Xolar
Ukraina
Spis treści
INSTRUKCJA DOTYCZĄCA STOSOWANIA LEKU KSOŁAR (XOLAIRÒ)
Skład:
substancja czynna: omalizumab;
1 przedwypełniona strzykawka z 0,5 ml roztworu zawiera 75 mg omalizumabu*;
substancje pomocnicze: L-argininy chlorowodorek, L-histydyny chlorowodorek monohydrat; L-histydyna, polisorbat 20, woda do wstrzykiwań;
lub
1 przedwypełniona strzykawka z 1 ml roztworu zawiera 150 mg omalizumabu*;
substancje pomocnicze: L-argininy chlorowodorek, L-histydyny chlorowodorek monohydrat; L-histydyna, polisorbat 20, woda do wstrzykiwań.
* Omalizumab – to ludzkie monoklonalne przeciwciało, wytwarzane metodą rekombinowanego DNA w komórkach jajnika chomika chińskiego (CHO).
Postać leku. Roztwór do wstrzykiwań.
Główne właściwości fizykochemiczne: roztwór od przejrzystego do lekko mętnego; od bezbarwnego do lekko żółtawobrunatnego.
Grupa farmakoterapeutyczna. Leki stosowane systemowo w chorobach obturacyjnych dróg oddechowych. Kod ATC R03D X05.
Właściwości farmakologiczne.
Farmakodynamika.
Omalizumab to humanizowany przeciwciało monoklonalne otrzymywane metodą rekombinacji DNA, które selektywnie wiąże ludzki immunoglobulinę E (IgE). Przeciwciało to jest IgG1 kappa zawierającym ludzką strukturę szkieletową z komplementarną domeną określającą pochodzącą z przeciwciała mysiego, które wiąże IgE. Leczenie Ksolarem hamuje zapalenie pośredniczone przez IgE, o czym świadczy obniżenie poziomu eozynofili w krwi i tkankach oraz zmniejszenie poziomu mediatorów zapalenia, w tym IL-4, IL-5 i IL-13, za pomocą komórek wrodzonych, adaptacyjnych i nielukocytowych.
Mechanizm działania
Omalizumab wiąże się z IgE i zapobiega jego wiązaniu się z FcεRI (receptorem o wysokiej powinności do IgE) na bazofilach i komórkach tucznych, zmniejszając w ten sposób ilość wolnego IgE zdolnego do uruchomienia kaskady alergicznej. Leczenie omalizumabem pacjentów z chorobami atopowymi prowadziło do istotnego hamowania receptora FcεRI na bazofilach.
Efekty farmakodynamiczne
Astma alergiczna
In vitro uwalnianie histaminy po stymulacji alergenem bazofilów uzyskanych od pacjentów leczonych Ksolarą było mniejsze o około 90% w porównaniu z wartością przed leczeniem.
W trakcie badań klinicznych u pacjentów z astmą alergiczną obserwowano zależne od dawki obniżenie poziomu wolnego IgE w surowicy krwi w ciągu jednej godziny po podaniu pierwszej dawki, które utrzymywało się w okresach między dawkami. Po jednym roku od zakończenia leczenia Ksolarą poziomy IgE wróciły do wartości sprzed leczenia, przy czym nie zaobserwowano reakcji odksztalnej w stosunku do poziomu IgE po wyeliminowaniu leku z organizmu.
Przewlekłe zapalenie zatok nosowych z polipami nosowymi (CRSwNP)
W badaniach klinicznych u pacjentów z CRSwNP leczenie Ksolarą prowadziło do zmniejszenia zawartości wolnego IgE w surowicy krwi (około 95%) oraz zwiększenia ogólnego poziomu IgE w surowicy krwi w podobnym stopniu, jak zaobserwowano u pacjentów z astmą alergiczną. Całkowity poziom IgE w surowicy wzrósł w wyniku tworzenia się kompleksów omalizumabu z IgE, które charakteryzują się wolniejszą szybkością wydalania w porównaniu z wolnym IgE.
Przewlekła pokrzywka samoistna (CSU)
Mechanizm działania
Omalizumab wiąże się z IgE i obniża poziomy wolnego IgE w surowicy. Następnie dochodzi do hamowania receptorów IgE (FcεRI) na komórkach. Mechanizm, w jaki sposób prowadzi to do ustąpienia objawów przewlekłej pokrzywki samoistnej, nie został w pełni wyjaśniony.
Efekty farmakodynamiczne
W badaniach klinicznych u pacjentów z CSU maksymalne hamowanie poziomu wolnego IgE obserwowano w ciągu trzech dni po pierwszym podaniu podskórnym. Po powtarzalnym podawaniu raz na 4 tygodnie poziomy wolnego IgE w surowicy krwi przed podaniem pozostawały stabilne między 12. a 24. tygodniem leczenia. Po zakończeniu przyjmowania leku Ksolarem poziomy wolnego IgE wzrastały, osiągając w ciągu 16 tygodni poziomy obserwowane przed leczeniem.
Doświadczenie kliniczne w astmie alergicznej
Dorośli i dzieci w wieku od 12 lat
Skuteczność i bezpieczeństwo stosowania Ksolara zostały zademonstrowane w trakcie 28-tygodniowego, podwójnie ślepego, placebo-kontrolowanego badania (badanie 1) z udziałem 419 pacjentów w wieku od 12 do 79 lat z ciężką astmą alergiczną, u których stwierdzono obniżoną funkcję płuc (FEV1 40–80% wartości należnej) i słabo kontrolowane objawy astmy mimo stosowania wysokich dawek kortykosteroidów wziewnych i długodziałających agonistów β2. Do badania zakwalifikowano pacjentów z wieloma przypadkami zaostrzeń astmy, których leczenie wymagało stosowania kortykosteroidów systemowych lub hospitalizacji, lub pomocy doraźnej z powodu zaostrzenia astmy oskrzelowej, mimo ciągłego leczenia wysokimi dawkami kortykosteroidów wziewnych i długodziałających agonistów β2. Ksolara lub placebo podawano podskórnie jako dodatek do terapii beklometazonu dipropionianem (lub równoważnikiem) w dawce > 1000 μg i długodziałającego agonisty β2. Dozwolone jako terapia wspomagająca były również kortykosteroidy doustne, teofilina i modyfikatory leukotrienów (odpowiednio 22%, 27% i 35% pacjentów).
Częstość zaostrzeń astmy wymagających leczenia kortykosteroidami systemowymi była punktem końcowym pierwotnym. Omalizumab zmniejszył częstość zaostrzeń astmy o 19% (p = 0,153). Dodatkowe oceny, które wykazały istotność statystyczną (p < 0,05) na korzyść Ksolara, obejmowały zmniejszenie częstości ciężkich zaostrzeń (w których funkcja płuc pacjenta była obniżona do poziomu poniżej 60% najlepszej wartości osobistej i wymagało się stosowania kortykosteroidów systemowych) oraz nieplanowanych wizyt związanych z astmą u lekarza (składających się z hospitalizacji, pomocy doraźnej i nieplanowanych wizyt u lekarza), a także poprawę ogólnej oceny przez lekarza skuteczności leczenia, jakości życia związanej z astmą (AQL), objawów astmy i funkcji płuc.
W analizie podgrupy pacjentów z ogólnym poziomem IgE ≥ 76 MIU/ml przed leczeniem stwierdzono większą klinicznie istotną korzyść z zastosowania Ksolara. U tych pacjentów w trakcie badania 1 Ksolarem zmniejszono częstość zaostrzeń o 40% (p = 0,002). Ponadto w programie badań zastosowania Ksolara w ciężkiej astmie odnotowano większą liczbę pacjentów z klinicznie istotną odpowiedzią w grupie z ogólnym poziomem IgE ≥ 76 MIU/ml, patrz tabela 1.
Tabela 1
Wyniki badania 1
| Populacja badania 1 ogółem |
||
| Badanie |
Xolar N = 209 |
Placebo N = 210 |
| Występ ostrej astmy |
||
| Częstość w okresie 28 tygodni, |
0,74 |
0,92 |
| % redukcji, wartość p dla współczynnika częstości |
19,4 %, p = 0,153 |
|
| Ciężkie występy astmy |
||
| Częstość w okresie 28 tygodni, |
0,24 |
0,48 |
| % redukcji, wartość p dla współczynnika częstości |
50,1 %, p = 0,002 |
|
| Wizyty u lekarza z powodu stanu nagłego |
||
| Częstość w okresie 28 tygodni, |
0,24 |
0,43 |
| % redukcji, wartość p dla współczynnika częstości |
43,9 %, p = 0,038 |
|
| Ogólna ocena lekarza |
||
| % pacjentów odpowiadających*, |
60,5 % |
42,8 % |
| wartość p ** |
<0,001 |
|
| Poprawa AQL |
||
| % pacjentów z poprawą ≥ 0,5, |
60,8 % |
47,8 % |
| wartość p |
0,008 |
|
* Znaczące polepszenie lub pełna kontrola.
** wartość p dla ogólnego rozkładu oceny.
W trakcie badania 2 oceniano skuteczność i bezpieczeństwo stosowania Xolaru w populacji 312 pacjentów z ciężką astmą alergiczną, których charakterystyka odpowiadała populacji badania 1. Leczenie Xolarem w trakcie tego otwartego badania doprowadziło do zmniejszenia częstości klinicznie istotnych zaostrzeń astmy o 61% w porównaniu z aktualnie stosowaną terapią astmy samodzielnie.
Cztery dodatkowe duże, placebo-kontrolowane badania pomocnicze Xolaru trwające od 28 do 52 tygodni miały na celu ocenę skuteczności i bezpieczeństwa stosowania Xolaru u 1722 dorosłych i dzieci w wieku od 12 lat (badania 3, 4, 5, 6) z ciężką astmą przewlekłą. Większość pacjentów miała niewystarczającą kontrolę astmy, ale otrzymywała mniej terapii wspomagającej astmy niż pacjenci w badaniach 1 lub 2. W trakcie badań 3–5 jako pierwotny punkt końcowy wykorzystano zaostrzenia, natomiast w badaniu 6 oceniano przede wszystkim zmniejszenie potrzeby stosowania kortykosteroidów inhalacyjnych.
W trakcie badań 3, 4 i 5 u pacjentów stosujących Xolar zmniejszono częstość zaostrzeń astmy odpowiednio o 37,5% (p = 0,027), 40,3% (p < 0,001) i 57,6% (p < 0,001) w porównaniu z placebo.
W trakcie badania 6 istotnie większa liczba pacjentów z ciężką astmą alergiczną stosujących Xolar była w stanie zmniejszyć dawkę flutykazonu do ≤ 500 μg/dzień bez pogorszenia kontroli astmy (60,3%) w porównaniu z grupą placebo (45,8%, p < 0,05).
