Xolar
Ukraine
Table of Contents
INSTRUCTIONS FOR MEDICAL USE OF THE MEDICINAL PRODUCT XOLAIR® (XOLAR)
Composition:
Active substance: omalizumab;
One pre-filled syringe containing 0.5 mL of solution contains 75 mg of omalizumab*;
Excipients: L-arginine hydrochloride, L-histidine hydrochloride monohydrate, L-histidine, polysorbate 20, water for injections.
or
One pre-filled syringe containing 1 mL of solution contains 150 mg of omalizumab*;
Excipients: L-arginine hydrochloride, L-histidine hydrochloride monohydrate, L-histidine, polysorbate 20, water for injections.
* Omalizumab is a humanized monoclonal antibody produced by recombinant DNA technology in Chinese hamster ovary (CHO) cells.
Pharmaceutical form. Solution for injection.
Main physicochemical properties: solution ranging from clear to slightly turbid; colorless to pale brownish-yellow.
Pharmacotherapeutic group. Agents for systemic use in obstructive respiratory diseases. ATC code R03DX05.
Pharmacological Properties.
Pharmacodynamics.
Omalizumab is a humanized monoclonal antibody produced by recombinant DNA technology that selectively binds to human immunoglobulin E (IgE). The antibody is a kappa-IgG1 containing a human framework region with the complementarity-determining region of a murine parent antibody that binds to IgE. Treatment with Xolair inhibits IgE-mediated inflammation, as evidenced by reductions in blood and tissue eosinophil levels and decreased levels of inflammatory mediators, including IL-4, IL-5, and IL-13, from innate, adaptive, and non-immune cells.
Mechanism of Action
Omalizumab binds to IgE and prevents its binding to FcεRI (the high-affinity IgE receptor) on basophils and mast cells, thereby reducing the amount of free IgE capable of triggering the allergic cascade. Treatment of patients with atopic diseases with omalizumab resulted in significant suppressive effects on basophil FcεRI receptors.
Pharmacodynamic Effects
Allergic Asthma
In vitro histamine release following allergen challenge of basophils obtained from patients treated with Xolair was reduced by approximately 90% compared to pre-treatment levels.
In clinical trials in patients with allergic asthma, dose-dependent reductions in serum free IgE levels were observed within one hour after the first dose, and these reductions were maintained between doses. One year after discontinuation of Xolair treatment, IgE levels returned to pre-treatment levels, with no rebound increase in IgE levels observed after the drug was eliminated from the body.
Chronic Rhinosinusitis with Nasal Polyps (CRSwNP)
In clinical trials in patients with CRSwNP, treatment with Xolair led to a reduction in serum free IgE levels (by approximately 95%) and an increase in total serum IgE levels to a similar extent as observed in patients with allergic asthma. The increase in total serum IgE was due to the formation of omalizumab-IgE complexes, which are cleared more slowly than free IgE.
Chronic Spontaneous Urticaria (CSU)
Mechanism of Action
Omalizumab binds to IgE and reduces serum free IgE levels. This is followed by downregulation of IgE receptors (FcεRI) on cells. The exact mechanism by which this leads to symptom control in chronic spontaneous urticaria is not fully understood.
Pharmacodynamic Effects
In clinical trials in patients with CSU, maximum suppression of free IgE levels was observed within three days after the first subcutaneous dose. After repeated dosing every 4 weeks, serum free IgE levels prior to dosing remained stable between weeks 12 and 24 of treatment. After discontinuation of Xolair, free IgE levels increased, reaching pre-treatment levels within 16 weeks.
Clinical Experience in Allergic Asthma
Adults and Children Aged 12 Years and Older
The efficacy and safety of Xolair were demonstrated in a 28-week, double-blind, placebo-controlled trial (Study 1) involving 419 patients aged 12–79 years with severe allergic asthma, who had reduced lung function (FEV1 40–80% of predicted) and poorly controlled asthma symptoms despite treatment with high-dose inhaled corticosteroids and long-acting beta2-agonists. Patients enrolled had a history of multiple asthma exacerbations requiring systemic corticosteroids or hospitalization, or emergency care due to asthma exacerbation, despite continuous treatment with high-dose inhaled corticosteroids and long-acting beta2-agonists. Xolair or placebo was administered subcutaneously as an add-on to therapy with beclomethasone dipropionate (or equivalent) at doses >1000 mcg and a long-acting beta2-agonist. Concomitant use of oral corticosteroids, theophylline, and leukotriene modifiers was permitted (in 22%, 27%, and 35% of patients, respectively).
The primary endpoint was the rate of asthma exacerbations requiring treatment with systemic corticosteroids. Omalizumab reduced the rate of asthma exacerbations by 19% (p = 0.153). Additional assessments showing statistical significance (p < 0.05) in favor of Xolair included reductions in the rate of severe exacerbations (defined as those resulting in lung function declining to below 60% of the patient’s best and requiring systemic corticosteroids) and asthma-related unplanned healthcare visits (comprising hospitalizations, emergency care, and unscheduled physician visits), as well as improvements in physician-rated overall treatment effectiveness, asthma-related quality of life (AQL), asthma symptoms, and lung function.
