Prothromplex total
UkraineTable of Contents
INSTRUCTIONS for medical use of the medicinal product PROTHROMPLEX TOTAL (PROTHROMPLEX TOTAL)
Composition:
active substance: human prothrombin complex;
1 vial of powder contains:
| Active substance |
Per vial [IU] |
After reconstitution in 17 ml of sterile water for injection [IU/ml] |
|
| Human prothrombin complex: human blood coagulation factor II human blood coagulation factor VII human blood coagulation factor IX human blood coagulation factor X |
375–708 417 500 500 |
22.5–42.5 25 30 30 |
|
| Each vial contains at least 333 IU of protein C, which has been co-purified with coagulation factors. |
|||
Excipients: sodium citrate, sodium chloride, sodium heparin, antithrombin III;
Solvent: water for injections – 17 ml.
The total protein content per vial is 250–625 mg. The specific activity of the medicinal product is at least 0.6 IU/mg, expressed as factor IX activity.
Factor IX activity (IU) was determined using the one-stage clotting assay described in the European Pharmacopoeia, calibrated against the World Health Organization (WHO) International Standard for factor IX concentrates.
Factor II, factor VII, and factor X activity (IU) were determined using the chromogenic assay described in the European Pharmacopoeia, calibrated against the World Health Organization (WHO) International Standards for factor II, factor VII, and factor X concentrates.
Protein C activity (IU) was determined using the chromogenic assay described in the European Pharmacopoeia, calibrated against the World Health Organization (WHO) International Standard for protein C concentrates.
Excipients with known effect: the medicinal product contains up to 68 mg of sodium per vial. In addition, each vial of the medicinal product contains sodium heparin (up to 0.5 IU per IU of factor IX) (see section "Special precautions for use").
Pharmaceutical form. Powder and solvent for solution for injection.
Key physicochemical properties:
Powder: lyophilized powder or compact dry substance, white to light yellow;
Solvent: sterile water for injections.
After reconstitution, the pH of the solution ranges from 6.5 to 7.5 and osmolality is not less than 240 mOsm/kg. The solution is clear or slightly opalescent.
Pharmacotherapeutic group. Antihemorrhagics. Coagulation factors. Combination of coagulation factors IX, II, VII, and X. ATC code B02BD01.
Pharmacological properties
Pharmacodynamics
Blood clotting factors II, VII, IX, and X, which are synthesized in the liver with the help of vitamin K, are commonly referred to as the prothrombin complex.
Factor VII is a zymogen of the active serine protease factor VIIa, which initiates the extrinsic pathway of blood coagulation. The tissue factor/factor VIIa complex activates coagulation factors X and IX, resulting in the formation of factors IXa and Xa. Further activation of the coagulation cascade leads to the conversion of prothrombin (factor II) into thrombin. Thrombin converts fibrinogen into fibrin, leading to clot formation. Normal thrombin generation is also critically important for platelet function as part of primary hemostasis.
Isolated severe factor VII deficiency leads to reduced thrombin generation and a tendency to bleeding due to impaired fibrin formation and defective primary hemostasis. Isolated factor IX deficiency is one form of classical hemophilia (hemophilia B). Isolated deficiencies of factors II or X are very rare, but in severe forms they cause a bleeding tendency similar to that observed in classical hemophilia.
Acquired deficiency of vitamin K-dependent blood clotting factors occurs during treatment with vitamin K antagonists. If the deficiency becomes severe, it may lead to a tendency to severe bleeding, predominantly retroperitoneal or cerebral, rather than joint or muscle bleeding. Severe liver failure also leads to a marked reduction in vitamin K-dependent clotting factors and predisposes to clinical bleeding, which, however, is often complex due to concomitant chronic intravascular coagulation, low platelet counts, deficiency of coagulation inhibitors, and impaired fibrinolysis.
Administration of human prothrombin complex concentrates provides an increase in plasma levels of vitamin K-dependent blood clotting factors and may temporarily correct coagulation abnormalities in patients with deficiency of one or more of these factors.
