | 0.0264 |
| Major non-procedure-related bleeding, PLATO | 3.1 | 2.3 | 0.0058 |
| Major + minor bleeding, PLATO | 16.1 | 14.6 | 0.0084 |
| Major + minor non-procedure-related bleeding, PLATO | 5.9 | 4.3 | <0.0001 |
| Major bleeding, TIMI definition | 7.9 | 7.7 | 0.5669 |
| Major + minor bleeding, TIMI definition | 11.4 | 10.9 | 0.3272 |
There were no differences between ticagrelor and clopidogrel in the incidence of PLATO-defined fatal/life-threatening major bleeding, overall PLATO-defined major bleeding, TIMI-defined major bleeding, or TIMI-defined minor bleeding (Table 2). However, there were more overall major and minor bleedings according to PLATO criteria in the ticagrelor group compared to the clopidogrel group. Fatal bleeding occurred in a very small number of patients in the PLATO study: 20 (0.2%) in the ticagrelor group and 23 (0.3%) in the clopidogrel group (see section 4.4).
Age, sex, body weight, race, geographic region, concomitant diseases, concomitant medications, and medical history, including prior stroke or transient ischemic attack, were not predictive factors for overall major bleeding or non-procedure-related major bleeding according to PLATO criteria. Therefore, no subgroup at increased risk for any category of bleeding was identified.
Bleeding related to CABG:
In the PLATO study, 42% of the 1584 patients (12% of the cohort) who underwent CABG surgery experienced PLATO-defined fatal/life-threatening major bleeding, with no difference observed between treatment groups. Fatal bleeding after CABG occurred in 6 patients in each treatment group (see section 4.4).
Non-CABG and non-procedure-related bleeding:
Ticagrelor and clopidogrel did not differ in terms of non-CABG-related, PLATO-defined fatal/life-threatening major bleeding. However, PLATO-defined overall major bleeding, TIMI-defined major bleeding, and TIMI-defined major + minor bleeding occurred more frequently in the ticagrelor group. Similarly, when all procedure-related bleedings were excluded, more bleeding events occurred in the ticagrelor group compared to the clopidogrel group (Table 2). Discontinuation of treatment due to non-procedure-related bleeding occurred more frequently in the ticagrelor group (2.9%) than in the clopidogrel group (1.2%; p<0.001).
Intracranial bleeding:
In the PLATO study, a higher number of non-procedure-related intracranial bleedings were observed with ticagrelor (n=27 bleedings in 26 patients, 0.3%) compared to clopidogrel (n=14 bleedings, 0.2%), including 11 fatal bleedings with ticagrelor and 1 with clopidogrel. No difference was observed in the overall number of fatal bleedings.
Bleeding results from the PEGASUS study
The overall bleeding event outcome from the PEGASUS study is presented in Table 3.
Table 3 – Analysis of all bleeding events, Kaplan-Meier estimated values at 36 months (PEGASUS)
| Tikagrelor 60 mg twice daily + ASA N=6958 | ASA as monotherapy N=6996 | |||
| Safety endpoints | KM% | Hazard ratio (95% CI) | KM% | p-value |
| Bleeding categories defined by TIMI | ||||
| Major TIMI bleeding | 2.3 | 2.32 (1.68, 3.21) | 1.1 | <0.0001 |
| Life-threatening bleeding | 0.3 | 1.00 (0.44, 2.27) | 0.3 | 1.0000 |
| Intracranial hemorrhage (ICH) | 0.6 | 1.33 (0.77, 2.31) | 0.5 | 0.3130 |
| Other major TIMI bleeding | 1.6 | 3.61 (2.31, 5.65) | 0.5 | <0.0001 |
| Major or minor TIMI bleeding | 3.4 | 2.54 (1.93, 3.35) | 1.4 | <0.0001 |
| Major or minor bleeding requiring medical attention according to TIMI | 16.6 | 2.64 (2.35, 2.97) | 7.0 | <0.0001 |
| Bleeding categories defined in the PLATO study | ||||
| Major bleeding in the PLATO study | 3.5 | 2.57 (1.95, 3.37) | 1.4 | <0.0001 |
| Life-threatening or fatal bleeding | 2.4 | 2.38 (1.73, 3.26) | 1.1 | <0.0001 |
| Other major bleeding in the PLATO study | 1.1 | 3.37 (1.95, 5.83) | 0.3 | <0.0001 |
| Major or minor bleeding in the PLATO study | 15.2 | 2.71 (2.40, 3.08) | 6.2 | <0.0001 |
In the PEGASUS study, the incidence of TIMI-defined major bleeding during treatment with ticagrelor 60 mg twice daily was higher than with ASA monotherapy. There was no increase in the risk of fatal bleeding, and only a slight increase was observed in the incidence of intracranial haemorrhage compared to ASA monotherapy. The number of fatal bleeding events observed was low: 11 (0.3%) with ticagrelor 60 mg and 12 (0.3%) with ASA monotherapy. The increased risk of TIMI-defined major bleeding with ticagrelor 60 mg was primarily due to a higher incidence of other TIMI-defined major bleeding events, mainly gastrointestinal adverse events.
