Cluvot 250 j.m.
Polonia
Contenido
- Folleto incluido en el envase: información para el paciente
- 1. Qué es Cluvot y para qué se utiliza
- 2. Información importante antes de utilizar Cluvot
- 3. Cómo utilizar Cluvot
- 4. Posibles efectos adversos
- 5. Cómo conservar Cluvot
- 6. Contenido del envase y otra información
- Información destinada exclusivamente al personal médico profesional:
Folleto incluido en el envase: información para el paciente
Cluvot 250 u.i.
polvo y disolvente para solución inyectable / perfusión
Factor XIII de la sangre humano
Lea todo el prospecto detenidamente antes de empezar a usar este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede volver a consultarlo.
- Si tiene alguna duda, debe consultar a su médico o farmacéutico.
- Este medicamento ha sido recetado exclusivamente para usted. No debe dárselo a otras personas. Este medicamento podría perjudicar a terceros, aunque sus síntomas sean iguales.
- Si experimenta cualquier efecto adverso, incluyendo aquellos no mencionados en este prospecto, debe informar inmediatamente a su médico o farmacéutico. Véase la sección 4.
Índice del prospecto
- Qué es Cluvot y para qué se utiliza
- Información importante antes de la utilización de Cluvot
- Cómo utilizar Cluvot
- Posibles efectos adversos
- Cómo conservar Cluvot
- Contenido del envase y otra información
1. Qué es Cluvot y para qué se utiliza
Qué es Cluvot
Cluvot es un medicamento que se presenta en forma de polvo blanco y disolvente. La solución preparada debe administrarse mediante inyección intravenosa.
Cluvot es el factor XIII de la coagulación sanguínea humano (F XIII) obtenido a partir del plasma humano (la parte líquida de la sangre) y desempeña un papel importante en el proceso de hemostasia (detención de la hemorragia).
Para qué se utiliza Cluvot
Cluvot está indicado para su uso en adultos, niños y adolescentes:
- para el tratamiento profiláctico de la deficiencia congénita del factor XIII, y
- para el manejo perioperatorio con el fin de tratar hemorragias durante intervenciones quirúrgicas en pacientes con deficiencia congénita del factor XIII.
2. Información importante antes de utilizar Cluvot
Esta sección del prospecto contiene información que debe tenerse en cuenta antes de comenzar la administración del medicamento Cluvot.
Cuándo no debe utilizarse Cluvot:
- si el paciente tiene alergia a la sustancia activa o a cualquiera de los demás componentes de este medicamento (indicados en el apartado 6). Debe informar al médico si existe hipersensibilidad a cualquier medicamento o alimento.
Advertencias y precauciones:
- si en el pasado se han presentado reacciones alérgicas tras la administración del factor XIII de la coagulación sanguínea. Como medida profiláctica, el médico puede decidir administrar antihistamínicos y corticosteroides.
- si aparecen síntomas alérgicos o reacciones de tipo anafiláctico (los síntomas alérgicos graves pueden provocar dificultad respiratoria importante o mareos). La administración de Cluvot debe interrumpirse inmediatamente (es decir, detener la inyección o infusión). En caso de shock, debe iniciarse un tratamiento conforme a los estándares médicos vigentes.
- si existe una trombosis reciente (coágulo sanguíneo). El paciente debe ser cuidadosamente vigilado debido al efecto estabilizador de la fibrina del factor XIII.
- la formación de inhibidores (anticuerpos neutralizantes) es una complicación conocida del tratamiento y significa que el tratamiento deja de ser eficaz. Si no se logra controlar la hemorragia con Cluvot, debe informarse inmediatamente al médico. El paciente debe ser monitorizado cuidadosamente para detectar el desarrollo (aparición) de inhibidores.
El médico debe evaluar siempre los beneficios del tratamiento frente al riesgo de posibles complicaciones.
