Idelvion 250 IU powder and solvent for solution for injection
Spain
Table of Contents
Package leaflet: Information for the user
Introduction
Package leaflet: information for the user
IDELVION 250 IU, powder and solvent for solution for injection
IDELVION 500 IU, powder and solvent for solution for injection
IDELVION 1,000 IU, powder and solvent for solution for injection
IDELVION 2,000 IU, powder and solvent for solution for injection
IDELVION 3,500 IU, powder and solvent for solution for injection
albutrepenonacog alfa (recombinant coagulation factor IX)
Read all of this leaflet carefully before you start using this medicine, because it contains important information for you.
-
Keep this leaflet. You may need to read it again.
-
If you have any questions, ask your doctor, pharmacist, or nurse.
-
This medicine has been prescribed for you only. Do not pass it on to others, even if they have the same symptoms as you, because it may harm them.
-
If you experience any adverse effects, talk to your doctor, pharmacist, or nurse, even if they are possible adverse effects not listed in this leaflet. See section 4.
Contents of the leaflet :
-
What IDELVION is and what it is used for
-
What you need to know before you use IDELVION
-
How to use IDELVION
-
Possible side effects
-
How to store IDELVION
-
Contents of the pack and other information
1. What IDELVION is and what it is used for
What is IDELVION?
IDELVION is a medicine for the treatment of haemophilia that replaces the natural blood clotting factor IX. The active substance in IDELVION is albutrepenonacog alfa (a recombinant fusion protein linking coagulation factor IX to albumin [rIX-FP]).
Factor IX plays a role in blood coagulation. Patients with haemophilia B lack this factor, meaning that their blood does not clot as quickly as it should, resulting in a tendency to bleed. IDELVION works by replacing factor IX in patients with haemophilia B, enabling their blood to clot properly.
What is IDELVION used for?
IDELVION is used to prevent or stop bleeding caused by a lack of sufficient factor IX in patients of all ages with haemophilia B (also known as congenital factor IX deficiency or Christmas disease).
2. What you need to know before using IDELVION
Do not use IDELVION
- if you are allergic to the active substance (albutrepenonacog alfa) or to any of the other ingredients of this medicine (listed in section 6).
- if you are allergic to hamster proteins.
Warnings and precautions
It is strongly recommended that each time you use IDELVION, you record the product name and batch number to keep track of the products and batches you have used.
Traceability
In order to improve the traceability of biological medicines, the name and batch number of the administered medicine must be clearly documented.
Talk to your doctor, pharmacist, or nurse before starting to use IDELVION.
-
Allergic reactions (hypersensitivity) may occur. The product contains traces of hamster proteins (see also "Do not use IDELVION"). If symptoms of allergic reactions occur, you must stop treatment immediately and contact your doctor or treatment centre where you are being monitored. Your doctor should inform you about the early signs of hypersensitivity reactions. These include hives, widespread skin rash, chest tightness, difficulty breathing, low blood pressure (hypotension), and anaphylaxis (a severe allergic reaction causing serious breathing problems or dizziness).
-
Due to the risk of allergic reactions with factor IX, the initial administration of IDELVION should be performed under medical supervision to ensure access to appropriate medical care in case of allergic reactions.
-
The development of inhibitors (neutralizing antibodies) is a known complication reported during treatment with IDELVION. Inhibitors prevent the treatment from working properly. If bleeding is not controlled with IDELVION, inform your doctor immediately. You should be regularly monitored for the development of inhibitors.
-
If you have liver or heart disease or have recently undergone major surgery, please inform your doctor, as there is an increased risk of blood clotting complications.
-
If a central venous access device (CVAD) is required for the administration of IDELVION, your doctor will consider the risk of CVAD-related complications, such as local infections, bacteria in the blood (bacteraemia), and formation of blood clots in blood vessels (thrombosis) at the catheter insertion site.
Use of IDELVION with other medicines
- Inform your doctor or pharmacist if you are taking, have recently taken, or might need to take any other medicines.
Pregnancy and breastfeeding
- If you are pregnant or breastfeeding, think you may be pregnant, or are planning to become pregnant, consult your doctor or pharmacist before using this medicine.