Children
There are insufficient data to recommend the use of PROTHROMPLEX TOTAL in children.
Pharmacokinetics
| Coagulation factor |
Half-life period |
| Factor II |
40–60 hours |
| Factor VII |
3–5 hours |
| Factor IX |
16–30 hours |
| Factor X |
30–60 hours |
Clinical characteristics
Indications
PROTROMPLEX TOTAL is indicated for:
- treatment of bleeding and perioperative prevention of bleeding in acquired deficiency of coagulation factors of the prothrombin complex, such as deficiency caused by vitamin K antagonist therapy or vitamin K antagonist overdose, when rapid correction of the deficiency is required;
- treatment and perioperative prevention of bleeding in congenital deficiency of vitamin K-dependent blood coagulation factors, when purified specific coagulation factor concentrate is unavailable.
PROTROMPLEX TOTAL is indicated for adults. There is insufficient pediatric data to recommend the use of PROTROMPLEX TOTAL in children.
Contraindications
Hypersensitivity to the active substance or to any of the excipients listed in the section "Composition".
Known allergy to heparin or history of heparin-induced thrombocytopenia.
Interaction with other medicinal products and other forms of interaction
Human prothrombin complex preparations neutralize the effect of vitamin K antagonist therapy. Interaction studies have not been performed.
Effect on biological assays. When performing coagulation assays sensitive to heparin in patients receiving high doses of human prothrombin complex, heparin should be taken into account as a component of the administered preparation.
Special precautions for use
Traceability
To improve the traceability of biological medicinal products, the name and batch number of the administered product should be clearly documented.
Advice should be sought from a specialist experienced in the treatment of coagulation disorders.
PROTROMPLEX TOTAL should be used in patients with acquired vitamin K-dependent coagulation factor deficiency (e.g., due to vitamin K antagonist therapy) only when rapid correction of the prothrombin complex level is required, such as in cases of major bleeding or emergency surgery. In other cases, dose reduction of the vitamin K antagonist and/or administration of vitamin K is usually sufficient.
Hypercoagulability may occur in patients receiving vitamin K antagonist therapy, which may be exacerbated by infusion of human prothrombin complex concentrate.
In patients with congenital deficiency of any vitamin K-dependent coagulation factor, specific factor replacement should be used if available.
Allergic hypersensitivity reactions, including anaphylactic reactions and anaphylactic shock, have been reported with the use of PROTROMPLEX TOTAL.
If hypersensitivity reactions of allergic or anaphylactic type occur, the injection/infusion should be stopped immediately. In the event of shock, standard anti-shock measures should be implemented.
Thromboembolism, DIC, fibrinolysis
When treating patients with congenital or acquired coagulation disorders using human prothrombin complex concentrates, including PROTROMPLEX TOTAL, there is a risk of thrombosis and disseminated intravascular coagulation (DIC), especially with repeated administration.
Cases of arterial and venous thromboembolic events have been reported during the use of PROTROMPLEX TOTAL, including myocardial infarction, stroke (cerebrovascular accident), pulmonary embolism, and DIC.
The risk may be increased when treating isolated factor VII deficiency, as other vitamin K-dependent coagulation factors with longer half-lives may accumulate to levels significantly exceeding normal. Patients receiving human prothrombin complex concentrates should be closely monitored for signs and symptoms of intravascular coagulation or thrombosis. Special careful monitoring is required in the following patients due to the risk of thromboembolic complications:
- patients with a history of ischemic heart disease;
- patients with liver disease;
- patients in the pre- and post-operative period;
- neonates;
- other patients at risk of thromboembolic events or disseminated intravascular coagulation.
In each of these cases, the potential benefit of treatment should be carefully weighed against the risk of such complications.
Viral safety
Standard measures to prevent transmission of infections with medicinal products prepared from human blood or plasma include donor selection, testing of individual donations and plasma pools for specific markers of infection, and implementation of effective manufacturing steps aimed at virus inactivation/removal. However, when using medicinal products prepared from human blood or plasma, it is not possible to completely exclude the risk of transmission of infectious agents. This also applies to unknown or recently discovered viruses and other pathogenic microorganisms.