Jakość życia związaną z astmą mierzono za pomocą kwestionariusza Junipera. We wszystkich sześciu badaniach zaobserwowano istotne statystycznie poprawy jakości życia u pacjentów otrzymujących Xolar w porównaniu z pacjentami z grupy placebo lub grupy kontrolnej.
Ogólna ocena skuteczności leczenia przez lekarza.
Ogólna ocena skuteczności leczenia była przeprowadzana przez lekarza w pięciu wyżej wymienionych badaniach jako kontrola astmy. Lekarz miał możliwość uwzględnienia PEF (maksymalnej prędkości wydechu), objawów w ciągu dnia i nocy, stosowania leków ratunkowych, danych z badania spirometrycznego oraz zaostrzeń astmy. We wszystkich pięciu badaniach istotnie większa część pacjentów stosujących Xolar została uznana za osiągnąwszą znaczące polepszenie lub pełną kontrolę astmy w porównaniu z pacjentami z grupy placebo.
Dzieci w wieku od 6 do 12 lat
Główne potwierdzenie bezpieczeństwa i skuteczności stosowania Xolaru w grupie wiekowej od 6 do 12 lat uzyskano w jednym randomizowanym, podwójnie ślepym, placebo-kontrolowanym badaniu wieloośrodkowym (badanie 7).
Badanie 7 było badaniem kontrolowanym placebo, które obejmowało specjalną podgrupę (n = 235) pacjentów, zgodnie z obecnymi wskazaniami, leczonych wysokimi dawkami kortykosteroidów inhalacyjnych (≥ 500 μg/dobę w postaci równoważnika flutykazonu) oraz agonistami β2 o długim działaniu.
Klinicznie istotne zaostrzenie definiowano jako pogorszenie objawów astmy według oceny klinicznej badacza, wymagające podwojenia początkowej dawki kortykosteroidów inhalacyjnych przez co najmniej 3 dni i/lub leczenia systemowymi kortykosteroidami (dawkowanie doustne lub dożylne) z powodu wskazań życiowych przez co najmniej 3 dni.
W specjalnej podgrupie pacjentów otrzymujących wysokie dawki kortykosteroidów inhalacyjnych grupa stosująca omalizumab wykazywała istotnie niższą częstość klinicznie istotnych zaostrzeń astmy niż grupa placebo. W 24. tygodniu różnica w częstości między grupami leczenia wyniosła 34% (stosunek częstości 0,662, p = 0,047) zmniejszenia w porównaniu z placebo u pacjentów otrzymujących omalizumab. W drugim, podwójnie ślepym okresie leczenia trwającym 28 tygodni różnica w częstości między grupami leczenia wyniosła 63% (stosunek częstości 0,37, p < 0,001) zmniejszenia w porównaniu z placebo u pacjentów otrzymujących omalizumab.
W trakcie 52-tygodniowego, podwójnie ślepego okresu leczenia (w tym 24-tygodniowa faza stosowania ustalonej dawki sterydów i 28-tygodniowa faza korekty dawki sterydów) różnica w częstości między grupami leczenia wyniosła 50% (stosunek częstości 0,504, p < 0,001) względnej redukcji częstości zaostrzeń u pacjentów otrzymujących omalizumab.
U grupy pacjentów otrzymujących omalizumab zaobserwowano większy spadek częstości stosowania agonistów β2 w trybie ratunkowym w porównaniu z grupą placebo na końcu 52-tygodniowego okresu leczenia, choć różnica między grupami nie była istotna statystycznie. Według ogólnej oceny skuteczności leczenia na końcu 52-tygodniowego, podwójnie ślepego okresu leczenia w podgrupie ciężko chorych pacjentów stosujących wysokie dawki kortykosteroidów inhalacyjnych oraz agonisty β2 o długim działaniu, odsetek pacjentów z oceną „doskonałą” skuteczności leczenia był wyższy, a stosunek pacjentów z oceną „zadowalającą” lub „złą” był niższy w grupie omalizumab w porównaniu z grupą placebo, przy czym różnica między grupami była istotna statystycznie (p < 0,001), jednocześnie nie zaobserwowano różnic między grupami stosującymi omalizumab i placebo w zakresie subiektywnych ocen jakości życia.
Doświadczenie kliniczne stosowania w przewlekłym zapaleniu zatok nosowych z polipami nosowymi (CRSwNP)
Bezpieczeństwo i skuteczność stosowania Xolaru oceniano w dwóch randomizowanych, podwójnie ślepych, placebo-kontrolowanych badaniach z udziałem pacjentów z CRSwNP (tabela 2). Pacjenci otrzymywali Xolar lub placebo podskórnie co 2 lub 4 tygodnie (patrz sekcja „Sposób stosowania i dawki”). Wszyscy pacjenci otrzymywali terapię wspomagającą w postaci mometazonu w nosie przez cały okres badania. Do włączenia do badania nie wymagano wcześniejszej operacji zatokowej ani wcześniejszego systemowego stosowania kortykosteroidów. Pacjenci otrzymywali Xolar lub placebo przez 24 tygodnie, po czym nastąpił 4-tygodniowy okres obserwacji. Dane demograficzne i wyjściowe charakterystyki, w tym współistniejące choroby alergiczne, opisano w tabeli 2.
Pierwotnymi punktami końcowymi były dwustronne wyniki oceny polipów nosowych (PPN) oraz średni dzienny wynik zatkania nosa (WZN) w 24. tygodniu. W obu badaniach dotyczących polipów nosowych (badanie 1 i 2) pacjenci otrzymujący Xolar wykazywali istotnie większe poprawy w porównaniu z poziomem wyjściowym w 24. tygodniu w zakresie PPN oraz średniego tygodniowego WZN niż pacjenci otrzymujący placebo. Wyniki badań 1 i 2 dotyczące polipów nosowych przedstawiono w tabeli 3.
Tabela 2
Dane demograficzne i wyjściowe charakterystyki w badaniach dotyczących polipów nosowych
| Parametr |
Badaie 1 dotyczące polipów nosowych N = 138 |
Badaie 2 dotyczące polipów nosowych N = 127 |
| Średnia wieku (lata) (SD) |
51,0 (13,2) |
50,1 (11,9) |
| % mężczyzn |
63,8 |
65,4 |
| Pacjenci przyjmujący systemowe kortykosteroidy w poprzednim roku (%) |
18,8 |
26,0 |
| Wskaznik podwójnej polipów nosowych (PPN): średnia (SD), skala 0–8 |
6,2 (1,0) |
6,3 (0,9) |
| Wskaźnik zatkania nosa (WZN): średnia (SD), skala 0–3 |
2,4 (0,6) |
2,3 (0,7) |
| Poziom odczuwania zapachu: średnia (SD), skala 0–3 |
2,7 (0,7) |
2,7 (0,7) |
| Suma punktów OCNPN-22: średnia (SD), skala 0–110 |
60,1 (17,7) |
59,5 (19,3) |
| Eozynofile we krwi (komórek/mkl): średnia (SD) |
346,1 (284,1) |
334,6 (187,6) |
| Całkowity IgE, MIU/ml: średnia (SD) |
160,9 (139,6) |
190,2 (200,5) |
| Astma (%) |
53,6 |
60,6 |
| Lekka (%) |
37,8 |
32,5 |
| Umiarkowana (%) |
58,1 |
58,4 |
| Ciężka (%) |
4,1 |
9,1 |
| Astma indukowana aspiryną (%) |
19,6 |
35,4 |
| Rinit aleryczny |
43,5 |
42,5 |
SD – odchylenie standardowe;
SNOT-22 – kwestionariusz SNOT-22 (22 pytania dotyczące objawów sinozonalnych);
IgE – immunoglobulina E;
MI – jednostki międzynarodowe.
Wyższe wyniki w skali NP, OZS i SNOT-22 wskazują na cięższy przebieg choroby.
Tabela 3
Zmiana względem wartości wyjściowej w 24. tygodniu oceny klinicznej w badaniach 1 i 2 dotyczących polipów nosowych oraz dane połączone
| Parametr |
Polipy nosowe, |
Polipy nosowe, |
Polipy nosowe, |
|||
| Placebo |
Xolar |
Placebo |
Xolar |
Placebo |
Xolar |
|
| N |
66 |
72 |
65 |
62 |
131 |
134 |
| Wskaźnik polipów nosa |
||||||
| Średnia wartość wyjściowa |
6,32 |
6,19 |
6,09 |
6,44 |
6,21 |
6,31 |
| Wartość średnia MNK na 24. tydzień |
0,06 |
-1,08 |
-0,31 |
-0,90 |
-0,13 |
-0,99 |
| Różnica (95%) CI |
-1,14 (-1,59, -0,69) |
-0,59 (-1,05, -0,12) |
-0,86 (-1,18, -0,54) |
|||
| p-wartość |
< 0,0001 |
0,0140 |
< 0,0001 |
|||
| Średnia wartość (przez 7 dni) codziennego PZN |
||||||
| Średnia wartość wyjściowa |
2,46 |
2,40 |
2,29 |
2,26 |
2,38 |
2,34 |
| Wartość średnia MNK na 24. tydzień |
-0,35 |
-0,89 |
-0,20 |
-0,70 |
-0,28 |
-0,80 |
| Różnica (95%) CI |
-0,55 (-0,84, -0,25) |
-0,50 (-0,80, -0,19) |
-0,52 (-0,73, -0,31) |
|||
| p-wartość |
0,0004 |
0,0017 |
< 0,0001 |
|||
| ZPNS |
||||||
| Średnia wartość wyjściowa |
9,33 |
8,56 |
8,73 |
8,37 |
9,03 |
8,47 |
| Wartość średnia MNK na 24. tydzień |
-1,06 |
-2,97 |
-0,44 |
-2,53 |
-0,77 |
-2,75 |
| Różnica (95%) |
-1,91 (-2,85, -0,96) |
-2,09 (-3,00, -1,18) |
-1,98 (-263, -1,33) |
|||
| p-wartość |
0,0001 |
< 0,0001 |
< 0,0001 |
|||
| OSNP-22 |
||||||
| Średnia wartość wyjściowa |
60,26 |
59,82 |
59,80 |
59,21 |
60,03 |
59,54 |
| Wartość średnia MNK na 24 tydzień |
-8,58 |
-24,70 |
-6,55 |
-21,59 |
-7,73 |
-23,10 |
| Różnica (95%) |
-16,12 (-21,86, -10,38) |
-15,04 (-21,26, -8,82) |
-15,36 (-19,57, -11,16) |
|||
| p-wartość |
< 0,0001 |
< 0,0001 |
< 0,0001 |
|||
| (MZP = 8,9) |
||||||
| TVZUP |
||||||
| Średnia wartość wyjściowa |
13,56 |
12,78 |
13,27 |
12,87 |
13,41 |
12,82 |
| Wartość średnia MNK na 24. tydzień |
0,63 |
4,44 |
0,44 |
4,31 |
0,54 |
4,38 |
| Różnica (95%) |
3,81 (1,38, 6,24) |
3,86 (1,57, 6,15) |
3,84 (2,17, 5,51) |
|||
| p-wartość |
0,0024 |
0,0011 |
< 0,0001 |
|||
MNK – metoda najmniejszych kwadratów; CI – przedział ufności; PZN – wskaźnik zatkania nosa; ZPNS – ogólny wskaźnik objawów nosowych; OSNP-22 – badanie z 22 pytaniami dotyczących objawów sinozonalnych; TVZUP – test wykrywania zapachu Uniwersytetu Pensylwanii; MZR – minimalnie istotna różnica.