An increased incidence of bleeding, similar to that observed for TIMI-defined major bleeding, was also seen for the categories of major or minor TIMI bleeding and major bleeding in the PLATO study, as well as for major or minor bleeding in the PLATO study (see Table 5). Discontinuation of treatment due to bleeding occurred more frequently with ticagrelor 60 mg than with ASA monotherapy (6.2% vs. 1.5%). Most of these bleeding events were of lower severity (classified as TIMI-defined bleeding requiring medical attention), such as epistaxis, bruising, and haematomas.
The bleeding profile associated with ticagrelor 60 mg was consistent across various predefined subgroups (e.g. by age, sex, body weight, race, geographic region, comorbid conditions, concomitant therapy, and medical history) for TIMI-defined major bleeding, TIMI-defined major or minor bleeding, and PLATO-defined major bleeding.
Intracranial haemorrhage (ICH):
Spontaneous intracranial haemorrhage (ICH) occurred at a similar frequency in patients receiving ticagrelor 60 mg and ASA monotherapy (n=13, 0.2% in both treatment groups). A slight increase in the frequency of traumatic and procedure-related ICH was observed in patients treated with ticagrelor 60 mg (n=15, 0.2%) compared to ASA monotherapy (n=10, 0.1%). There were 6 fatal intracranial haemorrhages in the ticagrelor 60 mg group and 5 in the ASA monotherapy group. The incidence of intracranial haemorrhage was low in both treatment groups, considering the high burden of comorbidities and cardiovascular risk factors in the studied population.
Dyspnoea
Patients treated with ticagrelor may experience dyspnoea or shortness of breath.
In the PLATO study, adverse events reported as dyspnoea (dyspnoea, dyspnoea at rest, exertional dyspnoea, paroxysmal nocturnal dyspnoea, or nocturnal dyspnoea), when combined, were reported by 13.8% of patients treated with ticagrelor and 7.8% of patients treated with clopidogrel. Dyspnoea was considered by the PLATO study investigators to be causally related to treatment in 2.2% of patients receiving ticagrelor and 0.6% receiving clopidogrel, with several cases of severe dyspnoea (0.14% with ticagrelor; 0.02% with clopidogrel) (see section 4.4). Most adverse events reported as dyspnoea were mild to moderate in intensity and typically occurred as a single episode early in treatment.
Compared to clopidogrel, patients with asthma/COPD treated with ticagrelor may have an increased risk of non-severe dyspnoea (3.29% ticagrelor vs. 0.53% clopidogrel) and severe dyspnoea (0.38% ticagrelor vs. 0.00% clopidogrel). In absolute terms, this risk is higher than in the overall PLATO study population. Caution should be exercised when using ticagrelor in patients with a history of asthma and/or COPD (see section 4.4).
Approximately 30% of dyspnoea episodes resolved within 7 days. The PLATO study included patients with a history of congestive heart failure, COPD, or asthma; these patients, as well as elderly patients, reported dyspnoea more frequently. Discontinuation of treatment due to dyspnoea occurred in 0.9% of patients in the ticagrelor group compared to 0.1% in the clopidogrel group. The increased frequency of dyspnoea episodes during ticagrelor treatment is not associated with new or worsening cardiac or pulmonary disease (see section 4.4). Ticagrelor does not affect pulmonary function tests.
In the PEGASUS study, dyspnoea was reported in 14.2% of patients receiving ticagrelor 60 mg twice daily and in 5.5% of patients receiving ASA monotherapy. As in the PLATO study, most reported cases of dyspnoea were mild to moderate in severity (see section 4.4). Patients who reported dyspnoea were generally older and more likely to have a history of dyspnoea, COPD, or asthma.
Diagnostic investigations
Increased uric acid levels: In the PLATO study, serum uric acid levels above the upper limit of normal occurred in 22% of patients receiving ticagrelor compared to 13% of patients receiving clopidogrel. Corresponding values in the PEGASUS study were 9.1%, 8.8%, and 5.5% for ticagrelor 90 mg, ticagrelor 60 mg, and placebo, respectively. Mean serum uric acid levels increased by approximately 15% in patients receiving ticagrelor compared to an increase of about 7.5% in those receiving clopidogrel. After discontinuation of treatment, a reduction in uric acid levels to approximately 7% was observed in ticagrelor-treated patients, whereas no such reduction was seen with clopidogrel. In the PEGASUS study, reversible increases in mean serum uric acid levels of 6.3% and 5.6% were observed with ticagrelor 90 mg and 60 mg, respectively, compared to a 1.5% decrease in the placebo group. In the PLATO study, the incidence of gouty arthritis was 0.2% in the ticagrelor group versus 0.1% in the clopidogrel group. Corresponding incidences of gout/gouty arthritis in the PEGASUS study were 1.6%, 1.5%, and 1.1% for ticagrelor 90 mg, ticagrelor 60 mg, and placebo, respectively.