The measures taken are considered effective against enveloped viruses such as HIV, hepatitis B virus, hepatitis C virus, and also against the non-enveloped hepatitis A virus.
The effectiveness of these measures may be limited against certain non-enveloped viruses, such as parvovirus B19. Parvovirus B19 infection may lead to severe disease in pregnant women (intrauterine infection) and in patients with immunodeficiency or increased erythropoiesis (e.g., hemolytic anemia).
It is strongly recommended that the name and batch number of PROTROMPLEX TOTAL be recorded at each administration to allow traceability between the patient and the product batch.
When a medicinal product prepared from human blood or plasma is administered regularly or repeatedly, appropriate vaccination (hepatitis A and B) should be considered.
Sodium
This medicinal product contains 68 mg of sodium per vial, or 0.14 mg of sodium per international unit, equivalent to 3.4% of the maximum daily sodium intake of 2 g recommended by WHO for adults.
Heparin
Heparin may cause allergic reactions and reduce the number of blood cells, which may adversely affect the blood coagulation system. Heparin-containing medicinal products should be avoided in patients with a history of heparin-induced allergic reactions.
Children
Available data are insufficient to recommend the use of PROTROMPLEX TOTAL in children.
Use during pregnancy or breastfeeding
The effect of PROTROMPLEX TOTAL on fertility has not been established in controlled clinical trials.
The safety of using human prothrombin complex during pregnancy and breastfeeding has not been established. There is insufficient data on the use of PROTROMPLEX TOTAL in pregnant or breastfeeding women.
Animal studies are not suitable for assessing the safety of the medicinal product with regard to fertility, embryonic/fetal development, parturition, or postnatal development. Therefore, PROTROMPLEX TOTAL should be used during pregnancy and breastfeeding only if clearly indicated.
For information on the risk of parvovirus B19 infection in pregnant women, see the section "Special precautions for use".
Ability to affect reaction speed when driving or operating machinery
Studies on the effect on reaction speed when driving or operating machinery have not been conducted.
Method of Administration and Dosage
Dosage
Except for the treatment of bleeding and perioperative prophylaxis of bleeding during treatment with vitamin K antagonists, the dosage recommendations provided below are general guidelines only. Treatment must be initiated under the supervision of a physician experienced in managing coagulation disorders.
The dose and duration of replacement therapy depend on the severity of the coagulation disorder, the location and extent of bleeding, and the patient's clinical condition. Dosage and frequency of administration should be individually adjusted for each patient. The intervals between doses should be adjusted according to the different half-lives of the various coagulation factors contained in the prothrombin complex (see "Pharmacokinetics").
Individual required doses can only be determined based on regular monitoring of individual plasma levels of the coagulation factors of interest, or on general tests assessing prothrombin complex levels (e.g., Quick's prothrombin time, INR, prothrombin time), along with continuous monitoring of the patient's clinical status.
During major surgical procedures, precise monitoring of replacement therapy is required using coagulation activity assays (specific assays for individual coagulation factors and/or general tests to determine prothrombin complex levels).
Treatment of Bleeding and Perioperative Prophylaxis of Bleeding During Treatment with Vitamin K Antagonists
In cases of severe bleeding or prior to surgical procedures associated with a high risk of bleeding, the goal should be to achieve normal coagulation parameters (Quick's prothrombin time 100%, international normalized ratio (INR) 1.0). The following empirical rule applies: 1 IU of factor IX per kg of body weight increases the Quick's prothrombin time by approximately 1%. If PROTHROMPLEX TOTAL is administered based on INR measurements, the dose will depend on the initial INR value and the target INR.
The dosing regimen outlined in Table 1 should be followed.