In a subgroup analysis of patients with a baseline total IgE level ≥76 IU/mL, a greater likelihood of clinically meaningful benefit from Xolair was demonstrated. In these patients, during Study 1, Xolair reduced the rate of exacerbations by 40% (p = 0.002). Furthermore, in the Xolair severe asthma development program, a higher proportion of patients with a baseline total IgE level ≥76 IU/mL showed a clinically meaningful response, see Table 1.
Table 1
Results of Study 1
| Overall population of Study 1 |
||
| Study |
Xolair N = 209 |
Placebo N = 210 |
| Asthma Exacerbations |
||
| Rate over 28 weeks, |
0.74 |
0.92 |
| % reduction, p-value for rate ratio |
19.4%, p = 0.153 |
|
| Severe Asthma Exacerbations |
||
| Rate over 28 weeks, |
0.24 |
0.48 |
| % reduction, p-value for rate ratio |
50.1%, p = 0.002 |
|
| Emergency Physician Visits |
||
| Rate over 28 weeks, |
0.24 |
0.43 |
| % reduction, p-value for rate ratio |
43.9%, p = 0.038 |
|
| Overall Investigator Assessment |
||
| % of patients who responded*, |
60.5% |
42.8% |
| p-value ** |
<0.001 |
|
| AQL Improvement |
||
| % of patients with ≥ 0.5 improvement, |
60.8% |
47.8% |
| p-value |
0.008 |
|
* Significant improvement or complete control.
** p-value for overall distribution score.
Study 2 evaluated the efficacy and safety of XOLAIR in a population of 312 patients with severe allergic asthma whose characteristics matched those of the population in Study 1. Treatment with XOLAIR in this open-label study resulted in a 61% reduction in the rate of clinically significant asthma exacerbations compared to current asthma therapy alone.
Four additional large placebo-controlled add-on studies of XOLAIR, lasting from 28 to 52 weeks, assessed the efficacy and safety of XOLAIR in 1722 adults and children aged 12 years and older (Studies 3, 4, 5, 6) with severe persistent asthma. Most patients had poorly controlled asthma but received less concomitant asthma therapy than patients in Studies 1 or 2. Studies 3–5 used exacerbations as the primary endpoint, whereas Study 6 primarily assessed reduction in the need for inhaled corticosteroids.
During Studies 3, 4, and 5, patients receiving XOLAIR experienced reductions in the rate of asthma exacerbations of 37.5% (p = 0.027), 40.3% (p < 0.001), and 57.6% (p < 0.001), respectively, compared to placebo.
In Study 6, a significantly higher proportion of patients with severe allergic asthma treated with XOLAIR were able to reduce their fluticasone dose to ≤ 500 mcg/day without worsening asthma control (60.3%) compared to the placebo group (45.8%, p < 0.05).
Asthma-related quality of life was measured using the Juniper questionnaire. For all six studies, a statistically significant improvement in quality of life was observed in patients receiving XOLAIR compared to those in the placebo or control group, relative to baseline.
Overall physician assessment of treatment efficacy.
Overall assessment of treatment efficacy was conducted by the physician in the five aforementioned studies as a measure of asthma control. The physician could consider peak expiratory flow (PEF), daytime and nighttime symptoms, rescue medication use, spirometry data, and asthma exacerbations. In all five studies, a statistically significantly greater proportion of patients receiving XOLAIR were considered to have achieved significant improvement or complete asthma control compared to patients in the placebo group.
Children aged 6 to 12 years
The primary confirmation of safety and efficacy of XOLAIR in the 6 to 12 years age group comes from one randomized, double-blind, placebo-controlled, multicenter study (Study 7).
Study 7 was a placebo-controlled trial that included a specific subgroup (n = 235) of patients, as defined in the current indications, who were receiving high-dose inhaled corticosteroids (≥ 500 mcg/day fluticasone equivalent) plus long-acting beta-agonists.
A clinically significant asthma exacerbation was defined as worsening asthma symptoms as assessed clinically by the investigator, requiring doubling of the initial inhaled corticosteroid dose for at least 3 days and/or treatment with systemic corticosteroids (oral or intravenous) for at least 3 days due to urgent medical need.
In the specific subgroup of patients receiving high-dose inhaled corticosteroids, the omalizumab treatment group had a statistically significantly lower rate of clinically significant asthma exacerbations compared to the placebo group. At Week 24, the difference in rates between treatment groups was 34% (rate ratio 0.662, p = 0.047) reduction compared to placebo in patients receiving omalizumab. During the second double-blind 28-week treatment period, the difference in rates between treatment groups was 63% (rate ratio 0.37, p < 0.001) reduction compared to placebo in patients receiving omalizumab.
During the 52-week double-blind treatment period (which included a 24-week fixed-dose steroid phase and a 28-week steroid dose-adjustment phase), the difference in rates between treatment groups was 50% (rate ratio 0.504, p < 0.001) relative reduction in exacerbation rates in patients receiving omalizumab.
In the omalizumab group, there was a greater reduction in the frequency of rescue beta-agonist use compared to the placebo group at the end of the 52-week treatment period, although the difference between groups was not statistically significant. In the overall assessment of treatment efficacy at the end of the 52-week double-blind treatment period in the subgroup of severe patients receiving high-dose inhaled corticosteroids plus long-acting beta-agonists, the proportion of patients with an "excellent" treatment efficacy rating was higher, and the proportions with a "fair" or "poor" rating were lower in the omalizumab group compared to the placebo group, with a statistically significant difference between groups (p < 0.001). However, there were no differences between the omalizumab and placebo groups regarding subjective quality-of-life assessments.