| Wartość wyjściowa |
| Wartość wyjściowa |
| Pierwsza analiza skuteczności |
| Analiza wtórna skuteczności |
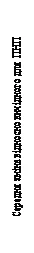
| Tydzien |
| Tydzień |
| Bad. 2 / placebo (N=65) Bad. 2 / omalizumab (N=62) |
| Bad. 1 / placebo (N=66) Bad. 1 / omalizumab (N=72) |
| Bad. 2 / placebo (N=65) Bad. 2 / omalizumab (N=62) |
| Bad. 1 / placebo (N=66) Bad. 1 / omalizumab (N=72) |
| Analiza wtórna skuteczności |
| Pierwotna analiza skuteczności |
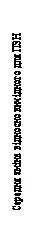
| -1,25 |
| -1,25 |
| -1,00 |
| -1.00 |
| -0.75 |
| -0.75 |
| -0.50 |
| -0,50 |
| -0.25 |
| -0.25 |
|
|
|
|
| 24 |
| 20 |
| 16 |
| 12 |
| 8 |
| 4 |
| 4 |
| 8 |
| 12 |
| 16 |
| 20 |
| 24 |
Rys. 1. Średnia zmiana wartości wskaźnika zatkania nosa (WZN) i średnia zmiana wartości wskaźnika polipów nosowych (WPN) w stosunku do wartości wyjściowych w badaniach 1 i 2 dotyczących polipów nosowych.
W zaplanowanej analizie połączonej leczenia nagłego (stosowanie kortykosteroidów ogólnoustrojowych przez 3 lub więcej kolejnych dni lub polipektomia nosowa) w okresie 24 tygodni leczenia, odsetek pacjentów wymagających leczenia nagłego był niższy w grupie stosującej Xolar w porównaniu z placebo (2,3% vs 6,2% odpowiednio). Stosunek szans potrzeby leczenia nagłego u pacjentów otrzymujących Xolar w porównaniu z placebo wynosił 0,38 (95% CI: 0,10; 1,49). W żadnym z badań nie odnotowano zabiegów chirurgicznych zatok nozowych.
Doświadczenie kliniczne stosowania w przewlekłym spontanicznym pokrzywce
Skuteczność i bezpieczeństwo stosowania Xolaru zostały wykazane w dwóch randomizowanych, kontrolowanych placebo badaniach fazy III (badanie 1 i 2) u pacjentów z PSP, u których objawy choroby utrzymywały się pomimo leczenia lekami przeciwhistaminowymi H1 w dawce zatwierdzonej. W trzecim badaniu (badanie 3) pierwotnie oceniano bezpieczeństwo Xolaru u pacjentów z PSP, u których objawy choroby utrzymywały się pomimo leczenia lekami przeciwhistaminowymi H1 w dawce około czterokrotnie przekraczającej zatwierdzoną dawkę, a także lekami przeciwhistaminowymi H2 i/lub antagonistami receptorów leukotrienów. W trzech badaniach wzięło udział 975 pacjentów w wieku od 12 do 75 lat (średni wiek 42,3 roku; 39 pacjentów w wieku 12–17 lat, 54 pacjentów w wieku ≥ 65 lat; 259 mężczyzn i 716 kobiet). Wszyscy pacjenci mieli objawy niekontrolowane odpowiednio, oceniane za pomocą 16-punktowej skali oceny ciężkości przebiegu pokrzywki (UAS7, zakres 0–42) oraz 8-punktowej skali oceny ciężkości przebiegu świądu (część UAS7; zakres 0–21) w ciągu 7 dni przed randomizacją, mimo przyjmowania leków przeciwhistaminowych przez co najmniej 2 tygodnie przed tym.
W badaniach 1 i 2 średnia wartość skali oceny ciężkości przebiegu świądu u pacjentów na początku badania wynosiła 13,7–14,5, a średnia wartość skali UAS7 wynosiła odpowiednio 29,5 i 31,7. W badaniu bezpieczeństwa 3 średnia wartość skali oceny ciężkości przebiegu świądu u pacjentów wynosiła 13,8, a średnia wartość skali UAS7 wynosiła 31,2 na początku badania. We wszystkich trzech badaniach pacjenci otrzymywali średnio 4–6 leków (w tym leki przeciwhistaminowe H1) na objawy PSP przed włączeniem do badania. Pacjenci otrzymywali lek Xolar w dawce 75 mg, 150 mg lub 300 mg, albo placebo, w postaci wstrzyknięć podskórnych co 4 tygodnie przez 24 i 12 tygodni odpowiednio w badaniach 1 i 2 oraz lek Xolar w dawce 300 mg lub placebo w postaci wstrzyknięć podskórnych co 4 tygodnie przez 24 tygodnie w badaniu 3. We wszystkich badaniach okres bez leczenia trwał 16 tygodni.
Pierwotnym punktem końcowym była zmiana wartości skali oceny ciężkości przebiegu świądu od wartości wyjściowej do 12. tygodnia. Omalizumab w dawce 300 mg zmniejszał wynik skali oceny ciężkości przebiegu świądu o 8,55–9,77 (p < 0,0001) w porównaniu z redukcją o 3,63–5,14 w grupie placebo (patrz tabela 4). Odsetek odpowiedzi według skali UAS7 ≤ 6 (w 12. tygodniu) był istotnie wyższy w grupach leczonych lekiem Xolar w dawce 300 mg (52–66%) (p < 0,0001) w porównaniu z 11–19% w grupie placebo, a pełna remisja (UAS7 = 0) osiągnięta została u 34–44% (p < 0,0001) pacjentów leczonych dawką 300 mg, w porównaniu z 5–9% w grupach placebo. Pacjenci leczeni dawką 300 mg osiągnęli najwyższy średni odsetek dni bez epizodów obrzęku naczynioruchowego od 4. do 12. tygodnia (91,0–96,1%; p < 0,001) w porównaniu z pacjentami z grupy placebo (88,1–89,2%). Średnia zmiana wartości ogólnego DLQI (dermatologiczny indeks jakości życia) od wartości wyjściowej do 12. tygodnia w grupach leczonych lekiem Xolar w dawce 300 mg była wyższa (p < 0,001) niż w grupach placebo, co wskazuje na poprawę o 9,7–10,3 punktu w porównaniu z 5,1–6,1 punktu w odpowiednich grupach placebo.
Tabela 4
Zmiana wartości skali oceny ciężkości przebiegu świądu od wartości wyjściowej do 12. tygodnia, badania 1, 2 i 3 (populacja mITT*)
| Wskaźniki |
Placebo |
Omalizumab |
| Badanie 1 |
||
| N |
80 |
81 |
| Średni wynik (SD) |
−3,63 (5,22) |
−9,40 (5,73) |
| Różnica średnich wartości metodą najmniejszych kwadratów w porównaniu do placebo 1 |
- |
−5,80 |
| 95 % CI dla różnicy |
- |
−7,49−4,10 |
| Wartość p w porównaniu do placebo 2 |
- |
< 0,0001 |
| Badanie 2 |
||
| N |
79 |
79 |
| Średni wynik (SD) |
−5,14 (5,58) |
−9,77 (5,95) |
| Różnica średnich wartości metodą najmniejszych kwadratów w porównaniu do placebo 1 |
- |
−4,81 |
| 95 % CI dla różnicy |
- |
−6,49−3,13 |
| Wartość p w porównaniu do placebo 2 |
- |
< 0,0001 |
| Badanie 3 |
||
| N |
83 |
252 |
| Średni wynik (SD) |
−4,01 (5,87) |
−8,55 (6,01) |
| Różnica średnich wartości metodą najmniejszych kwadratów w porównaniu do placebo 1 |
- |
|
| 95 % CI dla różnicy |
- |
−5,97 −3,08 |
| Wartość p w porównaniu do placebo 2 |
- |
< 0,0001 |
*Zmodyfikowana populacja (mITT) według zamiar leczenia: uwzględniono wszystkich pacjentów, którzy zostali randomizowani i otrzymali co najmniej jedną dawkę badanego leku.
BOCF (przeniesienie naprzód wartości ustalonej na początku) było stosowane do uzupełniania brakujących danych.
1 Średnia wartość najmniejszych kwadratów została obliczona za pomocą modelu ANCOVA. Zmiennymi były poziom wyjściowy według skali cotygodniowej oceny nasilenia świądu (< 13 w porównaniu z ≥ 13) oraz początkowa masa ciała (< 80 kg w porównaniu z ≥ 80 kg).
2 Wartość p uzyskano na podstawie testu t-Studenta ANCOVA.
Na rys. 2 przedstawiono średni wynik według skali cotygodniowej oceny nasilenia świądu w długim okresie w badaniu 1. Średni wynik według skali cotygodniowej oceny nasilenia świądu znacząco zmniejszył się osiągając maksymalny efekt w 12. tygodniu trwania 24-tygodniowego okresu leczenia. W badaniu 3 uzyskano analogiczne wyniki.
We wszystkich trzech badaniach średni wynik według skali cotygodniowej oceny nasilenia świądu stopniowo wzrastał w 16-tygodniowym okresie bez leczenia, zgodnie z pojawieniem się objawów. Średnie wartości na końcu okresu obserwacji były podobne do tych obserwowanych w grupie placebo, ale niższe niż odpowiednie średnie wartości na początku badania.
| Tydzień 12 Pierwotny punkt końcowy |
| Omalizumab w dawce 300 mg |
| Placebo |
| Tydzień |
| Zastosowano omalizumab lub placebo |
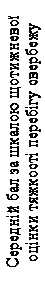 |
BOCF – przeniesienie pierwotnej ustalonej wartości do przodu; mITT – zmodyfikowana populacja według zamiaru leczenia.
Rys. 2. Średni wynik w skali cotygodniowej oceny nasilenia świądu w długim okresie czasu, badanie 1 (populacja mITT).