Seguridad antiviral
Cuando los medicamentos se fabrican a partir de sangre u plasma humano, se adoptan diversas medidas para proteger al paciente frente a la transmisión de agentes infecciosos. Estas medidas incluyen:
- la selección adecuada de donantes de sangre y plasma para excluir el riesgo de transmisión de agentes infecciosos;
- el análisis de cada donación y de los lotes de plasma en busca de marcadores de virus/infecciones;
- la inclusión en el proceso de fabricación de productos derivados de sangre o plasma de etapas que pueden inactivar o eliminar virus.
A pesar de la aplicación de los métodos mencionados anteriormente, no puede excluirse por completo la posibilidad de transmisión de agentes infecciosos tras la administración de un medicamento obtenido de sangre o plasma humano. Este riesgo incluye también virus desconocidos o recientemente descubiertos y otros agentes infecciosos.
Las medidas de protección adoptadas son eficaces frente a virus envueltos, como el virus de la inmunodeficiencia humana (VIH, causante del SIDA), el virus de la hepatitis B (VHB, causante de la hepatitis B), el virus de la hepatitis C (VHC, causante de la hepatitis C), así como frente a virus no envueltos, como el virus de la hepatitis A (VHA, causante de la hepatitis A) y el parvovirus B19.
En pacientes que reciben frecuentemente productos derivados del plasma humano, debe considerarse la vacunación frente a la hepatitis viral A y B.
Se recomienda que cada vez que se administre Cluvot, se registre en la documentación clínica la fecha de administración, el número de lote y el volumen infundido.
Cluvot y otros medicamentos
- Debe informar al médico o farmacéutico si está tomando, ha tomado recientemente o podría tener que tomar cualquier otro medicamento, incluidos aquellos disponibles sin receta médica.
- No se conocen interacciones entre el concentrado del factor de coagulación sanguínea FXIII y otros productos medicinales.
- Cluvot no debe mezclarse con otros productos medicinales, disolventes ni diluyentes, salvo los indicados en el apartado 6, y debe administrarse mediante conjuntos de perfusión separados.
Embarazo y lactancia
- Si la paciente está embarazada o en periodo de lactancia, sospecha que podría estar embarazada o tiene previsto tener un hijo, debe consultar con su médico o farmacéutico antes de utilizar este medicamento.
- Los datos limitados sobre el uso clínico de Cluvot durante el embarazo no han mostrado efectos negativos sobre el embarazo ni sobre el desarrollo embrionario y posnatal. Si fuera necesario, puede considerarse el uso de Cluvot durante el embarazo.
- No se conoce el grado de paso de Cluvot a la leche materna, pero dada su elevada masa molecular, es muy baja la probabilidad de que se excrete en la leche materna, y debido a su naturaleza proteica, la absorción de partículas intactas por el recién nacido es asimismo poco probable. Por tanto, Cluvot puede utilizarse durante la lactancia.
- No existen datos sobre el efecto de Cluvot sobre la fertilidad.
Conducción de vehículos y uso de máquinas
No se han realizado estudios sobre el efecto de este medicamento sobre la capacidad para conducir vehículos o manejar maquinaria.
Información importante sobre algunos componentes de Cluvot
Cluvot contiene sodio.
Debe tenerse en cuenta que Cluvot contiene sodio. Esto es importante en pacientes sometidos a una dieta baja en sodio. Cluvot contiene entre 124,4 y 195,4 mg (5,41 a 8,50 mmol) de sodio por dosis (40 UI/kg de peso corporal – para un peso corporal medio de 70 kg), cuando se administra la dosis recomendada (2800 UI = 44,8 ml).
3. Cómo utilizar Cluvot
- Cluvot generalmente es administrado por un médico.
- Cluvot está indicado exclusivamente para administración intravenosa.
Dosificación
El médico deberá calcular la dosis adecuada y decidirá la frecuencia con la que debe administrarse Cluvot al paciente, teniendo en cuenta el progreso del tratamiento.