Table 1
| Dosage regimen for PROTHROMPLEX TOTAL after determining the initial INR level |
|
| INR |
dose [IU/kg] (IU values refer to factor IX) |
| 2.0–3.9 |
25 |
| 4.0–6.0 |
35 |
| > 6.0 |
50 |
Correction of coagulation disorders caused by vitamin K antagonist administration lasts for approximately 6–8 hours. However, the effect of vitamin K when used concomitantly is usually achieved within 4–6 hours. Therefore, repeated treatment with human prothrombin complex concentrate is not always necessary if vitamin K has already been administered.
Since these recommendations are empirical and the degree and duration of activity restoration may vary, monitoring of INR during treatment is mandatory.
Treatment of bleeding and perioperative prophylaxis in congenital deficiency of any vitamin K-dependent coagulation factor when a specific coagulation factor concentrate is unavailable
Dose calculation for treatment is based on empirical data indicating that approximately 1 IU of factor IX per 1 kg of body weight increases factor IX activity in plasma by approximately 0.015 IU/mL; and 1 IU of factor VII per 1 kg of body weight increases factor VII activity in plasma by approximately 0.024 IU/mL. One IU of factor II or X per 1 kg of body weight increases factor II or X activity in blood plasma by 0.021 IU/mL.
The dose of the specific administered factor is expressed in International Units (IU) according to current WHO standards for each factor. The activity of a specific coagulation factor in blood plasma is expressed either as a percentage (relative to normal human plasma) or in International Units (relative to the international standard for specific coagulation factor concentrates).
One International Unit (IU) of coagulation factor activity is equivalent to the amount present in 1 mL of normal human plasma. For example, calculation of the required dose of factor X is based on empirical data where one International Unit (IU) of factor X per 1 kg of body weight increases factor X activity in blood plasma by 0.017 IU/mL. The required dose is determined using the following formula:
Required number of units (IU) = body weight (kg) × desired increase in factor X (IU/mL) × 60
Where 60 (mL/kg) is the reciprocal value of the calculated recovery rate.
If the individual recovery rate is known, this value should be used in the calculation.
Maximum single dose
For correction of INR values, doses exceeding 50 IU/kg are not required. If the severity of bleeding necessitates a higher dose, the treating physician must evaluate the risk/benefit ratio.
Children
The safety and efficacy of PROTHROMPLEX TOTAL in children have not been established in clinical trials.
Route of administration
Intravenous.
PROTHROMPLEX TOTAL should be administered intravenously slowly. A rate not exceeding 2 mL per minute (60 IU/min) is recommended.
Instructions for reconstitution of the medicinal product
General instructions
- Use only the reconstitution kit supplied.
- Check the expiry date and ensure that both the PROTHROMPLEX TOTAL powder and the water for injection (diluent) are at room temperature before preparation. Do not use after the expiry date stated on the label and packaging.
- Aseptic technique (cleanliness and low microbial contamination level) must be observed, and a flat surface should be used during preparation of the reconstituted solution. Wash hands and wear clean examination gloves (glove use is not mandatory).
- Warm the unopened diluent vial (sterile water for injection) to room temperature or body temperature (not exceeding 37°C).
- PROTHROMPLEX TOTAL should be reconstituted only immediately before administration. The solution is clear or slightly opalescent. Turbid solutions or solutions with sediment must be discarded.