Clinical experience in chronic rhinosinusitis with nasal polyps (CRSwNP)
The safety and efficacy of XOLAIR were evaluated in two randomized, double-blind, placebo-controlled studies involving patients with CRSwNP (Table 2). Patients received XOLAIR or placebo subcutaneously every 2 or 4 weeks (see section "Posology and method of administration"). All patients received background intranasal therapy with mometasone throughout the study. Prior sinonasal surgery or prior systemic corticosteroid use was not required for study entry. Patients received XOLAIR or placebo for 24 weeks followed by a 4-week observation period. Demographic and baseline characteristics, including allergic comorbidities, are described in Table 2.
The primary endpoints were bilateral nasal polyp scores (NPS) and mean daily nasal congestion score (NCS) at Week 24. In both nasal polyp studies (Study 1 and Study 2), patients receiving XOLAIR showed statistically significant greater improvements from baseline at Week 24 in NPS and mean weekly NCS compared to patients receiving placebo. Results from Studies 1 and 2 on nasal polyps are presented in Table 3.
Table 2
Demographic and baseline characteristics in nasal polyp studies
| Parameter |
Nasal Polyp Study 1 N = 138 |
Nasal Polyp Study 2 N = 127 |
| Mean age (years) (SD) |
51.0 (13.2) |
50.1 (11.9) |
| % male |
63.8 |
65.4 |
| Patients who received systemic corticosteroids in the prior year (%) |
18.8 |
26.0 |
| Bilateral nasal polyp score (NPS): mean (SD), scale 0–8 |
6.2 (1.0) |
6.3 (0.9) |
| Nasal congestion score (NCS): mean (SD), scale 0–3 |
2.4 (0.6) |
2.3 (0.7) |
| Smell sensation level: mean (SD), scale 0–3 |
2.7 (0.7) |
2.7 (0.7) |
| SNOT-22 total score: mean (SD), scale 0–110 |
60.1 (17.7) |
59.5 (19.3) |
| Blood eosinophil count (cells/µL): mean (SD) |
346.1 (284.1) |
334.6 (187.6) |
| Total IgE, IU/mL: mean (SD) |
160.9 (139.6) |
190.2 (200.5) |
| Asthma (%) |
53.6 |
60.6 |
| Mild (%) |
37.8 |
32.5 |
| Moderate (%) |
58.1 |
58.4 |
| Severe (%) |
4.1 |
9.1 |
| Aspirin-induced asthma (%) |
19.6 |
35.4 |
| Allergic rhinitis (%) |
43.5 |
42.5 |
SD – standard deviation;
SNOT-22 – 22-item Sino-Nasal Outcome Test;
IgE – immunoglobulin E;
IU – international units.
Higher scores on NPS, SNOT, and SNOT-22 indicate more severe disease.
Table 3
Change from baseline at Week 24 in clinical assessments in Studies 1 and 2 for nasal polyposis and pooled data
| Parameter |
Nasal Polyps, |
Nasal Polyps, |
Nasal Polyps, |
|||
| Placebo |
Xhance |
Placebo |
Xhance |
Placebo |
Xhance |
|
| N |
66 |
72 |
65 |
62 |
131 |
134 |
| Nasal Polyp Score |
||||||
| Mean baseline value |
6.32 |
6.19 |
6.09 |
6.44 |
6.21 |
6.31 |
| LS-mean value at Week 24 |
0.06 |
-1.08 |
-0.31 |
-0.90 |
-0.13 |
-0.99 |
| Difference (95%) CI |
-1.14 (-1.59, -0.69) |
-0.59 (-1.05, -0.12) |
-0.86 (-1.18, -0.54) |
|||
| p-value |
< 0.0001 |
0.0140 |
< 0.0001 |
|||
| Mean (over 7 days) daily NPS |
||||||
| Mean baseline value |
2.46 |
2.40 |
2.29 |
2.26 |
2.38 |
2.34 |
| LS-mean value at Week 24 |
-0.35 |
-0.89 |
-0.20 |
-0.70 |
-0.28 |
-0.80 |
| Difference (95%) CI |
-0.55 (-0.84, -0.25) |
-0.50 (-0.80, -0.19) |
-0.52 (-0.73, -0.31) |
|||
| p-value |
0.0004 |
0.0017 |
< 0.0001 |
|||
| RSS |
||||||
| Mean baseline value |
9.33 |
8.56 |
8.73 |
8.37 |
9.03 |
8.47 |
| LS-mean value at Week 24 |
-1.06 |
-2.97 |
-0.44 |
-2.53 |
-0.77 |
-2.75 |
| Difference (95%) |
-1.91 (-2.85, -0.96) |
-2.09 (-3.00, -1.18) |
-1.98 (-2.63, -1.33) |
|||
| p-value |
0.0001 |
< 0.0001 |
< 0.0001 |
|||
| SNOT-22 |
||||||
| Mean baseline value |
60.26 |
59.82 |
59.80 |
59.21 |
60.03 |
59.54 |
| LS-mean value at Week 24 |
-8.58 |
-24.70 |
-6.55 |
-21.59 |
-7.73 |
-23.10 |
| Difference (95%) |
-16.12 (-21.86, -10.38) |
-15.04 (-21.26, -8.82) |
-15.36 (-19.57, -11.16) |
|||
| p-value |
< 0.0001 |
< 0.0001 |
< 0.0001 |
|||
| (MCID = 8.9) |
||||||
| UPSIT |
||||||
| Mean baseline value |
13.56 |
12.78 |
13.27 |
12.87 |
13.41 |
12.82 |
| LS-mean value at Week 24 |
0.63 |
4.44 |
0.44 |
4.31 |
0.54 |
4.38 |
| Difference (95%) |
3.81 (1.38, 6.24) |
3.86 (1.57, 6.15) |
3.84 (2.17, 5.51) |
|||
| p-value |
0.0024 |
0.0011 |
< 0.0001 |
|||
MNK – method of least squares; CI – confidence interval; NSI – nasal congestion index; TNS – total nasal symptom score; SNOT-22 – 22-item sino-nasal outcome test; UPSIT – University of Pennsylvania Smell Identification Test; MIDS – minimal important difference.