Skuteczność po 24 tygodniach leczenia
Wyniki skuteczności obserwowane w 24. tygodniu leczenia porównywano z wynikami obserwowanymi w 12. tygodniu leczenia.
W przypadku dawki 300 mg w badaniach 1 i 3 średnie zmniejszenie wyniku na początku badania w skali cotygodniowej oceny nasilenia świądu wynosiło odpowiednio 9,8 i 8,6, odsetek pacjentów z UAS7 ≤ 6 wynosił 61,7 % i 55,6 %, a odsetek pacjentów z pełną odpowiedzią (UAS7 = 0) wynosił odpowiednio 48,1 % i 42,5 % (we wszystkich przypadkach p < 0,0001 w porównaniu z placebo).
Doświadczenie kliniczne dotyczące ponownego stosowania omalizumabu jest ograniczone.
Dane z badań klinicznych z udziałem nastolatków (w wieku od 12 do 17 lat) obejmowały informacje o 39 pacjentach, z których 11 otrzymało dawkę 300 mg. Wyniki dotyczące dawki 300 mg uzyskano u 9 pacjentów w 12. tygodniu i u 6 pacjentów w 24. tygodniu i wskazują na podobną odpowiedź terapeutyczną na leczenie omalizumabem jak u dorosłych pacjentów. Średnia zmiana wyniku na początku badania w skali cotygodniowej oceny nasilenia świądu wynosiła 8,25 w 12. tygodniu i 8,95 w 24. tygodniu. Szybkość odpowiedzi – 33 % w 12. tygodniu i 67 % w 24. tygodniu dla UAS7 = 0 oraz 56 % w 12. tygodniu i 67 % w 24. tygodniu dla UAS7 ≤ 6.
Farmakokinetyka.
Działanie omalizumabu badano u dorosłych pacjentów i dzieci w wieku od 12 lat z astmą alergiczną, u dorosłych pacjentów z APRzP, a także u dorosłych i nastolatków z CHS. Ogólne parametry farmakokinetyczne omalizumabu u tych pacjentów są podobne.
Wchłanianie
Po podaniu podskórnie omalizumab jest wchłaniany ze średnią biodostępnością absolutną wynoszącą 62 %. Po jednorazowym podaniu podskórnie dorosłym i dzieciom w wieku od 12 lat z astmą lub CHS omalizumab był wchłaniany powoli, osiągając maksymalne stężenie w surowicy średnio po 7–8 dniach. U pacjentów z astmą po wielokrotnym podawaniu omalizumabu pole pod krzywą „stężenie w surowicy – czas” od dnia 0 do dnia 14 w stanie równowagi było 6 razy wyższe niż po podaniu pierwszej dawki.
Farmakokinetyka omalizumabu jest liniowa w dawkach przekraczających 0,5 mg/kg. Po wielokrotnym podawaniu omalizumabu pole pod krzywą „stężenie w surowicy – czas” od dnia 0 do dnia 14 w stanie równowagi było nawet do 6 razy wyższe niż po podaniu pierwszej dawki.
Stosowanie Xolaru w postaci liofilizatu lub roztworu prowadziło do podobnych profili wskaźnika „stężenie w surowicy – czas” omalizumabu.
Rozkład
In vitro omalizumab tworzy kompleksy z IgE o ograniczonym rozmiarze. Kompleksy wytrącające się oraz kompleksy o masie cząsteczkowej przekraczającej milion daltonów nie zostały wykryte ani in vitro, ani in vivo. Biorąc pod uwagę farmakokinetykę w populacji, rozkład omalizumabu był podobny u pacjentów z astmą alergiczną i u pacjentów z CHS. Oczekiwany objętość rozkładu po podaniu podskórnej wynosiła 78 ± 32 ml/kg.
Eliminacja
W klirensie omalizumabu zaangażowane są procesy klirensu IgG, jak również eliminacja poprzez specyficzne wiązanie i tworzenie kompleksów z jego docelowym ligandem – IgE. Wątrobowe eliminowanie IgG obejmuje rozszczepienie w układzie siateczkowo-śródbłonkowym i w komórkach śródbłonka. Niezmieniony IgG jest również wydzielany z żółcią. U pacjentów z astmą średni okres półtrwania omalizumabu w surowicy wynosił 26 dni przy klirensie średnio 2,4 ± 1,1 ml/kg/dzień. Ponadto, przy masie ciała dwa razy większej, klirens zwiększał się o około połowę. U pacjentów z CHS, biorąc pod uwagę modele farmakokinetyczne, średni okres półtrwania omalizumabu w surowicy w stanie równowagi wynosił średnio 24 dni, a pozorny klirens w stanie równowagi u pacjentów o masie ciała 80 kg wynosił 3,0 ml/kg/dobę.
Charakterystyki w populacjach pacjentów
Wiek, przynależność rasowa/etniczna, płeć, wskaźnik masy ciała
Pacjenci z astmą i APRzP
Farmakokinetykę Xolaru w populacji analizowano w celu oceny wpływu cech demograficznych. Analiza tych ograniczonych danych wskazuje, że nie jest wymagana korekta dawki w zależności od wieku (6–76 lat u pacjentów z astmą alergiczną; 18–75 lat u pacjentów z APRzP), przynależności rasowej/etnicznej, płci ani wskaźnika masy ciała.
Pacjenci z CHS
Wpływ cech demograficznych i innych czynników na ekspozycję na Xolar oceniano na podstawie farmakokinetyki w populacji. Ponadto wpływ zmiennej niezależnej oceniano poprzez analizę stosunku stężenia omalizumabu do klinicznej odpowiedzi. Ta analiza wskazuje, że u pacjentów z CHS, niezależnie od wieku (12–75 lat), przynależności rasowej/etnicznej, płci, masy ciała, wskaźnika masy ciała, wyjściowego poziomu IgE, autoantyciał anty-FcεRI, jednoczesnego stosowania leków H2-blokujących histaminę lub antagonistów receptorów leukotrienów, korekta dawki nie jest wymagana.
Upośledzenie funkcji nerek i wątroby
Brak danych dotyczących farmakokinetyki lub farmakodynamiki u pacjentów z niewydolnością nerek lub wątroby.
Dane kliniczne.
Wskazania.
Astma alergiczna
Możliwość leczenia Xolarem należy rozważać wyłącznie u pacjentów z potwierdzoną astmą pośredniczoną przez IgE (immunoglobulinę E).
Dorośli i dzieci od 12. roku życia
Xolar jest wskazany jako terapia uzupełniająca w celu osiągnięcia lepszego kontroli astmy u pacjentów z ciężką, przewlekłą astmą alergiczną, u których test skórny lub badanie in vitro na reakcję na stale obecny w powietrzu alergen jest pozytywny, u których występuje zaburzona funkcja płuc (FEV1 (objętość życiowa wydechu wymuszonego) < 80 %), a także częste objawy w ciągu dnia lub częste przebudzenia w nocy oraz którzy mają dokumentację wielokrotnych ciężkich zaostrzeń astmy mimo stosowania wysokich dawek doustnych kortykosteroidów wziewnych w połączeniu z długodziałającymi agonistami β2.
Dzieci w wieku od 6 do 12 lat
Xolar jest wskazany jako terapia uzupełniająca w celu osiągnięcia lepszego kontroli astmy u pacjentów z ciężką, przewlekłą astmą alergiczną, u których test skórny lub badanie in vitro na reakcję na stale obecny w powietrzu alergen jest pozytywny, a także występują częste objawy w ciągu dnia lub przebudzenia w nocy oraz którzy mają dokumentację wielokrotnych ciężkich zaostrzeń astmy mimo stosowania wysokich dawek kortykosteroidów wziewnych w połączeniu z długodziałającymi agonistami β2.
Przewlekłe zapalenie zatok nosowych z polipami nosowymi (CRSwNP)
Xolar jest wskazany jako terapia uzupełniająca w połączeniu z kortykosteroidami do stosowania miejscowego w nosie (INS) w leczeniu dorosłych (od 18. roku życia) z ciężką postacią CRSwNP, u których terapia INS nie zapewnia odpowiedniej kontroli choroby.
Przewlekła pokrzywka samoistna (CSU), dawka 150 mg
Lek Xolar jest wskazany jako terapia uzupełniająca w leczeniu przewlekłej pokrzywki samoistnej u dorosłych i młodzieży (od 12. roku życia), u których odpowiedź na leczenie lekami przeciwhistaminowymi H1 była niewystarczająca.
Przeciwwskazania.
Podwyższona wrażliwość na substancję czynną lub na którykolwiek z innych składników leku.
Interakcje z innymi lekami i inne rodzaje interakcji.
Ponieważ IgE może uczestniczyć w odpowiedzi immunologicznej wywołanej przez niektóre infestacje pasożytnicze, lek Xolar może pośrednio obniżać skuteczność leków stosowanych w leczeniu infestacji pasożytniczych lub innych infekcji pasożytniczych.
Enzymy cytochromu P450, pompa wypompowująca i mechanizm wiązania z białkami nie są zaangażowane w klirens omalizumabu; dlatego możliwość interakcji z innymi lekami jest niewielka. Nie przeprowadzono badań interakcji Xolaru z lekami lub szczepionkami. Nie ma farmakologicznie uzasadnionych powodów do przypuszczeń, że leki stosowane zazwyczaj w leczeniu astmy będą oddziaływać z omalizumabem.
Astma alergiczna
W trakcie badań klinicznych Xolar stosowano zazwyczaj w połączeniu z kortykosteroidami wziewnymi i doustnymi, krótko- i długodziałającymi agonistami β2 wziewnymi, modyfikatorami leukotrienów, teofiliną oraz doustnymi lekami przeciwhistaminowymi. Nie zaobserwowano zmian w profilu bezpieczeństwa Xolaru pod wpływem tych powszechnie stosowanych leków w leczeniu astmy. Dane dotyczące stosowania Xolaru w połączeniu z immunoterapią swoistą (terapią hiposensybilizującą) są ograniczone. W trakcie badań klinicznych, w których Xolar stosowano łącznie z immunoterapią, bezpieczeństwo i skuteczność Xolaru w połączeniu z terapią swoistą nie różniły się od tych obserwowanych przy stosowaniu Xolaru jako monoterapii.
Przewlekła pokrzywka samoistna
W trakcie badań klinicznych u pacjentów z CSU Xolar stosowano jednocześnie z lekami przeciwhistaminowymi (przeciw-H1, przeciw-H2) oraz antagonistami receptorów leukotrienów. Nie ma danych potwierdzających, że bezpieczeństwo omalizumabu zmieniało się przy stosowaniu razem z tymi lekami w porównaniu z znanym profilem bezpieczeństwa w astmie alergicznej. Ponadto analiza populacyjna farmakokinetyki nie wskazywała na istotny wpływ leków przeciwhistaminowych H2 oraz antagonistów receptorów leukotrienów na farmakokinetykę omalizumabu.