Reporting suspected adverse reactions
It is important to report suspected adverse reactions after a medicinal product has been authorised. This enables continuous monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are required to report any suspected adverse reactions via the Department of Monitoring Adverse Drug Reactions, Office for Registration of Medicinal Products, Medical Devices and Biocidal Products
Al. Jerozolimskie 181C
PL-02 222 Warsaw
Tel.: + 48 22 49 21 301
Fax: + 48 22 49 21 309
Website: https://smz.ezdrowie.gov.pl
Suspected adverse reactions can also be reported directly to the marketing authorisation holder.
4.9 Overdose
Ticagrelor has been well tolerated following single doses of up to 900 mg. In a single ascending dose study, gastrointestinal toxicity was dose-dependent. Other clinically significant adverse effects that may occur following overdose include dyspnea and ventricular pauses (see section 4.8).
In case of overdose, the above potential adverse effects may occur and ECG monitoring should be considered.
There is currently no known antidote that reverses the effects of ticagrelor, and ticagrelor is not removed by dialysis (see section 5.2). Management of overdose should be conducted according to local standard medical practice. The expected effect of ticagrelor overdose is an increased risk of prolonged bleeding due to platelet inhibition. Platelet transfusion is unlikely to provide clinical benefit in patients experiencing bleeding (see section 4.4). If bleeding occurs, other appropriate supportive treatments should be initiated.
5. PHARMACOLOGICAL PROPERTIES
5.1 Pharmacodynamic properties
Pharmacotherapeutic group: Platelet aggregation inhibitors excluding heparin, ATC code: B01AC24
Mechanism of action
Ticagrelor Holsten contains ticagrelor, belonging to the chemical group of cyclopentyltriazolopyrimidines (CPTP). Ticagrelor is an oral, direct-acting, selective, and reversibly binding antagonist of the P2Y12 receptor, which prevents ADP-dependent activation and aggregation of platelets mediated through the P2Y12 receptor. Ticagrelor does not prevent ADP binding, but by binding to the P2Y12 receptor, it blocks ADP-induced signal transduction. Since platelets are involved in the initiation and/or progression of thrombotic complications of atherosclerosis, inhibition of platelet function has been shown to reduce the risk of cardiovascular events such as death, myocardial infarction, or stroke.
Ticagrelor also increases local concentrations of endogenous adenosine by inhibiting the equilibrative nucleoside transporter 1 (ENT-1).
Ticagrelor has been shown to enhance the following adenosine-dependent effects in healthy volunteers and patients with ACS: vasodilation (measured as increased coronary flow in healthy volunteers and ACS patients; headache), inhibition of platelet function (in human whole blood under in vitro conditions), and dyspnea. However, the relationship between the observed increase in adenosine levels and clinical outcomes (e.g., morbidity-mortality) has not been clearly established.
Pharmacodynamic action
Onset of action
In patients with stable coronary artery disease receiving acetylsalicylic acid, ticagrelor exhibits a rapid onset of pharmacological action, evidenced by a mean inhibition of platelet aggregation (IPA) of approximately 41% within 30 minutes after administration of a 180 mg loading dose, reaching a maximum IPA of 89% within 2 to 4 hours, sustained between 2 and 8 hours. In 90% of patients, the greatest degree of platelet inhibition exceeding 70% is observed within 2 hours after dosing.
Offset of action
If coronary artery bypass graft (CABG) surgery is planned, the risk of bleeding associated with ticagrelor is higher compared to clopidogrel if the drug is discontinued less than 96 hours before surgery.
Data on therapy switching
Switching therapy from clopidogrel 75 mg to ticagrelor 90 mg twice daily results in an absolute increase in IPA of 26.4%, while switching from ticagrelor to clopidogrel leads to an absolute decrease in IPA of 24.5%. Patients may switch from clopidogrel to ticagrelor without interruption of antiplatelet effect (see section 4.2).
Efficacy and safety data from clinical trials
Clinical data confirming the efficacy and safety of ticagrelor come from two phase 3 studies:
- The PLATO trial [ PLATelet Inhibition and Patient Outcomes ], in which ticagrelor was compared with clopidogrel, with both drugs administered in combination with ASA (acetylsalicylic acid) and other standard treatments;
- The PEGASUS TIMI-54 trial [ PrEvention with TicaGrelor of SecondAry Thrombotic Events in High-RiSk AcUte Coronary Syndrome Patients ], in which ticagrelor in combination with ASA was compared with ASA monotherapy.