| Initial value |
| Initial value |
| Primary analysis of efficacy |
| Secondary analysis of efficacy |
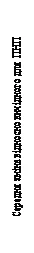
| Week |
| Week |
| Study 2 / Placebo (N=65) Study 2 / Omalizumab (N=62) |
| Placebo / Placebo (N=66) Placebo / Omalizumab (N=72) |
| Placebo / Placebo (N=65) Placebo / Omalizumab (N=62) |
| Placebo / Placebo (N=66) Placebo / Omalizumab (N=72) |
| Secondary efficacy analysis |
| Primary analysis of effectiveness |
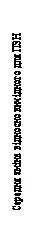
| -1.25 |
| -1.25 |
| -1.00 |
| -1.00 |
| -0.75 |
| -0.75 |
| -0.50 |
| -0.50 |
| -0.25 |
| -0.25 |
|
|
|
|
| 24 |
| 20 |
| 16 |
| 12 |
| 8 |
| 4 |
| 4 |
| 8 |
| 12 |
| 16 |
| 20 |
| 24 |
Fig. 1. Mean change from baseline in Nasal Congestion Score (NCS) and mean change from baseline in Nasal Polyp Score (NPS) in Studies 1 and 2 on nasal polyps.
In a pre-specified combined analysis of rescue medication use (systemic corticosteroids for 3 or more consecutive days or nasal polypectomy) during the 24-week treatment period, the proportion of patients requiring rescue medication was lower in the Xolair group compared to placebo (2.3% vs. 6.2%, respectively). The odds ratio for requiring rescue medication in patients receiving Xolair compared to placebo was 0.38 (95% CI: 0.10, 1.49). No sinus-nasal surgeries were reported in either study.
Clinical experience in chronic spontaneous urticaria
The efficacy and safety of Xolair were demonstrated in two randomized, placebo-controlled Phase III studies (Studies 1 and 2) in patients with CSU who continued to have symptoms despite treatment with approved doses of H1-antihistamines. A third study (Study 3) initially assessed the safety of Xolair in patients with CSU who continued to have symptoms despite treatment with H1-antihistamines at approximately four times the approved dose, as well as H2-antihistamines and/or leukotriene receptor antagonists. A total of 975 patients aged 12 to 75 years (mean age 42.3 years; 39 patients aged 12–17 years, 54 patients aged ≥65 years; 259 males and 716 females) were enrolled across the three studies. All patients had inadequately controlled symptoms as assessed by the 16-point Weekly Urticaria Activity Score (UAS7, range 0–42) and the 8-point Weekly Itch Severity Score (a component of UAS7; range 0–21) over the 7 days prior to randomization, despite taking antihistamines for at least 2 weeks prior.
In Studies 1 and 2, the mean baseline Weekly Itch Severity Score ranged from 13.7 to 14.5, and the mean baseline UAS7 score was 29.5 and 31.7, respectively. In the safety Study 3, the mean baseline Weekly Itch Severity Score was 13.8 and the mean baseline UAS7 score was 31.2. Across all three studies, patients were, on average, taking 4–6 medications (including H1-antihistamines) for CSU symptom control prior to study entry. Patients received Xolair at doses of 75 mg, 150 mg, or 300 mg, or placebo, administered subcutaneously every 4 weeks for 24 weeks and 12 weeks in Studies 1 and 2, respectively, and Xolair 300 mg or placebo administered subcutaneously every 4 weeks for 24 weeks in Study 3. In all studies, there was a 16-week post-treatment follow-up period.
The primary endpoint was the change from baseline to Week 12 in the Weekly Itch Severity Score. Omalizumab at 300 mg reduced the Weekly Itch Severity Score by 8.55–9.77 (p < 0.0001) compared to a reduction of 3.63–5.14 in the placebo group (see Table 4). The proportion of patients achieving an UAS7 score ≤6 at Week 12 was statistically significantly higher in the Xolair 300 mg treatment groups (52–66%) (p < 0.0001) compared to 11–19% in the placebo groups, and complete remission (UAS7 = 0) was achieved in 34–44% (p < 0.0001) of patients receiving 300 mg compared to 5–9% in the placebo groups. Patients treated with 300 mg achieved the highest mean percentage of days without angioedema episodes from Week 4 to Week 12 (91.0–96.1%; p < 0.001) compared to placebo groups (88.1–89.2%). The mean change from baseline in overall DLQI (Dermatology Life Quality Index) at Week 12 was greater (p < 0.001) in the Xolair 300 mg groups than in the placebo groups, indicating improvement by 9.7 to 10.3 points compared to 5.1–6.1 points in the corresponding placebo groups.