Przewlekłe zapalenie zatok nosowych z polipami nosowymi
W badaniach klinicznych Xolar stosowano zgodnie z protokołem w połączeniu z mometazonem w postaci sprayu do nosa. Inne często stosowane leki wspomagające obejmowały inne kortykosteroidy do nosa, leki rozszerzające oskrzela, leki przeciwhistaminowe, antagoniści receptorów leukotrienów, środki adrenergiczne/sympatymimetyki oraz miejscowe znieczulenia nosowe. Nie zaobserwowano żadnych oznak, że jednoczesne stosowanie innych często stosowanych leków wpływa na bezpieczeństwo Xolaru.
Dzieci
W badaniach klinicznych dotyczących CSU włączono kilku pacjentów w wieku od 12 do 17 lat, którzy przyjmowali Xolar jednocześnie z lekami przeciwhistaminowymi (przeciw-H1, przeciw-H2) oraz antagonistami receptorów leukotrienów. Badania u dzieci poniżej 12. roku życia nie były prowadzone.
Szczególne ostrzeżenia i środki ostrożności podczas stosowania.
Śledzenie
W celu poprawy śledzenia leków biologicznych należy dokładnie rejestrować nazwę handlową oraz numer serii podanego leku.
Ogólne
Xolar nie jest wskazany do leczenia napadów astmy, ostrego skurczu oskrzeli ani stanu astmatycznego.
Nie przeprowadzono badań dotyczących stosowania leku Xolar u pacjentów z zespołem podwyższonego stężenia IgE, alergicznym grzybiczym zapaleniem oskrzeli, ani w celu zapobiegania reakcjom anafilaktycznym, w tym wywołanym alergią pokarmową, atopowym zapaleniem skóry lub alergicznym nieżytą nosa. Xolar nie jest wskazany do leczenia tych stanów.
Nie przeprowadzono badań dotyczących stosowania leku Xolar u pacjentów z chorobami autoimmunologicznymi, stanami pośredniczonymi przez kompleksy immunologiczne, ani u pacjentów z już istniejącym zaburzeniem czynności nerek lub wątroby. Xolar należy stosować z ostrożnością u takich pacjentów.
Nie zaleca się nagłego odstawienia kortykosteroidów doustnych ani inhalacyjnych po rozpoczęciu terapii Xolarem w leczeniu astmy alergicznej lub PRZEzNP. Dawkę kortykosteroidów należy zmniejszać stopniowo pod bezpośrednim nadzorem lekarza, w razie potrzeby.
Zaburzenia ze strony układu odpornościowego
- Reakcje alergiczne typu I
Podczas stosowania omalizumabu mogą występować lokalne lub uogólnione reakcje alergiczne typu I, w tym anafilaksja i wstrząs anafilaktyczny; mogą one wystąpić nawet po długotrwałym leczeniu. Większość tych reakcji rozwija się w ciągu 2 godzin po pierwszej lub kolejnych dawkach leku Xolar, jednak czasem reakcje niepożądane pojawiają się później niż 2 godziny, a w niektórych przypadkach nawet po ponad 24 godzinach od wstrzyknięcia. Większość reakcji anafilaktycznych pojawia się podczas stosowania pierwszych trzech dawek Xolaru. Reakcje nadwrażliwości niezwiązane z omalizumabem w wywiadzie stanowią czynnik ryzyka rozwoju reakcji anafilaktycznych podczas stosowania Xolaru. Dlatego po podaniu Xolaru leki do leczenia reakcji anafilaktycznych muszą być zawsze dostępne do natychmiastowego zastosowania. W przypadku wystąpienia anafilaksji lub innej poważnej reakcji alergicznej należy natychmiast przerwać stosowanie Xolaru i rozpocząć odpowiednią terapię. Pacjentów należy poinformować, że takie reakcje są możliwe, i w przypadku ich wystąpienia należy niezwłocznie skontaktować się z lekarzem.
Przeciwciała przeciwko omalizumabowi wykryto u niewielkiej liczby pacjentów podczas badań klinicznych. Kliniczne znaczenie przeciwciał przeciwko Xolarowi nie jest dobrze poznane.
- Choroba surowicy
Choroba surowicy i reakcje podobne do choroby surowicy (opóźnione reakcje alergiczne typu III) rzadko występują u pacjentów przyjmujących humanizowane przeciwciała monoklonalne, w tym omalizumab. Prawdopodobny mechanizm patofizjologiczny obejmuje powstawanie i osadzanie się kompleksów immunologicznych w wyniku produkcji przeciwciał przeciwko omalizumabowi. Typowy czas wystąpienia to 1–5 dni po podaniu pierwszej lub kolejnych dawek, a także po długotrwałym leczeniu. Objawy wskazujące na chorobę surowicy obejmują artretyzm/artrologię, wysypkę (plamica czy inne postacie), gorączkę i limfadenopatię. W celu zapobiegania lub leczenia tych zaburzeń mogą być wskazane leki przeciwhistaminowe i kortykosteroidy, a pacjenci powinni informować lekarza o wszelkich podejrzanych objawach.
- Zespół Churga–Straussa i zespół hiperozynofilii
U pacjentów z ciężką astmą w pojedynczych przypadkach mogą występować zespół uogólnionej hiperozynofilii lub alergicznego granulomatycznego zapalenia naczyń (zespół Churga–Straussa), oba stany zazwyczaj leczy się za pomocą kortykosteroidów systemowych.
W pojedynczych przypadkach u pacjentów przyjmujących leki przeciwasmtatyczne, w tym omalizumab, może wystąpić lub rozwinąć się uogólniona eozynofilia i zapalenie naczyń. Zjawiska te są zazwyczaj związane ze zmniejszaniem dawki doustnych kortykosteroidów.
Podczas leczenia takich pacjentów należy pamiętać o rozwoju wyraźnej eozynofilii, wysypce zapalnej naczyń, pogorszeniu objawów płucnych, patologii zatok przynosowych, powikłaniach sercowych i/lub neuropatii.
W przypadku wszystkich ciężkich przypadków powyższych zaburzeń immunologicznych należy rozważyć możliwość przerwania stosowania omalizumabu.
Inwazje pasożytnicze (robacice)
IgE może uczestniczyć w odpowiedzi immunologicznej wywołanej przez niektóre inwazje robacice. Badanie kontrolowane placebo u pacjentów z wysokim, stałym ryzykiem inwazji robacicy wykazało nieznaczny wzrost częstości inwazji podczas stosowania omalizumabu, choć przebieg, ciężkość i odpowiedź na leczenie pozostały niezmienione. Występowanie inwazji robacicy podczas ogólnego programu klinicznego, który nie był specjalnie zaprojektowany do wykrywania takich inwazji, było mniejsze niż 1 przypadek na 1000 pacjentów. Jednak pacjenci z wysokim ryzykiem infekcji robaciczą powinni zachować ostrożność, szczególnie podczas podróży do regionów, w których inwazje robacice są endemiczne. Jeśli pacjent nie odpowiada na przepisane leczenie przeciwpasożytnicze, należy rozważyć odstawienie Xolaru.
Osoby wrażliwe na lateks
Kapsułka igły w strzykawce wstępnie napełnionej zawiera pochodne naturalnego kauczuku lateksowego. Obecnie nie wykryto lateksu z naturalnego kauczuku w kapsułce igły. Jednak stosowanie Xolaru, roztworu do wstrzykiwań w strzykawce wstępnie napełnionej, u osób wrażliwych na lateks nie było badane, a zatem istnieje potencjalne ryzyko reakcji nadwrażliwości, którego nie można całkowicie wykluczyć.
Stosowanie w czasie ciąży lub karmienia piersią.
Umiarkowana liczba danych dotyczących ciężarnych kobiet (od 300 do 1000 wyników ciąż) uzyskanych z rejestru ciąż i spontanicznych doniesień z okresu po marketingowym wskazuje na brak działania teratogennego oraz fetotoksycznego/neonatotoksycznego. Przyszłościowe badanie rejestru ciąż (EXPECT) u 250 ciężarnych kobiet z astmą, które przyjmowały Xolar, wykazało, że częstość występowania głównych wad wrodzonych była podobna (8,1% vs 8,9%) u pacjentek z EXPECT i pacjentek z odpowiadającymi chorobami (astma średnio ciężka i ciężka). Na interpretację danych mogą wpływać ograniczenia metodologiczne badania, w tym mała liczba badanych i nielosowy projekt.
Omalizumab przenika przez barierę łożyskową, jednak badania na zwierzętach nie wykazały bezpośredniego ani pośredniego szkodliwego wpływu na funkcję rozrodczą. Omalizumab był związany z wiekowym obniżeniem liczby płytek krwi u naczelnych niższych z większą względną wrażliwością u zwierząt niemowlęcych. W przypadku klinicznej potrzeby leczenia Xolarem w czasie ciąży może być rozważone.
IgG (immunoglobuliny G) występują w ludzkim mleku, dlatego można oczekiwać obecności omalizumabu w ludzkim mleku. Omalizumab wydzielany jest z mlekiem u naczelnych niższych.
Badanie EXPECT z udziałem 154 niemowląt narażonych na działanie leku Xolar w okresie wewnątrzmacicznym oraz podczas karmienia piersią wykazało brak reakcji niepożądanych u niemowląt karmionych piersią. Na interpretację danych mogą wpływać ograniczenia metodologiczne badania, w tym mała liczba badanych i nielosowy projekt.
Po przyjęciu doustnym białka immunoglobuliny G ulegają proteolizie jelitowej i mają niską biodostępność. Nie oczekuje się wpływu na noworodki/niemowlęta karmione piersią. Zatem, jeśli jest to klinicznie wskazane, leczenie Xolarem w czasie karmienia piersią może być rozważone.
Brak danych dotyczących wpływu omalizumabu na płodność człowieka. W specjalnie zaprojektowanych badaniach przedklinicznych dotyczących płodności, w tym badaniach rozrodu, nie zaobserwowano pogorszenia płodności męskiej ani żeńskiej po wielokrotnym stosowaniu omalizumabu w dawkach do 75 mg/kg. Ponadto nie zaobserwowano efektów genotoksycznych w oddzielnym badaniu przedklinicznym genotoksyczności.
Wpływ na zdolność prowadzenia pojazdów lub obsługi innych maszyn.
Xolar nie wpływa lub ma jedynie nieznaczny wpływ na zdolność prowadzenia samochodu lub obsługi innych maszyn.