PLATO study (acute coronary syndromes)
The PLATO study included 18,624 patients with acute coronary syndrome who presented within 24 hours of symptom onset of unstable angina (UA), non-ST-segment elevation myocardial infarction (NSTEMI), or ST-segment elevation myocardial infarction (STEMI), and who were initially managed medically, underwent percutaneous coronary intervention (PCI), or underwent CABG.
Clinical efficacy
In combination with daily ASA, ticagrelor 90 mg twice daily demonstrated superiority over clopidogrel 75 mg once daily in preventing the composite primary endpoint (cardiovascular death, myocardial infarction, or stroke), with the difference primarily driven by reductions in cardiovascular deaths and myocardial infarctions. Patients received either clopidogrel 300 mg loading dose (600 mg allowed in patients undergoing PCI) or ticagrelor 180 mg loading dose.
This benefit was evident early (absolute risk reduction [ARR] 0.6% and relative risk reduction [RRR] 12% at day 30), and efficacy was maintained throughout the 12-month period, achieving an ARR of 1.9% over one year and RRR of 16%. These results indicate that the appropriate duration of treatment with ticagrelor 90 mg twice daily is 12 months (see section 4.2).
Treating 54 patients with ACS with ticagrelor instead of clopidogrel prevents one cardiovascular event; treating 91 patients prevents one cardiovascular death (see Figure 1 and Table 4).
The superior outcomes with ticagrelor compared to clopidogrel were consistently observed across multiple patient subgroups, including body weight, sex, history of diabetes, transient ischemic attacks, or non-hemorrhagic stroke or revascularization; concomitant use of heparin, GpIIb/IIIa inhibitors, and proton pump inhibitors (see section 4.5); final clinical diagnosis (STEMI, NSTEMI, or UA); and planned management strategy at randomization (invasive or conservative).
There was a small but significant regional variation in treatment effect, with the hazard ratio (HR) for the primary endpoint indicating benefit of ticagrelor globally, except in North America, which represented approximately 10% of the overall study population, where the HR favored clopidogrel (interaction p=0.045).
Factorial analyses suggest a possible association with ASA dose, indicating reduced efficacy of ticagrelor with increasing ASA doses. Maintenance doses of ASA used in combination with ticagrelor should be 75–150 mg (see sections 4.2 and 4.4).
Figure 1 shows the estimated risk of first occurrence of any component of the composite primary efficacy endpoint.
Figure 1 – Analysis of the primary composite clinical endpoint of cardiovascular death, myocardial infarction, and stroke (PLATO)
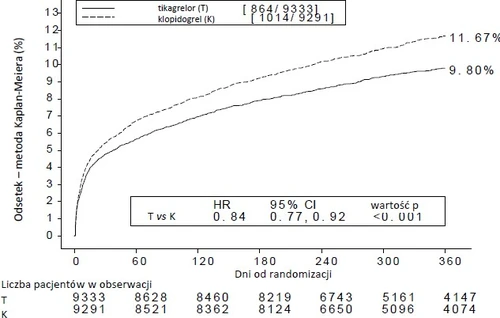
Ticagrelor reduced the incidence of the primary composite endpoint compared to clopidogrel in both the UA/NSTEMI and STEMI patient groups (Table 4). Therefore, the medicinal product Ticagrelor Holsten 90 mg twice daily in combination with low-dose ASA can be used in patients with ACS (unstable angina, non-ST-segment elevation myocardial infarction [NSTEMI], or ST-segment elevation myocardial infarction [STEMI]), including those managed medically and those undergoing percutaneous coronary intervention (PCI) or coronary artery bypass grafting (CABG).