Table 4
Change from baseline to Week 12 in Weekly Itch Severity Score, Studies 1, 2, and 3 (mITT* population)
| Parameters |
Placebo |
Omalizumab |
| Study 1 |
||
| N |
80 |
81 |
| Mean score (SD) |
−3.63 (5.22) |
−9.40 (5.73) |
| Least squares mean difference vs placebo1 |
- |
−5.80 |
| 95 % CI for difference |
- |
−7.49, −4.10 |
| P-value vs placebo2 |
- |
< 0.0001 |
| Study 2 |
||
| N |
79 |
79 |
| Mean score (SD) |
−5.14 (5.58) |
−9.77 (5.95) |
| Least squares mean difference vs placebo1 |
- |
−4.81 |
| 95 % CI for difference |
- |
−6.49, −3.13 |
| P-value vs placebo2 |
- |
< 0.0001 |
| Study 3 |
||
| N |
83 |
252 |
| Mean score (SD) |
−4.01 (5.87) |
−8.55 (6.01) |
| Least squares mean difference vs placebo1 |
- |
|
| 95 % CI for difference |
- |
−5.97, −3.08 |
| P-value vs placebo2 |
- |
< 0.0001 |
*Modified intent-to-treat population (mITT) by treatment assigned: includes all patients who were randomized and received at least one dose of the investigational medicinal product.
BOCF (baseline observation carried forward) was used to handle missing data.
1 Least squares mean was calculated using an ANCOVA model. Covariates were baseline weekly pruritus severity score (<13 vs. ≥13) and baseline body weight (<80 kg vs. ≥80 kg).
2 P-value was obtained from ANCOVA t-test.
Figure 2 shows the mean weekly pruritus severity score over time in Study 1. The mean weekly pruritus severity score significantly decreased, reaching maximum effect at Week 12 during the 24-week treatment period. Similar results were obtained in Study 3.
In all three studies, the mean weekly pruritus severity score gradually increased during the 16-week treatment-free period, consistent with symptom recurrence. Mean values at the end of the follow-up period were similar to those observed in the placebo group, but lower than the corresponding mean values at the beginning of the study.
| Week 12 Primary Endpoint |
| Omalizumab 300 mg dose |
| Placebo |
| Week |
| Administered omalizumab or placebo |
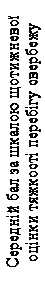 |
BOCF – baseline observation carried forward; mITT – modified intent-to-treat population.
Fig. 2. Mean weekly itch severity score over time, Study 1 (mITT population).
Efficacy at 24 weeks of treatment
Efficacy results observed at week 24 of treatment were compared with those observed at week 12 of treatment.
With the 300 mg dose in Studies 1 and 3, the mean reduction from baseline in the weekly itch severity score was 9.8 and 8.6, the percentage of patients achieving UAS7 ≤ 6 was 61.7% and 55.6%, and the percentage of patients achieving complete response (UAS7 = 0) was 48.1% and 42.5%, respectively (all p < 0.0001 versus placebo).
Clinical experience with repeated administration of omalizumab is limited.
Clinical trial data in adolescents (aged 12 to 17 years) included information from 39 patients, 11 of whom received the 300 mg dose. Results for the 300 mg dose were obtained from 9 patients at week 12 and 6 patients at week 24, demonstrating a similar therapeutic response to omalizumab as in adult patients. The mean change from baseline in the weekly itch severity score showed a reduction of 8.25 at week 12 and 8.95 at week 24. Response rates were 33% at week 12 and 67% at week 24 for UAS7 = 0, and 56% at week 12 and 67% at week 24 for UAS7 ≤ 6.
Pharmacokinetics.
The pharmacokinetics of omalizumab have been studied in adults and children aged 12 years and older with allergic asthma, in adults with CRSwNP, and in adults and adolescents with CSU. The overall pharmacokinetic parameters of omalizumab are similar across these patient populations.
Absorption
After subcutaneous administration, omalizumab is absorbed with a mean absolute bioavailability of 62%. Following a single subcutaneous dose in adults and children aged 12 years and older with asthma or CSU, omalizumab was slowly absorbed, reaching peak serum concentrations on average after 7–8 days. In patients with asthma, following multiple doses, the area under the serum concentration–time curve from day 0 to day 14 at steady state was 6-fold higher than after the first dose.
Omalizumab pharmacokinetics are linear at doses exceeding 0.5 mg/kg. Following multiple doses, the area under the serum concentration–time curve from day 0 to day 14 at steady state was up to 6-fold higher than after the first dose.
Administration of Xolair as either lyophilisate or solution resulted in similar omalizumab serum concentration–time profiles.
Distribution
In vitro, omalizumab forms IgE complexes of limited size. Precipitating complexes or complexes with a molecular mass exceeding one million daltons were not detected either in vitro or in vivo. Based on population pharmacokinetic analysis, the distribution of omalizumab is similar in patients with allergic asthma and those with CSU. The expected volume of distribution after subcutaneous administration is 78 ± 32 ml/kg.