Sposób stosowania i dawki
Xolar powinien przepisywać lekarz posiadający doświadczenie w rozpoznawaniu i leczeniu ciężkiej astmy przewlekłej, przewlekłego zapalenia zatok nosowych z polipami nosowymi (CRSwNP) lub przewlekłej pokrzywki samoistnej (CSU).
Dozowanie
Alergiczna astma i przewlekłe zapalenie zatok nosowych z polipami nosowymi (CRSwNP)
Zasady dozowania Xolaru w leczeniu alergicznej astmy i CRSwNP są takie same. Wymaganą dawkę i częstotliwość podawania Xolaru w tych stanach ustala się na podstawie stężenia IgE (j.m./ml) wyznaczonego przed rozpoczęciem leczenia oraz masy ciała pacjenta (kg). Przed rozpoczęciem stosowania leku w celu ustalenia dawki należy określić poziom IgE u pacjenta za pomocą ilościowego oznaczenia całkowitego IgE z wykorzystaniem dowolnej dostępnej komercyjnie metody. W zależności od wyników zalecana dawka Xolaru wynosi 75–600 mg. Dawka ta może być podzielona na 1–4 wstrzyknięcia.
Mniejsze prawdopodobieństwo uzyskania pozytywnego efektu obserwowano u pacjentów z alergiczną astmą, u których poziom IgE przed rozpoczęciem leczenia był niższy niż 76 j.m./ml. Przed rozpoczęciem terapii lekarz powinien upewnić się, że u dorosłych pacjentów oraz dzieci w wieku od 12 lat z poziomem IgE poniżej 76 j.m./ml oraz u dzieci w wieku od 6 do 12 lat z poziomem IgE poniżej 200 j.m./ml występuje jednoznaczna reaktywność in vitro (test RAST – radioalergosorbentowy test) na stale obecny alergen.
Szczegóły dotyczące przeliczenia dawki zamieszczono w tabeli 5, a sposób określania dawki – w tabelach 6 i 7.
Nie należy przepisywać Xolaru pacjentom, u których poziom IgE lub masa ciała przekraczają wartości podane w tabeli dawkowania.
Maksymalna zalecana dawka to 600 mg omalizumabu co dwa tygodnie.
Tabela 5
Przeliczenie dawki na liczbę strzykawek, liczbę wstrzyknięć i całkowitą objętość wstrzyknięcia dla każdego podania
| Dawka (mg) |
Liczba strzykawek |
Liczba iniekcji |
Całkowita objętość iniekcji (ml) |
|
| 75 mg |
150 mg |
|||
| 75 |
1 |
0 |
1 |
0,5 |
| 150 |
0 |
1 |
1 |
1,0 |
| 225 |
1 |
1 |
2 |
1,5 |
| 300 |
0 |
2 |
2 |
2,0 |
| 375 |
1 |
2 |
3 |
2,5 |
| 450 |
0 |
3 |
3 |
3,0 |
| 525 |
1 |
3 |
4 |
3,5 |
| 600 |
0 |
4 |
4 |
4,0 |
Tabela 6
PODANIE CO 4 TYGODNIE.
Dawki leku Xolar (mg/dawka) podawane podskórnie co 4 tygodnie
| Poziom wyjściowy IgE (j.m./ml) |
Masa ciała (kg) |
|||||||||
| > 20–25* |
> 25–30* |
> 30–40 |
> 40–50 |
> 50–60 |
> 60–70 |
> 70–80 |
> 80–90 |
> 90–125 |
> 125–150 |
|
| ≥ 30–100 |
75 |
75 |
75 |
150 |
150 |
150 |
150 |
150 |
300 |
300 |
| > 100–200 |
150 |
150 |
150 |
300 |
300 |
300 |
300 |
300 |
450 |
600 |
| > 200–300 |
150 |
150 |
225 |
300 |
300 |
450 |
450 |
450 |
600 |
|
| > 300–400 |
225 |
225 |
300 |
450 |
450 |
450 |
600 |
600 |
||
| > 400–500 |
225 |
300 |
450 |
450 |
600 |
600 |
||||
| > 500–600 |
300 |
300 |
450 |
600 |
600 |
|||||
| > 600–700 |
300 |
450 |
600 |
|||||||
| > 700–800 |
||||||||||
| > 800–900 |
PODAWANIE CO 2 TYGODNIE, |
|||||||||
| > 900–1000 |
patrz TABELA 5 |
|||||||||
| > 1000–1100 |
||||||||||
* W głównych badaniach dotyczących PRZP stosowanie u pacjentów o masie ciała poniżej 30 kg nie było badane.
Tabela 7
PODANIE CO 2 TYGODNIE.
Dawki leku Xolar (mg/dawka) podawane drogą podskórną co 2 tygodnie
| Poziom wyjściowy IgE (j.m./ml) |
Masa ciała (kg) |
||||||||||
| > 20–25* |
> 25–30* |
> 30–40 |
> 40–50 |
> 50–60 |
> 60–70 |
> 70–80 |
> 80–90 |
> 90–125 |
> 125– 150 |
||
| ≥ 30–100 |
PODAWANIE CO 4 TYGODNIE |
||||||||||
| > 100–200 |
patrz TABELA 4 |
||||||||||
| > 200–300 |
375 |
||||||||||
| > 300–400 |
450 |
525 |
|||||||||
| > 400–500 |
375 |
375 |
525 |
600 |
|||||||
| > 500–600 |
375 |
450 |
450 |
600 |
|||||||
| > 600–700 |
225 |
375 |
450 |
450 |
525 |
||||||
| > 700–800 |
225 |
225 |
300 |
375 |
450 |
450 |
525 |
600 |
|||
| > 800–900 |
225 |
225 |
300 |
375 |
450 |
525 |
600 |
||||
| > 900–1000 |
225 |
300 |
375 |
450 |
525 |
600 |
|||||
| > 1000–1100 |
225 |
300 |
375 |
450 |
600 |
Brak wystarczających danych, aby zalecić dawkę. |
|||||
| > 1100–1200 |
300 |
300 |
450 |
525 |
600 |
||||||
| > 1200–1300 |
300 |
375 |
450 |
525 |
|||||||
| > 1300–1500 |
300 |
375 |
525 |
600 |
|||||||
* W głównych badaniach dotyczących przewlekłego zapalenia zatok naczyniakowych z polipami nosowymi (CRSwNP) nie badano stosowania leku u pacjentów o masie ciała poniżej 30 kg.
Podanie
Tylko do podania podskórnie. Nie podawać dożylnie ani domięśniowo.
Dawki przekraczające 150 mg (tabela 5) należy podzielić na dwie lub więcej iniekcji.
Pacjentom, którzy nie mieli w wywiadzie przypadków anafilaksji, możliwe jest samodzielne podawanie Xolaru lub podawanie przez osobę opiekującą się pacjentem, począwszy od 4. dawki i dalej, jeśli lekarz uzna to za stosowne. Pacjent lub osoba opiekująca się nim musi zostać przeszkolona w zakresie prawidłowej techniki wstrzykiwania oraz rozpoznawania wczesnych objawów i objawów ciężkich reakcji alergicznych.
Pacjenci lub osoby opiekujące się nimi muszą zostać poinstruowane, aby podawać pełną dawkę Xolaru zgodnie z instrukcją dotyczącą stosowania medycznego.
Dzieciom w wieku od 6 do 12 lat nie należy samodzielnie podawać iniekcji leku Xolar, jednak jeśli lekarz uzna to za stosowne, osoba opiekująca się dzieckiem może podać mu tę iniekcję po odpowiednim przeszkoleniu.
Trwanie leczenia, kontrola i korekta dawki
Astma alergiczna
Xolar przeznaczony jest do długotrwałego leczenia. Badania kliniczne potwierdziły skuteczność leczenia Xolarem stosowanym przez co najmniej 12–16 tygodni. Na 16. tygodniu terapii Xolarem należy ocenić stan pacjenta pod kątem skuteczności leczenia przed podaniem kolejnych iniekcji. Decyzja o kontynuowaniu terapii Xolarem powinna opierać się na stwierdzeniu istotnego postępu w kontroli astmy.
Przewlekłe zapalenie zatok naczyniakowych z polipami nosowymi (CRSwNP)
W badaniach klinicznych dotyczących CRSwNP po 4 tygodniach obserwowano zmiany wskaznika polipów nosowych (WPN) i wskaznika zatkania nosa (WZN). Należy okresowo ponownie oceniać potrzebę kontynuowania terapii w zależności od ciężkości choroby pacjenta i poziomu kontroli objawów.
Astma alergiczna i przewlekłe zapalenie zatok naczyniakowych z polipami nosowymi (CRSwNP)
Przerwanie leczenia Xolarem w większości przypadków prowadzi do odwrotnej regresji poziomu wolnego IgE i rozwoju odpowiednich objawów. Poziom ogólnego IgE jest podwyższony w trakcie leczenia i pozostaje podwyższony przez rok po zakończeniu leczenia. W związku z tym powtarzające się oznaczanie poziomu IgE w trakcie leczenia Xolarem nie może być wykorzystywane do ustalenia wymaganej dawki leku. Oznaczanie dawki po przerwie w leczeniu trwającej krócej niż rok należy przeprowadzać na podstawie poziomu IgE uzyskanego przy wstępnym ustalaniu dawki. Poziom ogólnego IgE surowicy może być ponownie oznaczany w celu wyboru dawki, jeśli leczenie Xolarem zostało przerwane ponad rok temu.
W przypadku istotnych zmian masy ciała konieczna jest korekta dawki.
Przewlekła pokrzywka samoistna (CSU), dawkowanie 150 mg/1 ml
Zalecana dawka to 300 mg podawane podskórnie co 4 tygodnie.
Należy okresowo ponownie oceniać potrzebę kontynuowania przyjmowania leku.
Dane z badań klinicznych dotyczące leczenia trwającego dłużej niż 6 miesięcy wskazanego w tym przypadku są ograniczone.
Osobliwe populacje
Osoby starsze (65 lat i więcej)
Dostępne są ograniczone dane dotyczące stosowania leku Xolar u pacjentów w wieku powyżej 65 lat, jednak nie ma podstaw do przypuszczeń, że osoby starsze wymagają innego podejścia do dawkowania niż młodsze osoby dorosłe.
Pacjenci z zaburzeniami funkcji nerek lub wątroby
Nie przeprowadzono badań wpływu zaburzeń funkcji nerek lub wątroby na farmakokinetykę leku Xolar. Ponieważ klirens omalizumabu w dawkach klinicznych odbywa się głównie za pośrednictwem układu siateczkowo-śródbłonkowego (RES), mało prawdopodobne jest jego zmiany związane z zaburzeniami funkcji nerek lub wątroby. Choć nie ma specyficznych zaleceń dotyczących korekty dawki, Xolar należy stosować u tych pacjentów z ostrożnością.