Table 4 – Analysis of primary and secondary efficacy endpoints (PLATO)
| Ticagrelor 90 mg twice daily (% of patients in whom | Clopidogrel 75 mg once daily (% of patients in whom | ARRa (%/year) | RRRa (%) (95% CI) | p-value | |
| event occurred) N=9333 | event occurred) N=9291 | ||||
| CV death (cardiovascular), MI (myocardial infarction, excluding silent MI) or stroke | 8.5 | 10.0 | 1.7 | 16 (6, 25) | 0.0025 |
| Invasive treatment strategy | 8.5 | 10.0 | 1.7 | 16 (6, 25) | 0.0025 |
| Conservative treatment strategy | 11.3 | 13.2 | 2.3 | 15 (0.3, 27) | 0.0444d |
| CV death | 3.8 | 4.8 | 1.1 | 21 (9, 31) | 0.0013 |
| MI (excluding silent MI)b | 5.4 | 6.4 | 1.1 | 16 (5, 25) | 0.0045 |
| Stroke | 1.3 | 1.1 |
|
| 0.2249 |
| Death from any cause, MI (excluding silent MI) or stroke | 9.7 | 11.5 | 2.1 | 16 (8, 23) | 0.0001 |
| CV death, all MI, stroke, SRI, RI, TIA or other ATE | 13.8 | 15.7 | 2.1 | 12 (5, 19) | 0.0006 |
| Death from any cause | 4.3 | 5.4 | 1.4 | 22 (11, 31) | 0.0003d |
| Stent thrombosis | 1.2 | 1.7 | 0.6 | 32 (8, 49) | 0.0123d |
Genetic Subanalysis in the PLATO Study
Genotyping for CYP2C19 and ABCB1, performed in the PLATO study in 10,285 patients, enabled the
assessment of relationships between genotypic groups and outcomes in the PLATO study.
The superiority of ticagrelor over clopidogrel in reducing the number of major cardiovascular events was not
significantly dependent on CYP2C19 or ABCB1 genotype. As in the overall PLATO study, the total number of
PLATO-defined major bleeding events did not differ between the ticagrelor and clopidogrel groups, regardless of
CYP2C19 or ABCB1 genotype. PLATO-defined major non-CABG-related bleeding occurred more frequently in
the ticagrelor group compared with clopidogrel in patients with loss of one or more functional CYP2C19 alleles,
but was similar to the clopidogrel group in patients without loss of functional alleles.
Overall Assessment of Efficacy and Safety
The overall assessment of efficacy and safety (cardiovascular death, myocardial infarction, stroke, or PLATO-defined major bleeding) indicates that the benefits of ticagrelor efficacy compared with clopidogrel were not offset by the number of major bleeding events (ARR 1.4%, RRR 8%, HR 0.92; p=0.0257) over 12 months following an acute coronary event.
Clinical Safety
Holter Subgroup
To evaluate the occurrence of ventricular pauses and other arrhythmias during the PLATO study, investigators performed Holter monitoring in a subgroup of nearly 3,000 patients, with recordings obtained in approximately 2,000 patients during the acute phase of acute coronary syndrome and after one month. The primary observed variable was the occurrence of ventricular pauses ≥3 seconds. A higher incidence of ventricular pauses was observed in the ticagrelor group (6.0%) compared to the clopidogrel group (3.5%) during the acute phase of acute coronary syndrome; after one month, the respective rates were 2.2% and 1.6% (see section 4.4). The increased frequency of ventricular pauses during the acute phase was more pronounced in patients treated with ticagrelor who had a history of congestive heart failure (9.2% vs. 5.4% in patients without a history of congestive heart failure; for clopidogrel, 4.0% vs. 3.6%). This disparity was not observed after one month: 2.0% vs. 2.1% in ticagrelor-treated patients with or without a history of congestive heart failure, respectively; and 3.8% vs. 1.4% for clopidogrel. No adverse clinical consequences (including pacemaker implantation) associated with these abnormalities were observed in this patient group.
PEGASUS Study (Prior Myocardial Infarction)
The PEGASUS TIMI-54 study was a randomized, double-blind, placebo-controlled, parallel-group, international, multicenter, event-driven trial involving 21,162 patients, evaluating the prevention of cardiovascular events with ticagrelor at two doses (either 90 mg twice daily or 60 mg twice daily) in combination with low-dose ASA (75–150 mg), compared to ASA monotherapy, in patients with a history of myocardial infarction and additional risk factors for such events.
Patients were eligible for inclusion if they were at least 50 years old, had experienced a prior myocardial infarction (1 to 3 years before randomization), and had at least one of the following risk factors for atherosclerotic thrombotic events: age ≥65 years, pharmacologically treated diabetes, a second prior myocardial infarction, multivessel coronary artery disease, or chronic moderate renal dysfunction.
Patients were excluded if P2Y12 receptor antagonist, dipyridamole, cilostazol, or anticoagulant therapy was planned during the study period; if they had a bleeding disorder or a history of ischemic stroke, intracranial hemorrhage, central nervous system tumor, or intracranial vascular malformation; if they had gastrointestinal bleeding within the past 6 months or had undergone major surgery within the past 30 days.