Elimination
Omalizumab clearance involves IgG clearance mechanisms as well as elimination via specific binding and complex formation with its target ligand, IgE. Hepatic elimination of IgG includes degradation in the reticuloendothelial system and endothelial cells. Unchanged IgG is also excreted in bile. In patients with asthma, the mean serum half-life of omalizumab is approximately 26 days, with a mean clearance of 2.4 ± 1.1 ml/kg/day. Additionally, when body weight is doubled, clearance increases approximately twofold. In patients with CSU, based on pharmacokinetic modeling, the mean serum half-life at steady state is 24 days, and the apparent clearance at steady state in patients with a body weight of 80 kg is 3.0 ml/kg/day.
Characteristics in patient populations
Age, race/ethnicity, gender, body mass index
Patients with asthma and CRSwNP
Population pharmacokinetic analysis of Xolair was performed to assess the impact of demographic characteristics. Analysis of these limited data indicates that dose adjustment based on age (6–76 years for patients with allergic asthma; 18–75 years for patients with CRSwNP), race/ethnicity, gender, or body mass index is not required.
Patients with CSU
The impact of demographic characteristics and other factors on Xolair exposure was evaluated through population pharmacokinetic analysis. Additionally, the effect of covariates was assessed by analyzing the relationship between omalizumab concentration and clinical response. This analysis indicates that for patients with CSU, regardless of age (12–75 years), race/ethnicity, gender, body weight, body mass index, baseline IgE levels, anti-FcεRI autoantibodies, or concomitant use of H2-antihistamines or leukotriene receptor antagonists, dose adjustment is not required.
Renal and hepatic impairment
There are no data on pharmacokinetics or pharmacodynamics in patients with renal or hepatic impairment.
Clinical characteristics.
Indications.
Allergic asthma
The possibility of treatment with Xolair should be considered only for patients with confirmed IgE (immunoglobulin E)-mediated asthma.
Adults and children aged 12 years and older
Xolair is indicated as add-on therapy for achieving better asthma control in patients with severe persistent allergic asthma who have a positive skin test or in vitro test for sensitivity to a perennial aeroallergen, who have impaired lung function (FEV1 (forced expiratory volume in 1 second) < 80%), and who experience frequent daytime symptoms or frequent nocturnal awakenings, and who have documented evidence of multiple severe asthma exacerbations despite treatment with high daily doses of inhaled corticosteroids plus inhaled long-acting beta2-agonists.
Children aged 6 to 12 years
Xolair is indicated as add-on therapy for achieving better asthma control in patients with severe persistent allergic asthma who have a positive skin test or in vitro test for sensitivity to a perennial aeroallergen, and who experience frequent daytime symptoms or nocturnal awakenings, and who have documented evidence of multiple severe asthma exacerbations despite treatment with high daily doses of inhaled corticosteroids plus inhaled long-acting beta2-agonists.
Chronic rhinosinusitis with nasal polyps (CRSwNP)
Xolair is indicated as add-on therapy together with intranasal corticosteroids (INS) for the treatment of adults (aged 18 years and older) with severe CRSwNP for whom treatment with INS does not provide adequate disease control.
Chronic spontaneous urticaria (CSU), 150 mg dosage
The medicinal product Xolair is indicated as add-on therapy for chronic spontaneous urticaria in adults and adolescents (aged 12 years and older) who have an inadequate response to treatment with H1-antihistamines.
Contraindications.
Hypersensitivity to the active substance or to any of the other components of the medicinal product.
Interaction with other medicinal products and other forms of interaction.
Since IgE may be involved in the immune response to certain helminthic infestations, Xolair may indirectly reduce the efficacy of medicinal products used to treat helminthic or other parasitic infections.
Cytochrome P450 enzymes, efflux pumps, and protein binding mechanisms are not involved in the clearance of omalizumab; therefore, the potential for interaction with other drugs is negligible. No studies on interactions between Xolair and other medicinal products or vaccines have been conducted. There are no pharmacologically justified reasons to expect that medicinal products commonly used for the treatment of asthma would interact with omalizumab.
Allergic asthma
During clinical trials, Xolair was generally used in combination with inhaled and oral corticosteroids, short- and long-acting inhaled beta-agonists, leukotriene modifiers, theophylline, and oral antihistamines. No evidence of altered safety of Xolair due to these widely used asthma medications has been observed. Limited data are available regarding the use of Xolair in combination with specific immunotherapy (hyposensitization therapy). In clinical trials where Xolair was used concomitantly with immunotherapy, the safety and efficacy of Xolair in combination with specific immunotherapy did not differ from those observed when Xolair was used as monotherapy.
Chronic spontaneous urticaria
During clinical trials, patients with CSU received Xolair concomitantly with antihistamine agents (H1 and H2 antagonists) and leukotriene receptor antagonists. There are no data confirming that the safety of omalizumab was altered when used with these medicinal products compared to its known safety profile in allergic asthma. Furthermore, population pharmacokinetic analysis did not indicate a relevant effect of H2-antihistamines and leukotriene receptor antagonists on the pharmacokinetics of omalizumab.