- During pregnancy and breastfeeding, IDELVION should only be administered if clearly necessary.
Driving and using machines
IDELVION does not affect your ability to drive or operate machinery.
IDELVION contains sodium
This medicine contains up to 8.6 mg of sodium (main component of table/cooking salt) per vial. This corresponds to 0.4% of the maximum daily recommended sodium intake for an adult.
3. How to use IDELVION
Your treatment should be initiated and supervised by a physician experienced in the treatment of blood coagulation disorders. Follow your doctor's instructions exactly for using this medicine. Consult your doctor if you have any doubts.
Your doctor will calculate the dose of IDELVION you need. The amount of IDELVION required and the duration of treatment depend on:
- the severity of your condition
- the location and severity of the bleeding
- your clinical condition and clinical response
- your body weight
IDELVION is administered as an injection into a vein (intravenous, IV) after reconstitution of the powder with the solvent provided by your doctor or nurse. You or another person may also administer IDELVION as an intravenous injection, but only after receiving proper training.
If you use more IDELVION than you should
Contact your doctor immediately if you inject more IDELVION than prescribed.
If you stop treatment with IDELVION
Do not stop using IDELVION without first consulting your doctor.
Reconstitution and administration
General instructions
- The powder must be mixed with the solvent (liquid) and withdrawn from the vial while maintaining sterility (germ-free). Your doctor will show you how to prepare the solution and how to correctly withdraw the solution from the vial.
- IDELVION must not be mixed with other medicines or solvents except those mentioned in section 6.
- The solution should be transparent or slightly opalescent, yellow to colorless; that is, it may shimmer when exposed to light but must not contain any visible particles. After withdrawing or filtering the solution (see below), it should be visually inspected before use. Do not use the solution if it is cloudy or contains flakes or particles.
- Disposal of unused product and all residual materials must be carried out in accordance with local regulations and your doctor's instructions.
Reconstitution
Without opening any vials, warm the IDELVION powder and the solvent to room temperature or body temperature. This can be achieved by leaving the vials at room temperature for about one hour or by holding them in your hands for several minutes.
DO NOT expose the vials to direct heat. The vials must not be heated above body temperature (37°C).
Carefully remove the protective caps from the vials, then clean the exposed portions of the rubber stoppers with an alcohol swab. Allow the vials to dry before opening the Mix2Vial package (which contains the transfer device with filter), and then follow the instructions below.
|
|
|
|
|
|
|
|
|
Discard the solvent vial with the attached blue Mix2Vial adapter. |
|
|
|
|
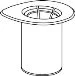 1
1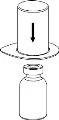 2
2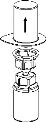 3
3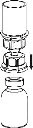 4
4 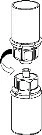 5
5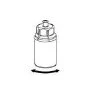 6
6 7
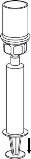
7Transfer and administration
|
|
9 |
|
 8
8Use the venipuncture kit supplied with the product and insert the needle into a vein. Allow the blood to flow until the end of the tube. Attach the syringe to the threaded locking end of the venipuncture kit. Slowly inject the reconstituted solution (at a rate comfortable for you, up to a maximum of 5 mL/min) into the vein, as instructed by your doctor. Try to prevent blood from entering the syringe containing the product.
Check whether you experience any adverse effects immediately after the injection. If you experience any adverse effect that may be related to the administration of IDELVION, the injection must be stopped (see also sections 2 and 4).
If you have any further questions about the use of this medicine, ask your doctor, pharmacist, or nurse.
4. Possible adverse effects
Like all medicines, this medicine can cause adverse effects, although not everyone will experience them.
Contact your doctor:
-
if you notice symptoms of allergic reactions (see below);
-
if you notice that the medicine stops working properly.
The following adverse effects have been observed with factor IX-containing medicines:
-
Hypersensitivity allergic-type reactions (common) may occur, including the following symptoms: erythema, skin itching (generalized urticaria), chest tightness, difficulty breathing, low blood pressure (hypotension), and anaphylaxis (a serious reaction causing severe breathing difficulty and dizziness). If this occurs, you must immediately stop administering the medicine and contact your doctor.