Para recomendaciones más detalladas, véase el apartado: „Información destinada únicamente al personal médico o de salud”.
Sobredosis
No se han notificado casos de sobredosis y no se esperan cuando el medicamento es administrado por personal médico.
4. Posibles efectos adversos
Como todos los medicamentos, este medicamento puede producir efectos adversos, aunque no siempre ocurren en todas las personas.
Los siguientes efectos adversos se han observado raramente (ocurrieron en más de 1 de cada 10 000 pacientes
y menos de 1 de cada 1 000):
- Reacciones alérgicas tales como urticaria generalizada (habón con picor en la piel), erupción cutánea, descenso de la presión arterial (que puede provocar sensación de desmayo o mareo) y dificultad para respirar.
- Aumento de la temperatura corporal.
Los siguientes efectos adversos se han observado muy raramente (ocurrieron en menos de 1 de cada 10 000 pacientes):
- Formación de inhibidores del factor FXIII.
Si aparecen reacciones alérgicas, la administración del medicamento Cluvot debe interrumpirse inmediatamente
y debe iniciarse el tratamiento adecuado. Deben aplicarse los estándares médicos vigentes para el tratamiento del shock.
Efectos adversos en niños y adolescentes
Se espera que los efectos adversos en niños sean los mismos que en adultos.
Notificación de efectos adversos
Si aparecen cualquier síntoma adverso, incluyendo aquellos no mencionados en este prospecto, debe informarlos al médico o farmacéutico. Los efectos adversos pueden notificarse directamente al Departamento de Vigilancia de Reacciones Adversas a Medicamentos de la Oficina de Registro de Productos Medicinales, Productos Médicos y Productos Biocidas
Al. Jerozolimskie 181C,
02-222 Varsovia
Tel.: + 48 22 49 21 301
Fax: + 48 22 49 21 309
Página web: https://smz.ezdrowie.gov.pl
Los efectos adversos también pueden notificarse al titular de la autorización.
Gracias a la notificación de efectos adversos, se puede recopilar más información sobre la seguridad del uso de este medicamento.
5. Cómo conservar Cluvot
- Conservar en el refrigerador (2 ⁰C - 8 ⁰C).
- No congelar.
- Conservar en el envase exterior para protegerlo de la luz.
- Cluvot no contiene conservantes. El medicamento debe utilizarse inmediatamente después de la reconstrucción. Si no se utiliza inmediatamente, el tiempo de conservación a temperatura ambiente no debe superar las 4 horas. No conservar en el refrigerador ni congelar la solución después de la reconstrucción.
- El medicamento debe guardarse en un lugar fuera de la vista y del alcance de los niños.
- No utilizar Cluvot después de la fecha de caducidad indicada en la etiqueta y en el envase exterior tras la abreviatura EXP.
- El número de lote del medicamento aparece en el envase exterior y en la etiqueta, tras la abreviatura: Lot.
6. Contenido del envase y otra información
Qué contiene Cluvot
Principio activo:
Concentrado del factor XIII de la coagulación del plasma humano (FXIII) que contiene 250 UI por vial.
Excipientes:
Albúmina humana, glucosa monohidratada, cloruro de sodio, hidróxido de sodio (en pequeñas cantidades para ajustar el pH).
Disolvente: Agua para preparaciones inyectables
Aspecto del medicamento y contenido del envase
Cluvot se presenta en forma de un polvo blanco y un disolvente adjunto, que es agua para preparaciones inyectables.
La solución reconstituida debe ser incolora, transparente o ligeramente opalescente. Vista a la luz, no debe estar turbia ni contener partículas ni residuos (grumos).