Chronic rhinosinusitis with nasal polyps
In clinical trials, Xolair was administered according to protocol in combination with mometasone, an intranasal spray. Other commonly used concomitant medicinal products included other intranasal corticosteroids, bronchodilators, antihistamines, leukotriene receptor antagonists, adrenergic agents/sympathomimetics, and local nasal anesthetics. There was no indication that concomitant use of these commonly used medicinal products altered the safety of Xolair.
Children
Clinical trials in CSU included some patients aged 12 to 17 years who received Xolair concomitantly with antihistamines (H1 and H2 antagonists) and leukotriene receptor antagonists. Studies in children under 12 years of age have not been conducted.
Special precautions for use.
Traceability
To improve the traceability of biological medicinal products, the name and batch number of the administered product should be clearly recorded.
General
Xolair is not indicated for the treatment of acute asthma attacks, acute bronchospasm, or status asthmaticus.
The use of Xolair has not been studied in patients with hyper-IgE syndrome or allergic bronchopulmonary aspergillosis, or for the prevention of anaphylactic reactions, including those triggered by food allergy, atopic dermatitis, or allergic rhinitis. Xolair is not indicated for the treatment of such conditions.
The use of Xolair has not been studied in patients with autoimmune diseases, immune complex-mediated disorders, or pre-existing renal or hepatic impairment. Xolair should be used with caution in such patients.
Rapid discontinuation of systemic or inhaled corticosteroids after initiation of Xolair therapy in the treatment of allergic asthma or CRSwNP is not recommended. Dose reduction of corticosteroids should be performed under close medical supervision and, if necessary, gradually.
Immune system disorders
- Type I hypersensitivity reactions
Local or systemic type I hypersensitivity reactions, including anaphylaxis and anaphylactic shock, may occur during treatment with omalizumab. Such reactions may occur even after prolonged treatment. Most of these reactions develop within 2 hours after the first or subsequent injections of Xolair, although adverse reactions may sometimes occur more than 2 hours after injection, and in some cases even later than 24 hours post-injection. Most anaphylactic reactions occur during administration of the first three doses of Xolair. A history of hypersensitivity reactions unrelated to omalizumab is a risk factor for developing anaphylactic reactions during Xolair treatment. Therefore, medications for the treatment of anaphylactic reactions must always be readily available immediately after Xolair administration. If anaphylaxis or another serious allergic reaction occurs, Xolair must be discontinued immediately and appropriate therapy initiated. Patients should be informed that such reactions are possible and must seek immediate medical attention if an allergic reaction occurs.
Antibodies to omalizumab have been detected in a small number of patients during clinical trials. The clinical significance of anti-Xolair antibodies is not well established.
- Serum sickness
Serum sickness and serum sickness-like reactions (delayed type III hypersensitivity reactions) have been rarely observed in patients receiving humanized monoclonal antibodies, including omalizumab. The probable pathophysiological mechanism involves formation and deposition of immune complexes due to the production of antibodies against omalizumab. The typical onset is 1–5 days after administration of the first or subsequent injections, as well as after prolonged treatment. Symptoms suggestive of serum sickness include arthritis/arthralgia, rash (urticaria or other forms), fever, and lymphadenopathy. Antihistamines and corticosteroids may be indicated for prevention or treatment of these disorders, and patients should be instructed to report any suspicious symptoms to their physician.
- Churg-Strauss syndrome and hypereosinophilic syndrome
In patients with severe asthma, systemic hypereosinophilia or allergic granulomatous vasculitis (Churg-Strauss syndrome) may occur in isolated cases. Both conditions are usually treated with systemic corticosteroids.
In isolated cases, systemic eosinophilia and vasculitis may occur or develop in patients receiving anti-asthma treatments, including omalizumab. These phenomena are usually associated with reduction in the dose of oral corticosteroids being used.
When treating such patients, clinicians should be aware of the potential development of marked eosinophilia, vasculitic rash, worsening of pulmonary symptoms, paranasal sinus abnormalities, cardiac complications, and/or neuropathy.
In all severe cases of the aforementioned immune disorders, discontinuation of omalizumab should be considered.
Parasitic (helminthic) infections
IgE may be involved in the immune response triggered by certain helminthic infections. A placebo-controlled study in patients with a consistently high risk of helminthic infections showed a slight increase in the frequency of infections during omalizumab treatment, although the course, severity, and response to treatment remained unchanged. The incidence of helminthic infections during the overall clinical program, which was not specifically designed to detect such infections, was less than 1 case per 1000 patients. However, patients at high risk of helminthic infection should exercise caution, especially when traveling to areas where helminthic infections are endemic. If a patient does not respond to prescribed anti-helminthic treatment, discontinuation of Xolair should be considered.
Latex-sensitive individuals
The needle cap of the pre-filled syringe contains a derivative of natural rubber latex. Currently, no natural rubber latex has been detected in the needle cap. However, the use of Xolair solution for injection in a pre-filled syringe has not been studied in individuals sensitive to latex, and therefore a potential risk of hypersensitivity reactions cannot be completely ruled out.
Use during pregnancy or breastfeeding
Moderate data from pregnant women (from 300 to 1,000 pregnancy outcomes), obtained from the pregnancy registry and spontaneous post-marketing reports, indicate no evidence of malformative or fetal/neonatal toxicity. A prospective study of the pregnancy registry (EXPECT) in 250 pregnant women with asthma who received Xolair showed that the prevalence of major congenital anomalies was similar (8.1% vs. 8.9%) in EXPECT patients and patients with comparable diseases (moderate to severe asthma). Interpretation of the data may be affected by methodological limitations of the study, including small sample size and non-randomized design.