Clinical Efficacy
Figure 2 – Analysis of the primary composite clinical endpoint of cardiovascular death, myocardial infarction, and stroke (PEGASUS)
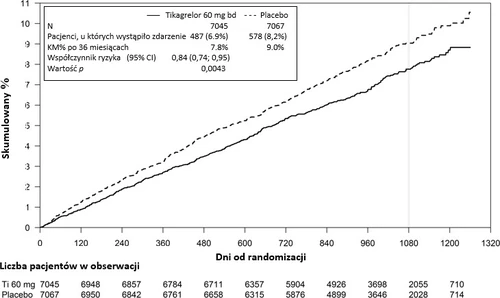
Table 5 – Analysis of primary and secondary efficacy endpoints (PEGASUS)
| Ticagrelor 60 mg twice daily + ASA N = 7045 | ASA monotherapy N = 7067 | p-value | ||||
| Characteristic | Patients with event | KM % | HR (95% CI) | Patients with event | KM % | |
| Primary endpoint | ||||||
| Composite endpoint of CV death/MI/stroke | 487 (6.9%) | 7.8% | 0.84 (0.74, 0.95) | 578 (8.2%) | 9.0% | 0.0043 (s) |
| CV death | 174 (2.5%) | 2.9% | 0.83 (0.68, 1.01) | 210 (3.0%) | 3.4% | 0.0676 |
| MI | 285 (4.0%) | 4.5% | 0.84 (0.72, 0.98) | 338 (4.8%) | 5.2% | 0.0314 |
| Stroke | 91 (1.3%) | 1.5% | 0.75 (0.57, 0.98) | 122 (1.7%) | 1.9% | 0.0337 |
| Ticagrelor 60 mg twice daily + ASA N = 7045 | ASA monotherapy N = 7067 | p-value | ||||
| Characteristic | Patients with event | KM % | HR (95% CI) | Patients with event | KM % | |
| Secondary endpoint | ||||||
| CV death | 174 (2.5%) | 2.9% | 0.83 (0.68, 1.01) | 210 (3.0%) | 3.4% | |
| Death from any cause | 289 (4.1%) | 4.7% | 0.89 (0.76, 1.04) | 326 (4.6%) | 5.2% | |
.
Both ticagrelor treatment regimens in combination with ASA (using ticagrelor doses of 60 mg twice daily and 90 mg twice daily) demonstrated superiority over ASA monotherapy in preventing cardiovascular events (composite endpoint: cardiovascular death, myocardial infarction, and stroke), with consistent treatment effect throughout the study period, resulting in 16% RRR and 1.27% ARR in the ticagrelor 60 mg group, and 15% RRR and 1.19% ARR in the ticagrelor 90 mg group.
Although the efficacy profiles of the 90 mg and 60 mg doses were similar, data indicate that the lower dose demonstrates a more favorable tolerability and safety profile with respect to the risk of bleeding and dyspnea. Therefore, only the medicinal product Ticagrelor Holsten at a dose of 60 mg twice daily in combination with ASA is recommended for the prevention of cardiovascular events (cardiovascular death, myocardial infarction, and stroke) in patients with a history of myocardial infarction and high risk of atherosclerotic thrombotic events.
Compared to ASA monotherapy, the use of ticagrelor at a dose of 60 mg twice daily led to a statistically significant reduction in the incidence of the primary composite endpoint of cardiovascular death, myocardial infarction, and stroke. Each component contributed to the reduction in the incidence of the composite endpoint (cardiovascular death 17% RRR, myocardial infarction 16% RRR, and stroke 25% RRR).
The RRR for the composite endpoint was similar from day 1 to day 360 (17% RRR) and from day 361 onwards (16% RRR). Data on the efficacy and safety of ticagrelor use beyond 3 years of extended treatment are limited. There are no data indicating benefit (lack of reduction in the primary composite endpoint of cardiovascular death, myocardial infarction, and stroke, with an increase in major bleeding) of ticagrelor 60 mg twice daily in clinically stable patients beyond 2 years after myocardial infarction or when more than one year has elapsed since discontinuation of a previous ADP inhibitor (see section 4.2).
Clinical safety
The frequency of discontinuing ticagrelor 60 mg treatment due to bleeding and dyspnea was higher in patients over 75 years of age (42%) than in younger patients (23 to 31%), with a difference compared to placebo greater than 10% (42% vs 29%) in patients >75 years of age.
Children and adolescents
In a randomized, double-blind, parallel-group Phase III study (HESTIA 3), 193 children and adolescents (aged 2 to less than 18 years) with sickle cell anemia were randomly assigned to receive either placebo or ticagrelor at doses ranging from 15 mg to 45 mg twice daily, depending on body weight.
Ticagrelor resulted in platelet inhibition with a median of 35% before dose and 56% 2 hours after dose at steady state.
Compared to placebo, no benefit of ticagrelor treatment was observed regarding the frequency of vaso-occlusive crises.
The European Medicines Agency has waived the obligation to submit results of studies with the medicinal product Ticagrelor Holsten in all pediatric subpopulations for acute coronary syndrome (ACS) and prior myocardial infarction (information on use of the product in children and adolescents is provided in section 4.2).