-
Inhibitors: the medicine stops working properly (ongoing bleeding). You may develop a neutralizing antibody (inhibitor) to factor IX (frequency not known), meaning that factor IX will no longer work effectively. If this occurs, you must immediately stop administering the medicine and contact your doctor.
The following adverse effects have been observed frequently with IDELVION (may affect up to 1 in 10 people):
- Headache
- Reactions at the injection site
- Dizziness
- Skin rash
The following adverse effect has been observed uncommonly (may affect up to 1 in 100 people):
- Eczema
Adverse effects in children and adolescents
Adverse effects in children are expected to be the same as those in adults.
Reporting of adverse effects
If you experience any adverse effect, consult your doctor, pharmacist, or nurse, even if it is a possible adverse effect not listed in this leaflet. You may also report them directly via the Spanish Pharmacovigilance System for Human Medicines: www.notificaRAM.es. By reporting adverse effects, you can help provide more information on the safety of this medicine.
5. Storage of IDELVION
-
Keep this medicine out of the sight and reach of children.
-
Do not use this medicine after the expiry date stated on the label and the carton.
-
Do not store above 25 °C.
-
Do not freeze.
-
Keep the vial in its carton to protect from light.
-
After reconstitution, the product should preferably be used immediately.
If the reconstituted product is not administered immediately, the storage times and conditions prior to use are the responsibility of the user.
6. Contents of the pack and other information
Composition of IDELVION
The active substance is:
250 IU per vial; after reconstitution with 2.5 ml of water for injections, the solution contains 100 IU/ml of albutrepenonacog alfa.
500 IU per vial; after reconstitution with 2.5 ml of water for injections, the solution contains 200 IU/ml of albutrepenonacog alfa.
1,000 IU per vial; after reconstitution with 2.5 ml of water for injections, the solution contains 400 IU/ml of albutrepenonacog alfa.
2,000 IU per vial; after reconstitution with 5 ml of water for injections, the solution contains 400 IU/ml of albutrepenonacog alfa.
3,500 IU per vial; after reconstitution with 5 ml of water for injections, the solution contains 700 IU/ml of albutrepenonacog alfa.
The other components are:
Sodium citrate, polysorbate 80, mannitol, sucrose, and hydrochloric acid (for pH adjustment).
See the last paragraph of section 2.
Solvent: water for injections
Appearance of IDELVION and contents of the pack
IDELVION is presented as a white to yellowish powder and is supplied with a solvent in the form of water for injections.
The reconstituted solution should be clear or slightly opalescent, yellow to colourless; that is, it may shimmer when exposed to light but must not contain any visible particles.
Presentations
A pack with 250, 500 or 1,000 IU containing:
1 vial of powder
1 vial with 2.5 ml of water for injections
1 transfer device with 20/20 filter
Administration set (inner box):
1 disposable 5 ml syringe
1 venous access device
2 alcohol-impregnated wipes
1 non-sterile dressing
A pack with 2,000 or 3,500 IU containing:
1 vial of powder
1 vial with 5 ml of water for injections
1 transfer device with 20/20 filter
Administration set (inner box):
1 disposable 10 ml syringe
1 venous access device
2 alcohol-impregnated wipes
1 non-sterile dressing
Only certain pack sizes may be marketed.