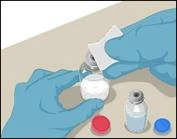
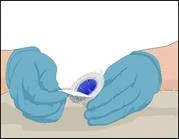
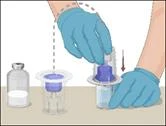
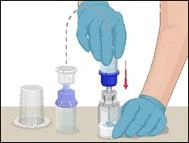
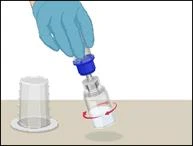
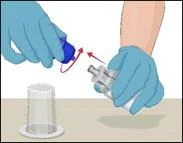
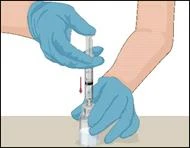
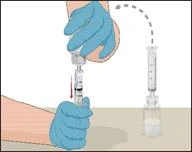
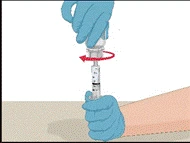
Instructions for reconstitution of the powder for solution for injection
| Steps |
Image |
|
| 1 |
|
|
| 2 |
|
|
| 3 |
|
|
| 4 |
|
|
| 5 |
|
|
| 6 |
|
|
| 7 |
|
|
| 8 |
|
|
| 9 |
|
|
| 10 |
|
|
| 11 |
|
|
| 12 |
|
|

Instructions for Use
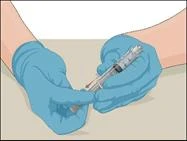
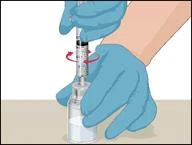
Before administration, check the prepared solution in the syringe for the presence of foreign particles or discoloration. The solution should be clear, colorless, and free from foreign particles. The Mix2Vial filter included in the system completely removes such particles. Filtration does not affect the dose calculation. The solution in the syringe must not be used if it is cloudy or contains flakes or particles after filtration.
- Attach the infusion needle to the syringe containing the prepared solution of PROTROMPLEX TOTAL. For convenience, it is recommended to use an infusion set with a "butterfly" needle. Holding the syringe with the needle pointing upwards, remove air bubbles by gently tapping the syringe and slowly and carefully expel air from the syringe and needle.
- Apply a tourniquet and prepare the injection site by thoroughly cleaning the skin with a sterile alcohol swab (or another appropriate sterile solution).
- Insert the needle into the vein and remove the tourniquet. Slowly administer the PROTROMPLEX TOTAL solution. Administer at a rate no faster than 2 mL per minute. Disconnect the empty syringe.
Note:
Do not remove the butterfly needle until the entire contents of the syringe have been administered, and do not touch the Luer port connecting to the syringe.
- Remove the needle from the vein and apply a sterile gauze pad to the injection site for several minutes.
Do not replace the protective needle cap. Dispose of the needle, syringe, empty vials of PROTROMPLEX TOTAL, and diluent into a puncture-resistant sharps container for proper disposal. Do not discard these items with household waste.
Children
The safety and efficacy of PROTROMPLEX TOTAL in children have not been established in clinical studies.
Overdose
Administration of high doses of human plasma-derived prothrombin complex concentrates has been associated with cases of myocardial infarction, disseminated intravascular coagulation, venous thrombosis, and pulmonary thromboembolism. Therefore, in the event of overdose, the risk of thromboembolic complications or disseminated intravascular coagulation is increased.
Adverse reactions
Summary of safety profile
Disorders of the immune system
Replacement therapy with human prothrombin complex concentrates, including treatment with the medicinal product PROTROMPLEX TOTAL, may lead to the development of circulating antibodies that inhibit one or more factors of the human prothrombin complex. If such inhibitors develop, the clinical manifestation of this condition will be considered as inadequate clinical response.
Allergic reactions or anaphylactic-type reactions have been frequently observed.
General disorders and administration site reactions
Increased body temperature has been frequently observed.
Vascular disorders
Following administration of human prothrombin complex, there is a risk of thromboembolic events (see section "Special precautions").
Information on exclusion of risk of transmission of infectious agents is provided in section "Special precautions".
List of adverse reactions in tabular form
Acute myocardial infarction, venous thrombosis and pyrexia, listed in the adverse reactions table below (Table 2), were reported in one clinical trial using PROTROMPLEX TOTAL during reversal of oral anticoagulation in patients (n = 61) with acquired deficiency of coagulation factors of the prothrombin complex (II, VII, IX, X). Other adverse reactions included in Table 2 are known only from post-marketing experience, and the frequency category was statistically estimated assuming that each adverse reaction could have occurred in a clinical trial involving 61 patients.