5.2 Pharmacokinetic Properties
Ticagrelor exhibits linear pharmacokinetics, and exposure to ticagrelor and its active metabolite (AR-C124910XX) is approximately dose-proportional over the range up to 1260 mg.
Absorption
Ticagrelor is rapidly absorbed, with a median tmax of approximately 1.5 hours. Formation of the major circulating metabolite AR-C124910XX (also active) from ticagrelor is rapid, with a median tmax of approximately 2.5 hours. After administration of a single 90 mg oral dose of ticagrelor on an empty stomach to healthy volunteers, Cmax is 529 ng/ml and AUC is 3451 ng*h/ml. For the metabolite, the parent substance-related ratios are 0.28 for Cmax and 0.42 for AUC.
The pharmacokinetics of ticagrelor and AR-C124910XX in patients with a history of myocardial infarction were essentially similar to those observed in the population of patients with ACS. According to population pharmacokinetic analysis in the PEGASUS study, median ticagrelor Cmax was 391 ng/ml and AUC was 3801 ng*h/ml at steady state after administration of ticagrelor 60 mg. For ticagrelor 90 mg, Cmax was 627 ng/ml and AUC was 6255 ng*h/ml at steady state.
The mean absolute bioavailability of ticagrelor is estimated to be 36%. Consumption of a high-fat meal results in a 21% increase in AUC of ticagrelor and a 22% decrease in Cmax of the active metabolite, but does not cause changes in Cmax of ticagrelor or AUC of the active metabolite. These minor changes are considered to have minimal clinical significance; therefore, ticagrelor may be taken with or without food. Both ticagrelor and the active metabolite are substrates of P-glycoprotein (P-gp).
Ticagrelor administered as crushed tablets mixed with water, given orally or via nasogastric tube, has bioavailability comparable to that of intact tablets in terms of AUC and Cmax for both ticagrelor and the active metabolite. Initial exposure (0.5 and 1 hour post-dose) to ticagrelor administered as a crushed tablet mixed with water was greater than that observed with administration of the intact (un-crushed) tablet, with essentially identical concentration profiles at later time points (from 2 to 48 hours).
Distribution
The volume of distribution at steady state is 87.5 L. Ticagrelor and the active metabolite are highly bound to human plasma proteins (>99.0%).
Biotransformation
CYP3A4 is the main enzyme responsible for the metabolism of ticagrelor and for the formation of the active metabolite, and its interactions with other substrates of the CYP3A isoenzyme include both induction and inhibition.
The main metabolite of ticagrelor, AR-C124910XX, is also active, as determined in in vitro studies, where it binds to the platelet ADP P2Y receptor.
Systemic exposure to the active metabolite is approximately 30–40% of exposure to ticagrelor.
Elimination
The primary route of elimination of ticagrelor is hepatic metabolism. After administration of radiolabeled ticagrelor, mean recovery of radioactivity was approximately 84% (57.8% in faeces and 26.5% in urine). Recovered ticagrelor and active metabolite in urine each accounted for less than 1% of the administered dose. The main route of elimination of the active metabolite is likely biliary excretion. The mean elimination half-life was approximately 7 hours for ticagrelor and 8.5 hours for the active metabolite.
Special Patient Groups
Elderly
In pharmacokinetic analyses across populations, increased exposure to ticagrelor (approximately 25% higher for Cmax and AUC) and to the active metabolite was observed in elderly subjects (≥75 years) with ACS compared to younger patients. These differences are considered not clinically significant (see section 4.2).
Children and Adolescents
Limited data are available in children and adolescents with sickle cell anaemia (see sections 4.2 and 5.1).
In the HESTIA 3 study, patients aged from 2 to less than 18 years, weighing ≥12 to ≤24 kg, >24 to ≤48 kg, and >48 kg, received ticagrelor as 15 mg orodispersible tablets for children at doses of 15, 30, and 45 mg twice daily, respectively. Population pharmacokinetic analysis showed that mean AUC ranged from 1095 ng*h/ml to 1458 ng*h/ml, and mean Cmax ranged from 143 ng/ml to 206 ng/ml at steady state.
Gender
Higher exposure to ticagrelor and to the active metabolite was observed in women compared to men. These differences are considered not clinically significant.
Renal Impairment
In patients with severe renal impairment (creatinine clearance <30 ml/min), exposure to ticagrelor was approximately 20% lower and exposure to the active metabolite was approximately 17% higher than in patients with normal renal function.
In patients with end-stage renal disease undergoing haemodialysis, AUC and Cmax values of ticagrelor at a dose of 90 mg administered on a non-dialysis day were 38% and 51% higher, respectively, than in patients with normal renal function. A similar increase in exposure was observed when ticagrelor was administered immediately before dialysis (49% and 61%, respectively), indicating that ticagrelor is not dialysed. Exposure to the active metabolite increased to a lesser extent (AUC by 13–14% and Cmax by 17–36%). The antiplatelet effect of ticagrelor was independent of dialysis in patients with end-stage renal disease and was similar to that in patients with normal renal function (see section 4.2).