Marketing Authorisation Holder and Manufacturer
CSL Behring GmbH
Emil-von-Behring-Straße 76
35041 Marburg
Germany
For further information about this medicinal product, please contact the local representative of the Marketing Authorisation Holder:
Belgium/Belgium/Belgium CSL Behring NV Tel/Tel: +32 15 28 89 20 | Lithuania CentralPharma Communications UAB Tel: +370 5 243 0444 |
| Luxembourg/Luxembourg CSL Behring NV Tél/Tel: +32 15 28 89 20 |
Czech Republic CSL Behring s.r.o. Tel: +420 702 137 233 | Hungary CSL Behring Kft. Tel.: +36 1 213 4290 |
Denmark CSL Behring AB Tlf: +46 8 544 966 70 | Malta AM Mangion Ltd. Tel: +356 2397 6333 |
Germany CSL Behring GmbH Tel: +49 6190 75 84810 | Netherlands CSL Behring BV Tel: +31 85 111 96 00 |
Estonia CentralPharma Communications OÜ Tel: +3726015540 | Norway CSL Behring AB Tlf: +46 8 544 966 70 |
Greece CSL Behring ΕΠΕ Tel: +30 210 7255 660 | Austria CSL Behring GmbH Tel: +43 1 80101 1040 |
Spain CSL Behring S.A. Tel: +34 933 67 1870 | Poland CSL Behring Sp.z o.o. Tel: +48 22 213 22 65 |
France CSL Behring S.A. Tél: +33 –(0)-1 53 58 54 00 | Portugal CSL Behring Lda Tel: +351 21 782 62 30 |
Croatia Marti Farm d.o.o. Tel: +385 1 5588297 | Romania Prisum Healthcare S.R.L. Tel: +40 21 322 0171 |
Ireland CSL Behring GmbH Tel: +49 6190 75 84700 Iceland CSL Behring AB Sími: +46 8 544 966 70 | Slovenia Emmes Biopharma Global s.r.o. podružnica v Sloveniji Tel: +386 41 42 0002 Slovakia CSL Behring Slovakia s.r.o. Tel: +421 911 653 862 |
Italy CSL Behring S.p.A. Tel: +39 02 34964 200 | Finland/Sweden CSL Behring AB Puh/Tel: +46 8 544 966 70 |
Cyprus CSL Behring ΕΠΕ Tel: +30 210 7255 660 | Sweden CSL Behring AB Tel: +46 8 544 966 70 |
Latvia CentralPharma Communications SIA Tel: +371 6 7450497 | |

Date of the most recent review of this summary
Detailed information on this medicine is available on the website of the European Medicines Agency: http://www.ema.europa.eu, and on the website of the Spanish Agency of Medicines and Health Products (AEMPS) (http://www.aemps.gob.es/).
This information is intended for healthcare professionals only:
Dosage
The dose and duration of replacement therapy depend on the severity of factor IX deficiency, the location and severity of bleeding, and the patient's clinical condition.
The number of units of factor IX administered is expressed in International Units (IU), relative to the current WHO standard for products containing factor IX. Plasma factor IX activity is expressed as a percentage (relative to normal human plasma) or in International Units (relative to an international standard for plasma factor IX).
One International Unit (IU) of factor IX activity corresponds to the amount of factor IX present in 1 ml of normal human plasma.
On-demand treatment
The calculation of the required dose of factor IX is based on the empirical finding that 1 IU of factor IX per kg of body weight increases plasma factor IX activity by a mean of 1.3 IU/dl in patients ≥ 12 years of age and by 1.0 IU/dl in patients < 12 years of age. The required dose is determined using the following formula:
Required dose (IU) = body weight (kg) × desired increase in factor IX (% of normal level or IU/dl) × {reciprocal of observed recovery (IU/kg per IU/dl)}
Predicted increase in factor IX (IU/dl or % of normal level) = dose (IU) × recovery (IU/dl per IU/kg) / body weight (kg)
The dose and frequency of administration should always be adjusted according to the observed clinical efficacy in each individual case.
Patients < 12 years of age
In case of an incremental recovery of 1 IU/dl per 1 IU/kg, the dose is calculated as follows:
Required dose (IU) = body weight (kg) × desired increase in factor IX (IU/dl) × 1 dl/kg
Examples:
-
A maximum level of 50% of normal is required in a 20 kg patient with severe hemophilia B. The appropriate dose would be 20 kg × 50 IU/dl × 1 dl/kg = 1,000 IU.
-
A dose of 1,000 IU of IDELVION administered to a 25 kg patient is expected to produce a peak factor IX increase after injection of 1,000 IU / 25 kg × 1.0 (IU/dl per IU/kg) = 40 IU/dl (40% of normal level).