Adverse reactions that may occur during administration of the medicinal product PROTROMPLEX TOTAL are classified by system organ class according to the Medical Dictionary for Regulatory Activities (MedDRA) terminology (version 15.1). Within each frequency category, adverse reactions are listed in order of decreasing severity. Frequencies are defined as very common (≥ 1/10), common (≥ 1/100 to < 1/10), uncommon (≥ 1/1,000 to < 1/100), rare (≥ 1/10,000 to < 1/1,000), very rare (< 1/10,000), and not known (cannot be estimated from available data).
Table 2
| System Organ Class (SOC) |
Adverse Reaction |
Frequency |
| Blood and lymphatic system disorders |
Disseminated intravascular coagulation |
Common |
| Development of inhibitors to one or more factors of the prothrombin complex (factors II, VII, IX, X)* |
||
| Immune system disorders |
Anaphylactic shock |
Common |
| Anaphylactic reaction |
||
| Hypersensitivity |
||
| Nervous system disorders |
Acute cerebrovascular accident |
Common |
| Headache |
||
| Cardiac disorders |
Heart failure |
Common |
| Acute myocardial infarction** |
||
| Tachycardia |
||
| Vascular disorders |
Arterial thrombosis |
Common |
| Vein thrombosis** |
||
| Arterial hypotension |
||
| Flushing |
||
| Respiratory, thoracic and mediastinal disorders |
Pulmonary embolism |
Common |
| Dyspnea |
||
| Wheezing |
||
| Gastrointestinal disorders |
Vomiting |
Common |
| Nausea |
||
| Skin and subcutaneous tissue disorders |
Urticaria |
Common |
| Erythematous rash |
||
| Pruritus |
||
| Renal and urinary disorders |
Nephrotic syndrome |
Common |
| General disorders and administration site reactions |
Pyrexia** |
Common |
* Occurrence in patients with congenital factor deficiencies.
** Based on clinical trial data.
Adverse reactions characteristic of the class of medicinal products
Skin and subcutaneous tissue disorders: angioedema, paresthesia.
General disorders and administration site conditions: injection site reaction.
Nervous system disorders: lethargy.
Psychiatric disorders: restlessness.
Pediatric population
For information on use in children, see sections "Dosage and administration" and "Children".
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorization of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the product. Healthcare professionals and patients are encouraged to report any suspected adverse reactions and lack of efficacy via the automated pharmacovigilance information system at: https://aisf.dec.gov.ua.
Shelf life. 3 years.
Within the stated shelf life, the product may be stored at room temperature (not above 25 °C) for a single period of up to six months. The start and end dates of room temperature storage must be recorded on the packaging. After storage at room temperature, PROTROMPLEX TOTAL must not be returned to the refrigerator (2 to 8 °C); it must be used within six months or disposed of.
Chemical and physical in-use stability has been demonstrated for 3 hours at 20–25 °C after reconstitution.
From a microbiological standpoint, PROTROMPLEX TOTAL should be used immediately after reconstitution, as it contains no preservatives. If not used immediately, the user is responsible for the duration and conditions of storage prior to use after reconstitution. The reconstituted solution must not be returned to the refrigerator.
Storage conditions
Store in a refrigerator (2 to 8 °C). Do not freeze.
Store in the original packaging to protect from light.
Storage conditions after reconstitution are specified in the section "Shelf life".
Keep out of the reach of children.
Incompatibilities
This medicinal product must not be mixed with other medicinal products except those specified in the section "Dosage and administration". Only the provided reconstitution kit should be used for reconstitution.
As with all coagulation factor products, the efficacy and tolerability of this medicinal product may be compromised by mixing with other medicinal products. It is recommended to flush a common venous catheter with isotonic sodium chloride solution before and after administration of PROTROMPLEX TOTAL.
Packaging
1 vial of powder for solution for injection (500 IU) with 1 vial of solvent (water for injections, 17 ml) and a Mix2Vial filter for reconstitution, packed in a cardboard box.
Prescription status
Prescription only.
Manufacturer
Takeda Manufacturing Austria AG / Takeda Manufacturing Austria AG.
Manufacturer's address and location of operations
Industriestrasse 67, 1221 Vienna, Austria / Industriestrasse 67, 1221 Vienna, Austria.