Hepatic Impairment
Cmax and AUC of ticagrelor were 12% and 23% higher, respectively, in patients with mild hepatic impairment compared to matched healthy subjects, but the antiplatelet effect of ticagrelor was similar in both groups. Dose adjustment is not necessary in patients with moderate hepatic impairment. Studies on the use of ticagrelor in patients with severe hepatic impairment have not been conducted, and information on its pharmacokinetics in patients with moderate hepatic impairment is not available. In patients with baseline moderate or severe elevations in one or two liver function tests, plasma concentrations of ticagrelor were on average similar or slightly higher than in patients without baseline elevations in these parameters. Dose adjustment is not required in patients with moderate hepatic impairment (see sections 4.2 and 4.4).
Racial Differences
Patients of Asian origin show a 39% higher mean bioavailability compared to Caucasian patients. In patients identifying their race as Black, ticagrelor bioavailability is 18% lower than in Caucasian patients. In clinical pharmacology studies among Japanese subjects, exposure to ticagrelor (Cmax and AUC) was approximately 40% higher (and 20% higher after adjustment for body weight) compared to Caucasian subjects. Exposure in patients identifying their race as Latino was similar to that in Caucasian patients.
5.3 Preclinical safety data
Preclinical data from conventional pharmacological studies of ticagrelor and its major metabolite regarding safety in pharmacotherapy, as well as single and repeated dose toxicity studies and potential genotoxicity, revealed no unacceptable risk of adverse effects in humans.
Gastrointestinal irritation was observed in several animal species exposed under clinically relevant conditions (see section 4.8).
In female rats administered high doses of ticagrelor, an increased incidence of uterine tumors (adenocarcinomas) and an increased incidence of liver adenomas were observed. The mechanism underlying the development of uterine tumors in rats likely involves disruption of hormonal balance, which may lead to tumor formation in rats. The mechanism of liver adenoma formation is likely rodent-specific induction of hepatic enzyme activity. Therefore, these findings of carcinogenicity are considered unlikely to be relevant to humans.
In rats, minor developmental abnormalities were observed following administration to pregnant females at toxic doses (safety margin 5.1). In rabbit fetuses, minor delays in liver maturation and skeletal development were observed when pregnant females were administered high doses without signs of maternal toxicity (safety margin 4.5).
Studies in rats and rabbits showed toxic effects on reproduction, including slight reduction in body weight gain of pregnant females, reduced neonatal survival, lower birth weight, and delayed growth.
Ticagrelor caused irregular cycles (mostly prolonged) in female rats, but did not affect overall fertility in male and female rats. Pharmacokinetic studies using radiolabeled ticagrelor demonstrated that both the active substance and its metabolites are excreted into rat milk (see section 4.6).
6. PHARMACEUTICAL DATA
6.1 List of excipients
Tablet core
Hypromellose (E464)
Mannitol (E421)
Microcrystalline cellulose (E460)
Sodium carboxymethyl starch
Magnesium stearate (E470b)
Tablet coating
Hypromellose (E464)
Titanium dioxide (E171)
Macrogol 400 (E1521)
Talc (E553b)
Iron oxide red (E172)
6.2 Pharmaceutical incompatibilities
Not applicable.
6.3 Shelf life
3 years
6.4 Special precautions during storage
This medicinal product does not require storage at a specific temperature; store in the original packaging to protect from light.
6.5 Type and contents of the container
Transparent blisters of PVC/PVDC/Aluminium and/or transparent blisters of
PVC/PE/PVDC/Aluminium in cardboard boxes.
Blisters (with or without sun/moon symbols) in cardboard boxes containing 14, 15, 20, 28, 30,
56, 60, 90, 98, 100, 168, 195, 196 and 200 film-coated tablets.
Not all pack sizes may be marketed.
6.6 Special precautions for disposal
Any unused residues of the medicinal product or its waste must be disposed of in accordance with local regulations.
7. MARKETING AUTHORISATION HOLDER
Holsten Pharma GmbH
Hahnstraße 31-35
60528 Frankfurt am Main
Germany
8. MARKETING AUTHORISATION NUMBERS
Authorisation number:
9. DATE OF FIRST AUTHORISATION OR RENEWAL OF THE AUTHORISATION
AND DATE OF RENEWAL OF THE AUTHORISATION
Date of first authorisation:
10. DATE OF ADOPTION OR PARTIAL CHANGE OF THE TEXT
SUMMARY OF PRODUCT CHARACTERISTICS