Patients ≥ 12 years of age
In case of an incremental recovery of 1.3 IU/dl per 1 IU/kg, the dose is calculated as follows:
Required dose (IU) = body weight (kg) × desired increase in factor IX (IU/dl) × 0.77 dl/kg
Examples:
-
A maximum level of 50% of normal is required in an 80 kg patient with severe hemophilia B. The appropriate dose would be 80 kg × 50 IU/dl × 0.77 dl/kg = 3,080 IU.
-
A dose of 2,000 IU of IDELVION administered to an 80 kg patient is expected to produce a peak factor IX increase after injection of 2,000 IU × 1.3 (IU/dl per IU/kg) / 80 kg = 32.5 IU/dl (32.5% of normal level).
In the case of the following bleeding events, factor IX activity should not fall below the established plasma activity level (in % of normal or IU/dl) during the corresponding period. The following table may be used as a dosing guide for bleeding episodes and surgery:
Degree of bleeding/ type of surgical procedure | Required factor IX level (% or IU/dl) | Dosing frequency (hours)/duration of treatment (days) |
Minor to moderate bleeding Minor or moderate hemarthrosis, muscle bleeding (except iliopsoas), or oral cavity bleeding | 30-60 | A single dose should be sufficient in most bleeds. A maintenance dose should be administered 24 - 72 hours later if additional evidence of bleeding occurs. |
Major bleeding Potentially life-threatening bleeding, deep muscle bleeding, including iliopsoas bleeding | 60-100 | Should be repeated every 24 - 72 hours during the first week, and then a maintenance dose should be administered weekly until bleeding stops and wound heals. |
Minor surgery For example, (including uncomplicated dental extractions) | 50-80 (pre- and postoperative) | A single dose should be sufficient in most minor procedures. If needed, a maintenance dose may be administered 24 - 72 hours later until bleeding stops and wound heals. |
Major surgery | 60-100 (pre- and postoperative) | Should be repeated every 24 - 72 hours during the first week, and then a maintenance dose should be administered 1 - 2 times per week until bleeding stops and wound heals. |
Prophylactic treatment
For long-term prophylaxis to prevent bleeding in patients with severe hemophilia B, the usual dose is 35 to 50 IU/kg once weekly. Patients well-controlled on a once-weekly regimen may be treated with up to 75 IU/kg every 10 or 14 days. In patients > 18 years of age, a further extension of the treatment interval may be considered.
In some cases, especially in younger patients, it may be necessary to shorten the administration intervals or use higher doses.
After a bleeding episode during prophylaxis, patients should maintain their prophylactic regimen as much as possible, with administration of 2 doses of IDELVION at least 24 hours apart, but longer intervals when considered appropriate for the patient.
Pediatric population
For long-term prophylactic treatment, the recommended dosing regimen is 35 to 50 IU/kg once weekly. For adolescents aged 12 years or older, dosage recommendations are the same as for adults (see above).
Warnings and special precautions for use
Inhibitors
Following repeated treatment with human coagulation factor IX products, patients should be monitored for the development of neutralizing antibodies (inhibitors), which should be quantified in Bethesda Units (BU) using appropriate biological assays.
Published literature has reported cases demonstrating a correlation between the development of a factor IX inhibitor and allergic reactions. Therefore, patients experiencing allergic reactions should be evaluated for the presence of an inhibitor. It should be noted that patients with factor IX inhibitors may be at increased risk of anaphylaxis upon subsequent exposure to factor IX.
Monitoring of treatment
During treatment, it is recommended to adequately monitor factor IX levels to determine the dose to be administered and the frequency of infusions. Patient responses to factor IX may vary, reflecting differences in half-life and recovery. Dose based on body weight may need to be adjusted in patients who are underweight or overweight. In the special case of major surgical procedures, accurate monitoring of replacement therapy using coagulation tests (plasma factor IX activity) is essential.
When using a one-stage coagulation assay based on activated partial thromboplastin time (aPTT) in vitro to determine factor IX activity in patient blood samples, plasma factor IX activity results may be significantly affected by the aPTT reagent and reference standard used in the assay. Measurement using a one-stage coagulation assay employing a kaolin-based aPTT reagent or an aPTT reagent with Actin FS is likely to result in underestimation of the activity level. This is particularly important when changing laboratories or reagents used in the assay.