Dzieci
Bezpieczeństwo i skuteczność Xolaru w leczeniu astmy alergicznej u pacjentów w wieku poniżej 6 lat nie zostały ustalone. Brak danych.
Bezpieczeństwo i skuteczność Xolaru w leczeniu CRSwNP u pacjentów w wieku poniżej 18 lat nie zostały ustalone.
Bezpieczeństwo i skuteczność Xolaru w leczeniu CSU u pacjentów w wieku poniżej 12 lat nie zostały ustalone.
Specjalne wskazówki dotyczące stosowania
Opakowanie zawiera wypełniony strzykawkę zapakowaną w plastikowy pojemnik (rys. 3).
| Ochronna osłona strzykawki |
| Skrzydełka zabezpieczające strzykawki |
| Ochrona igły |
| Okienko przeglądowe Oznakowanie i termin ważności |
| Uchwyty na palce |
| Porusz |
| Głowa tłokowa |
Rys. 3. Wygląd zewnętrzny Xolaru, roztworu do wstrzykiwań, 75 mg/0,5 ml lub 150 mg/1 ml, w szprycie wstępnie napełnionej (niebieski dla 75 mg/0,5 ml; fioletowy dla 150 mg/1 ml).
- Nie otwieraj uszczelnionego zewnętrznego opakowania plastikowego, dopóki nie będziesz gotowy(-wa) na zastosowanie Xolaru, roztworu do wstrzykiwań.
- Nie stosuj leku, jeśli uszczelnienie na zewnętrznej skrzynce lub uszczelnienie tacki plastikowej jest uszkodzone, ponieważ może to stanowić zagrożenie.
- Nie wstrząsaj szprycy.
- Bądź ostrożny(-na), aby nie dotykać skrzydełek ochronnych szprycy przed użyciem. Dotknięcie skrzydełek może spowodować zbyt wczesne aktywowanie szprycy ochronnej.
- Zdejmij osłonkę igły bezpośrednio tuż przed wykonaniem wstrzyknięcia.
- Szprycy nie można ponownie używać. Zużytą szprycę należy bezwzględnie usunąć natychmiast po użyciu.
- Przed użyciem należy wyjąć szprycę z lodówki i pozwolić jej osiągnąć temperaturę pokojową (25°C) przed przygotowaniem do wstrzyknięcia (zajmie to około 20 minut). Szprycę należy pozostawić w opakowaniu, aby chronić ją przed światłem. W razie potrzeby szprycę można ponownie umieścić w lodówce. Całkowity czas, przez który szprycę wolno przechowywać w temperaturze pokojowej (25°C) przed użyciem, nie powinien przekraczać 48 godzin.
Miejsce wstrzyknięcia
Jeśli zastrzyk wykonuje osoba opiekująca się pacjentem, można również użyć zewnętrznej części barku. |
Przygotowanie Xolaru, roztworu do wstrzykiwań (75 mg/0,5 ml lub 150 mg/1 ml) w szprycie wstępnie napełnionej, przed wstrzyknięciem
Uwaga: W zależności od dawki przepisanej przez lekarza może być konieczne przygotowanie jednej lub kilku szpryc wstępnie napełnionych i wstrzyknięcie zawartości wszystkich przygotowanych szpryc. W Tabeli 8 przedstawiono przykłady liczby wstrzyknięć każdej dawki potrzebnych do osiągnięcia przepisanej dawki.
Tabela 8
| Liczba strzykawek potrzebnych do podania dawki |
| Dawka |
|
- Wyjmij opakowanie ze strzykawką z lodówki i pozostaw je otwarte na około 20 minut, aby osiągnęło temperaturę pokojową (pozostaw strzykawkę w opakowaniu, aby chronić ją przed światłem).
- Gdy będziesz gotowy do użycia strzykawki, dokładnie umyj ręce wodą z mydłem.
- Przetrzyj miejsce zastrzyku tamponem alkoholowym.
- Wyjmij plastikowy pojemnik z opakowania i odklej papierową warstwę. Wyjmij strzykawkę z pojemnika, chwytając ją za środkową część osłony ochronnej.
- Sprawdź strzykawkę. Roztwór powinien być od przezroczystego do nieco mętnego. Kolor roztworu może wahać się od bezbarwnego do lekko brązowozółtego. Możesz zauważyć pęcherzyk powietrza, co jest normalne. NIE UŻYWAJ, jeśli strzykawka jest uszkodzona lub jeśli ciecz jest wyraźnie mętna, wyraźnie brązowa lub zawiera cząstki.
- Trzymając strzykawkę poziomo, sprawdź przez okienko kontrolne datę ważności wydrukowaną na etykiecie. Uwaga: Można obrócić wewnętrzną część zespołu strzykawki, aby zobaczyć etykietę w okienku kontrolnym. NIE UŻYWAJ, jeśli minął termin ważności.
Jak stosować Xolar, roztwór do wstrzykiwań (75 mg/0,5 ml lub 150 mg/1 ml) w strzykawce wstępnie napełnionej
|
|
Delikatnie zdejmij osłonkę igły ze strzykawki. Odrzuć osłonkę igły. Na końcu igły może pojawić się kropla płynu. Jest to normalne. |
|
|
|
Delikatnie naciśnij skórę w miejscu wstrzyknięcia i całkowicie wsuń igłę, jak pokazano, aby zapewnić pełne wprowadzenie leku. |
|
|
|
Trzymaj strzykawkę, jak pokazano. Wolno naciskaj tłoczek do oporu, aby głowica tłoczka całkowicie znalazła się między ochronnymi skrzydełkami strzykawki. |
|
|
|
Trzymaj tłok całkowicie wciśnięty, podczas ostrożnego wyjmowania igły z miejsca iniekcji. |
|
|
|
Wolno puścić tłoczek, po czym osłona ochronna strzykawki automatycznie zakryje igłę. W miejscu wstrzyknięcia może pojawić się niewielka ilość krwi. Można przyłożyć watę lub gazę do miejsca wstrzyknięcia i przytrzymać przez 30 sekund. Nie należy wcierać w miejsce wstrzyknięcia. W razie potrzeby miejsce wstrzyknięcia można zakleić małym plasterkiem. |
Każdy nie wykorzystany lek lub odpady należy usunąć zgodnie z lokalnymi przepisami.
Dzieci.
Astma alergiczna
Xolar można stosować u dzieci od 6. roku życia, które już przyjmują leki na astmę, ale objawy astmy nie są wystarczająco dobrze kontrolowane przez takie leki, jak sterydowe inhalatory w wysokich dawkach oraz inhalatory agonistów beta.
Dzieci w wieku od 6 do 11 lat nie powinny samodzielnie stosować Xolaru w wstępnie napełnionej strzykawce. Jeśli lekarz uzna to za stosowne, osoba opiekująca się dzieckiem może wykonać wstrzyknięcie Xolaru po odpowiednim przeszkoleniu.
Przewlekłe zapalenie zatok nosowych z polipami nosowymi (CRSwNP)
Xolar nie powinien być stosowany u dzieci (do 18. roku życia) w przypadku tego wskazania.
Przewlekła spontaniczna pokrzywka (CSU)
Xolar można stosować u dzieci od 12. roku życia, które już przyjmują leki przeciwhistaminowe, ale objawy CSU nie są odpowiednio kontrolowane przez te leki. Dawkowanie dla nastolatków od 12. roku życia jest takie samo jak u dorosłych.
Przedawkowanie.
Maksymalna dawka tolerowana Xolaru nie została określona. Podanie dożylnie pacjentom pojedynczej dawki do 4 000 mg nie powodowało objawów toksyczności granicznej dawki. Najwyższa kumulacyjna dawka podana pacjentowi wyniosła 44 000 mg w ciągu 20-tygodniowego okresu i nie spowodowała żadnych ostrych niepożądanych skutków.
W przypadku podejrzenia przedawkowania należy monitorować stan pacjenta pod kątem wszelkich nietypowych objawów. Należy odpowiednio rozpoznać stan i zastosować leczenie.
Efekty niepożądane.
Astma alergiczna i przewlekłe zapalenie nosa z zatokami i polipami nosowymi (CRSwNP)
Podczas badań klinicznych z udziałem dorosłych i dzieci w wieku od 12 lat z astmą alergiczną najczęściej występującymi efektami niepożądanymi były ból głowy oraz reakcje w miejscu wstrzyknięcia, w tym ból, obrzęk, zaczerwienienie i świąd w miejscu wstrzyknięcia. W trakcie badań klinicznych u dzieci w wieku od 6 do 12 lat najczęściej występującymi efektami niepożadanymi związanymi z lekiem były ból głowy, gorączka i ból w górnej części brzucha. Większość reakcji miała charakter łagodny lub umiarkowany. Podczas badań klinicznych u pacjentów w wieku od 18 lat z CRSwNP najczęściej występującymi efektami niepożądanymi związanymi z lekiem były ból głowy, zawroty głowy, artrealgia, ból w górnej części brzucha oraz reakcje w miejscu wstrzyknięcia.
W tabeli 9 wymieniono efekty niepożądane zaobserwowane podczas badań klinicznych w ogólnej populacji badanej pod kątem bezpieczeństwa w przypadku astmy alergicznej i CRSwNP, pogrupowane według klas układów narządów MedDRA i częstości występowania. W ramach każdej grupy według częstości efekty niepożądane zostały uporządkowane według malejącej ciężkości. Częstość określono następująco: bardzo często (≥ 1/10), często (≥ 1/100 – < 1/10), nieczęsto (≥ 1/1000 – < 1/100), rzadko (≥ 1/10000 – < 1/1000) i bardzo rzadko (< 1/10000). Częstość zdarzeń, o których zgłoszono w okresie postmarketingowym, określono jako nieznaną (nie można oszacować na podstawie dostępnych danych).