Tamaños de envases
Un envase de 250 UI contiene:
1 vial con polvo
1 vial con 4 ml de agua para preparaciones inyectables
1 sistema de transferencia 20/20 con filtro (Mix2Vial)
Juego de administración (envase interior):
1 jeringa desechable de 5 ml
1 juego de punción
2 torundas impregnadas con alcohol
1 esparadrapo no estéril
Titular del permiso de comercialización y Fabricante
CSL Behring GmbH
Emil-von-Behring-Strasse 76
35041 Marburg
Alemania
Información destinada exclusivamente al personal médico profesional:
Dosificación
1 ml equivale aproximadamente a 62,5 U.I., y 100 U.I. equivalen respectivamente a 1,6 ml.
Importante:
La cantidad necesaria para la administración y la frecuencia de administración deben adaptarse siempre a la eficacia clínica en cada paciente individual.
Dosificación
La dosificación debe ajustarse individualmente según el peso corporal, los resultados de laboratorio y el estado clínico del paciente.
Esquema habitual de dosificación en profilaxis
Dosis inicial
- 40 unidades internacionales (U.I.) por kilogramo de peso corporal.
- La velocidad de infusión no debe superar los 4 ml por minuto.
Dosis siguientes
- La dosificación debe determinarse en función del nivel actual de actividad del FXIII, administrando las dosis cada 28 días (4 semanas) con el fin de mantener una actividad mínima del FXIII en torno al 5-20%.
- Se recomienda un ajuste de la dosis de ± 5 U.I. por kg de peso corporal, calculado en función del nivel mínimo de actividad del FXIII indicado en la tabla nº 1 y del estado clínico del paciente.
- El ajuste de la dosis debe realizarse basándose en una prueba específica y sensible para la determinación del nivel de FXIII. A continuación, en la tabla nº 1, se muestran ejemplos de ajuste de dosis utilizando la prueba estándar de actividad Berichrom.
Tabla 1: Ajuste de la dosificación utilizando la prueba de actividad Berichrom
| Nivel mínimo de actividad del factor XIII (%) | Ajuste de la dosis |
| Un nivel mínimo < 5% | Aumentar en 5 unidades por kg. |
| Nivel mínimo entre 5% y 20% | Sin cambios |
| Dos niveles mínimos > 20% | Disminuir en 5 unidades por kg. |
| Un nivel mínimo > 25% | Disminuir en 5 unidades por kg. |
La actividad expresada en unidades se determina mediante la prueba de actividad Berichrom, referida al
Estándar Internacional actual para el Factor XIII de Coagulación Sanguínea en plasma. Por lo tanto,
una unidad corresponde a una Unidad Internacional.
Profilaxis preoperatoria.
Después de la última dosis administrada en la profilaxis rutinaria, en caso de intervención quirúrgica programada:
- Entre el día 21 y el 28 desde la última dosis: se debe administrar al paciente una dosis completa inmediatamente antes de la intervención quirúrgica, y la siguiente dosis profiláctica debe administrarse 28 días después.
- Entre el día 8 y el 21 desde la última dosis: puede administrarse una dosis adicional (completa o parcial) antes de la intervención quirúrgica. La dosis debe ajustarse según el nivel de actividad del FXIII del paciente, su estado clínico y debe establecerse de acuerdo con la semivida del producto medicinal Cluvot.
- Dentro de los 7 días siguientes a la última dosis: puede no ser necesaria una administración adicional.
El ajuste de la dosis puede diferir de la dosis recomendada y debe individualizarse según el nivel de actividad del Factor XIII y el estado clínico del paciente. Todos los pacientes deben ser estrechamente monitorizados durante y después de la intervención quirúrgica.
Por lo tanto, se recomienda monitorizar el aumento del nivel de actividad del Factor XIII mediante una prueba específica del factor XIII. En el caso de intervenciones quirúrgicas graves y hemorragias importantes, el objetivo debe ser alcanzar valores cercanos a los normales (personas sanas: 70%-140%).
Niños y adolescentes
La dosis y la vía de administración en niños y adolescentes se basan en el peso corporal y, por lo tanto, generalmente no difieren de las indicaciones para adultos. La dosis y/o la frecuencia de administración para cada paciente deben ajustarse siempre según la eficacia clínica y el nivel de actividad del FXIII.