Tabela 9
Efekty niepożądane zaobserwowane podczas badań klinicznych w przypadku astmy alergicznej i CRSwNP
| Częstość |
Reakcje niepożądane |
| Infekcje i inwazje |
|
| Nieczęsto |
Farągryt |
| Rzadko |
Inwazje pasożytnicze |
| Z układy krwi i chłonnego |
|
| Częstość nieznana |
Idiopatyczna trombocytopenia, w tym przypadki ciężkie |
| Z układy odpornościowego |
|
| Rzadko |
Reakcja anafilaktyczna, inne ciężkie stany alergiczne, rozwój przeciwciał przeciwko lekowi |
| Częstość nieznana |
Choroba surowicy, która może obejmować gorączkę i limfadenopatię |
| Z układy nerwowego |
|
| Często |
Ból głowy* |
| Nieczęsto |
Syncope, parestezje, senność, zawroty głowy*** |
| Z układy naczyniowego |
|
| Nieczęsto |
Obniżenie ciśnienia ortostatycznego, napływy krwi |
| Z układy oddechowego, klatki piersiowej i przestrzeni międzywyrostkowej |
|
| Nieczęsto |
Alergiczny skurcz oskrzeli, kaszel |
| Rzadko |
Obniżenie krtani |
| Częstość nieznana |
Alergiczny gruźliczy zapalenie naczyń (zespoł Charga – Straussa) |
| Z układy pokarmowego |
|
| Często |
Ból w górnej części brzucha**,*** |
| Nieczęsto |
Oznaki lub objawy niestrawności, biegunka, nudności |
| Z układy skóry i tkanki podskórnej |
|
| Nieczęsto |
Światłoczułość, pokrzywka, wysypka, swędzenie |
| Rzadko |
Przyśrodkowy obrzęk naczynioruchowy |
| Częstość nieznana |
Alopepsja |
| Z układy mięśniowo-szkieletowego i tkanki łącznej |
|
| Nieczęsto Rzadko Częstość nieznana |
Artrologia**** Układowe toczeńcze zapalenie skóry (SLE) Mialgia, obrzęk stawów |
| Ogólne zaburzenia i stanów związane ze sposobem stosowania leku |
|
| Bardzo często |
Piroksja** |
| Często |
Reakcje w miejscu wstrzyknięcia, takie jak obrzęk, zaczerwienienie, ból, swędzenie |
| Nieczęsto |
Choroba podobna do grypy, obrzęk kończyn górnych, przyrost masy ciała, zmęczenie |
* Bardzo często u dzieci w wieku od 6 do 12 lat.
** U dzieci w wieku od 6 do 12 lat.
*** Często w badaniach dotyczących polipów nosowych.
**** Częstotliwość nieznana w badaniach dotyczących astmy alergicznej.
Przewlekła pokrzywka samoistna
Bezpieczeństwo i tolerancję omalizumabu oceniano po podaniu dawek 75 mg, 150 mg i 300 mg co 4 tygodnie u 975 pacjentów z przewlekłą pokrzywką samoistną (PPS), z których 242 otrzymywało placebo. Ogółem 733 pacjentów otrzymywało omalizumab przez okres do 12 tygodni, a 490 pacjentów – do 24 tygodni. Wśród nich 412 pacjentów otrzymywało omalizumab przez okres do 12 tygodni, a 333 pacjentów – do 24 tygodni w dawce 300 mg.
Efekty niepożądane w przebiegu PPS występowały u pacjentów otrzymujących różne dawki, u których stwierdzano istotne czynniki ryzyka, choroby współistniejące, różny wiek oraz stosowanie leków współadministracyjnych (np. badania dotyczące astmy obejmowały dzieci w wieku od 6 do 12 lat), patrz tabela 10.
W tabeli 10 wymieniono efekty niepożądane (zdarzenia występujące u ≥1% pacjentów w dowolnej grupie leczenia, które występowały co najmniej o 2% częściej w dowolnej grupie leczenia omalizumabem niż w grupie placebo (po ocenie lekarskiej)), obserwowane po podaniu dawki 300 mg w trzech połączonych badaniach fazy III. Wymienione efekty niepożądane podzielono na dwie grupy: reakcje występujące w trakcie 12- i 24-tygodniowego okresu leczenia.
Efekty niepożądane wymieniono według klas układów narządów MedDRA. W ramach każdej klasy układów narządów efekty niepożądane uporządkowano według częstości występowania, zaczynając od najczęściej występujących. Kategorie częstości zdefiniowano następująco: bardzo często (≥1/10); często (≥1/100 do <1/10); nieczęsto (≥1/1000 do <1/100); rzadko (≥1/10000 do <1/1000); bardzo rzadko (<1/10000); nieznana (częstości nie można ustalić na podstawie dostępnych danych).
| Tabela 10 |
|||
| Odczyny niepożądane według połączonych danych dotyczących bezpieczeństwa w przypadku XSK (dzień 1 – tydzień 24) przy stosowaniu omalizumabu w dawce 300 mg |
|||
| Leczenie 12-tygodniowe |
Połączone badania 1, 2 i 3 omalizumabu |
Kategoria częstości |
|
| Placebo N = 242 |
300 mg N = 412 |
||
| Infekcje i inwazje |
|||
| Zatkanie nosa |
5 (2,1 %) |
20 (4,9 %) |
Często |
| Zaburzenia ze strony układu nerwowego |
|||
| Ból głowy |
7 (2,9 %) |
25 (6,1 %) |
Często |
| Zaburzenia ze strony układu mięśniowo-szkieletowego i tkanki łącznej |
|||
| Artrodynia |
1 (0,4 %) |
12 (2,9 %) |
Często |
| Zaburzenia ogólne i w miejscu podania |
|||
| Reakcje w miejscu wstrzyknięcia* |
2 (0,8 %) |
11 (2,7 %) |
Często |
| Leczenie 24-tygodniowe |
Połączone badania 1 i 3 omalizumabu |
Kategoria częstości |
|
| Placebo N = 163 |
300 mg N = 333 |
||
| Infekcje i inwazje |
|||
| Infekcja dróg oddechowych górnych |
5 (3,1 %) |
19 (5,7 %) |
Często |
* Chociaż różnica w porównaniu z placebo większa niż 2 % nie została wykazana, reakcje w miejscu wstrzyknięcia zostały uwzględnione, ponieważ we wszystkich przypadkach stwierdzono związek przyczynowo-skutkowy związany z leczeniem w ramach badania.
Opis poszczególnych działań niepożądanych
Zaburzenia ze strony układu odpornościowego
W celu uzyskania dodatkowych informacji należy zapoznać się z sekcją „Szczególne ostrzeżenia i środki ostrożności”.
Anafilaksja
Reakcje anafilaktyczne podczas badań klinicznych obserwowano bardzo rzadko. Jednak w wyniku łącznego przeszukania bazy danych dotyczącej bezpieczeństwa w badaniach pozarejestrowych stwierdzono łącznie 898 przypadków anafilaksji. Na podstawie obliczonej ekspozycji wynoszącej 566 923 pacjentolata leczenia odpowiada to stopniowi notowania około 0,20 %.
Zatorowość tętnicza (ATE)
W trakcie kontrolowanych badań klinicznych oraz w bieżącym analizie badań obserwacyjnych zaobserwowano liczbową nierównowagę ATE. Definicja łącznego punktu końcowego ATE obejmowała udar mózgu, przemijające ataki niedokrwienne, zawał mięśnia sercowego, niestabilną dławicę bolesną oraz śmierć sercowo-naczyniową (w tym śmierć z nieznanej przyczyny). W końcowej analizie badania obserwacyjnego częstość ATE na 1000 pacjentolat wynosiła 7,52 (115/15286 pacjentolat) w grupie pacjentów przyjmujących lek Xolar oraz 5,12 (51/9963 pacjentolat) w grupie kontrolnej. W analizie wieloczynnikowej z uwzględnieniem początkowych czynników ryzyka sercowo-naczyniowego stosunek ryzyka wynosił 1,32 (95 % przedział ufności 0,91–1,91). W oddzielnej analizie połączonych badań klinicznych, które obejmowały wszystkie randomizowane, podwójnie ślepe, kontrolowane placebo badania kliniczne trwające 8 lub więcej tygodni, częstość ATE na 1000 pacjentolat wynosiła 2,69 (5/1856 pacjentolat) w grupie pacjentów przyjmujących lek Xolar oraz 2,38 (4/1680 pacjentolat) w grupie placebo (stosunek ryzyka 1,13, 95 % przedział ufności 0,24–5,71).
Płytki krwi
Podczas badań klinicznych u kilku pacjentów stwierdzono liczbę płytek krwi poniżej dolnej granicy normy laboratoryjnej. W żadnym z tych przypadków zmiana ta nie prowadziła do krwawień ani obniżenia stężenia hemoglobiny. U ludzi (pacjenci w wieku od 6 lat) nie obserwowano przypadków trwałej trombocytopenii podobnych do tych obserwowanych u małp, choć w okresie pozarejestrowym otrzymano pojedyncze doniesienia o przypadkach trombocytopenii idiopatycznej, w tym ciężkiej.
Inwazje pasożytnicze
Badanie kontrolowane placebo wykazało nieznaczny wzrost zachorowań u pacjentów z trwałym wysokim ryzykiem inwazji nicieni na tle przyjmowania omalizumabu, co nie było statystycznie istotne. Przebieg, nasilenie i odpowiedź na leczenie inwazji nie uległy zmianie.
Zgłaszanie działań niepożądanych po rejestracji leku ma istotne znaczenie. Umożliwia to monitorowanie stosunku korzyści do ryzyka stosowania tego leku. Osoby pracujące w zawodach medycznych i farmaceutycznych, a także pacjenci lub ich ustawowi przedstawiciele powinni zgłaszać wszystkie przypadki podejrzewanych działań niepożądanych oraz braku skuteczności leku poprzez Zautomatyzowany System Informacyjny Nadzoru Farmakologicznego pod adresem: https://aisf.dec.gov.ua
Okres ważności. 18 miesięcy.
Lek może być przechowywany w temperaturze 25 °C przez 48 godzin (w całości). W razie potrzeby lek można ponownie umieścić w lodówce do dalszego użytku.
Warunki przechowywania.
Przechowywać w temperaturze 2–8 °C w miejscu niedostępnym dla dzieci. Nie zamarzać. Przechowywać w oryginalnym opakowaniu.
Opakowanie.
Roztwór do wstrzykiwań, 75 mg/0,5 ml
Po 0,5 ml roztworu do wstrzykiwań w strzykawce wstępnie napełnionej z zamocowaną igłą i osłoną na igłę. Po 1 strzykawce wstępnie napełnionej w blisterze; 1 blister w pudełku kartonowym.
Roztwór do wstrzykiwań, 150 mg/1 ml
Po 1 ml roztworu do wstrzykiwań w strzykawce wstępnie napełnionej z zamocowaną igłą i osłoną na igłę. Po 1 strzykawce wstępnie napełnionej w blisterze; 1 blister w pudełku kartonowym.
Kategoria wydania. Na receptę.
Producent.
Novartis Pharma GmbH, Niemcy / Novartis Pharma GmbH, Germany.
Miejsce położenia producenta oraz adres miejsca prowadzenia działalności.
Roonstrasse 25, Gostenhof, Nuremberg, Bavaria, 90429, Niemcy /
Roonstrasse 25, Gostenhof, Nuremberg, Bavaria, 90429, Germany.