Pacientes de edad avanzada
La dosis y la vía de administración en pacientes de edad avanzada (> 65 años) no han sido documentadas en estudios clínicos.
Vía de administración
Instrucciones generales
La solución debe ser transparente o ligeramente opalescente. Tras la filtración/la extracción del contenido del vial (ver más abajo), el producto reconstituido debe examinarse visualmente antes de la administración; debe comprobarse la ausencia de partículas extrañas y cualquier cambio de color.
No utilizar soluciones turbias ni aquellas que contengan flóculos o partículas.
La reconstitución y la extracción del vial deben realizarse en condiciones asépticas.
Reconstitución
Llevar el disolvente a temperatura ambiente. Retirar las tapas de plástico de los viales que contienen el polvo y el disolvente, y limpiar los tapones con una solución aséptica. Tras su secado, abrir el sistema que contiene el conector Mix2Vial.
1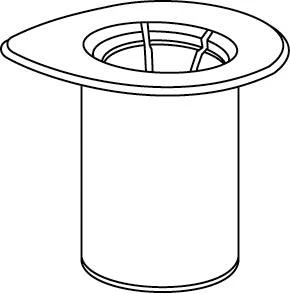 |
|
2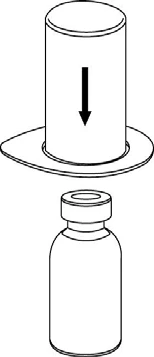 |
|
3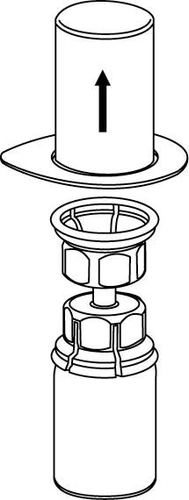 |
|
4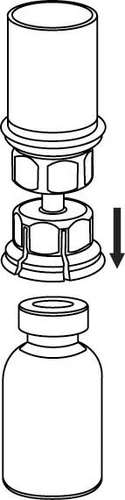 |
|
5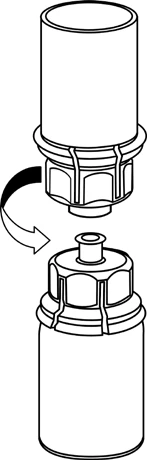 |
|
6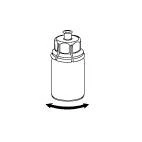 |
|
7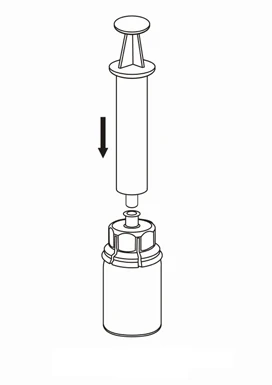 |
|
Ingesta y forma de administración
8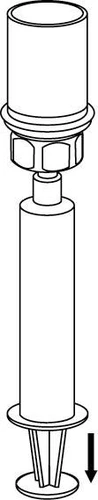 |
|
9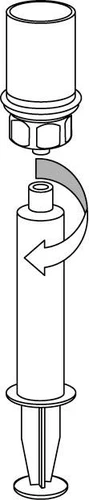 |
|
Se debe tener cuidado para que la sangre no entre en la jeringa rellena con el medicamento, ya que existe el riesgo de que la sangre pueda coagularse en la jeringa y de que se administren al paciente coágulos de fibrina.
La solución, tras la reconstitución, debe administrarse mediante un conjunto separado para inyecciones/infusiones (suministrado con el producto), mediante una inyección intravenosa lenta, a una velocidad que no supere los 4 ml por minuto.
Cualquier medicamento no utilizado y sus residuos deben eliminarse de acuerdo con los requisitos locales.