Omalizumab crosses the placental barrier; however, animal studies have not revealed any direct or indirect harmful effects on reproductive function. Omalizumab has been associated with age-related decreases in platelet counts in lower primates, with greater relative sensitivity in immature animals. Xolair treatment during pregnancy may be considered if clinically necessary.
Immunoglobulin G (IgG) is present in human breast milk; therefore, omalizumab is expected to be present in human breast milk. Omalizumab is excreted in breast milk of lower primates.
The EXPECT study involving 154 infants exposed to Xolair during the in utero period and during breastfeeding found no adverse reactions in breastfed infants. Interpretation of the data may be affected by methodological limitations of the study, including small sample size and non-randomized design.
When administered orally, immunoglobulin G proteins undergo intestinal proteolysis and have low bioavailability. No effect on breastfed newborns/infants is expected. Therefore, if clinically necessary, Xolair treatment during breastfeeding may be considered.
There are no data on the effect of omalizumab on human fertility. In dedicated preclinical fertility studies, including mating studies, no impairment of male or female fertility was observed after repeated administration of omalizumab at doses up to 75 mg/kg. Furthermore, no genotoxic effects were observed in a separate preclinical genotoxicity study.
Effect on the ability to drive and use machines
Xolair has no effect or has a negligible effect on the ability to drive a vehicle or operate machinery.
Method of Administration and Dosage
Xolair should only be prescribed by a physician experienced in the diagnosis and treatment of severe persistent asthma, chronic rhinosinusitis with nasal polyps (CRSwNP), or chronic spontaneous urticaria (CSU).
Dosage
Allergic Asthma and Chronic Rhinosinusitis with Nasal Polyps (CRSwNP)
The same principles apply when determining dosage for the treatment of allergic asthma and CRSwNP. The required dose and frequency of Xolair administration in these conditions are determined based on the patient’s IgE concentration (IU/mL), measured prior to initiating treatment, and the patient’s body weight (kg). Prior to starting treatment, the patient’s IgE level must be determined using a quantitative assay for total IgE with any commercially available serum test. Depending on these parameters, the recommended dose of Xolair ranges from 75 to 600 mg. This dose may be divided into 1–4 injections.
A lower likelihood of a positive response has been observed in patients with allergic asthma who have baseline IgE levels below 76 IU/mL. Prior to initiating therapy, the physician should confirm in vitro reactivity (radioallergosorbent test [RAST]) to a perennial allergen in adult patients and in patients aged 12 years and older with IgE levels below 76 IU/mL, as well as in patients aged 6 to 12 years with IgE levels below 200 IU/mL.
Dose calculation is provided in Table 5, and dose determination is outlined in Tables 6 and 7.
Xolair should not be prescribed to patients whose IgE levels or body weight exceed the values specified in the dosage tables.
The maximum recommended dose is 600 mg of omalizumab every two weeks.
Table 5
Dose conversion into number of syringes, number of injections, and total injection volume per administration
| Dose (mg) |
Number of syringes |
Number of injections |
Total injection volume (ml) |
|
| 75 mg |
150 mg |
|||
| 75 |
1 |
0 |
1 |
0.5 |
| 150 |
0 |
1 |
1 |
1.0 |
| 225 |
1 |
1 |
2 |
1.5 |
| 300 |
0 |
2 |
2 |
2.0 |
| 375 |
1 |
2 |
3 |
2.5 |
| 450 |
0 |
3 |
3 |
3.0 |
| 525 |
1 |
3 |
4 |
3.5 |
| 600 |
0 |
4 |
4 |
4.0 |
Table 6
ADMINISTRATION EVERY 4 WEEKS.
Doses of the medicinal product Xolar (mg/dose) administered by subcutaneous injection every 4 weeks
| Baseline IgE level (IU/mL) |
Body weight (kg) |
|||||||||
| > 20–25* |
> 25–30* |
> 30–40 |
> 40–50 |
> 50–60 |
> 60–70 |
> 70–80 |
> 80–90 |
> 90–125 |
> 125–150 |
|
| ≥ 30–100 |
75 |
75 |
75 |
150 |
150 |
150 |
150 |
150 |
300 |
300 |
| > 100–200 |
150 |
150 |
150 |
300 |
300 |
300 |
300 |
300 |
450 |
600 |
| > 200–300 |
150 |
150 |
225 |
300 |
300 |
450 |
450 |
450 |
600 |
|
| > 300–400 |
225 |
225 |
300 |
450 |
450 |
450 |
600 |
600 |
||
| > 400–500 |
225 |
300 |
450 |
450 |
600 |
600 |
||||
| > 500–600 |
300 |
300 |
450 |
600 |
600 |
|||||
| > 600–700 |
300 |
450 |
600 |
|||||||
| > 700–800 |
||||||||||
| > 800–900 |
ADMINISTER EVERY 2 WEEKS, |
|||||||||
| > 900–1000 |
see TABLE 5 |
|||||||||
| > 1000–1100 |
||||||||||
* In the main studies on CRSwNP, use in patients with body weight below 30 kg has not been studied.
Table 7
ADMINISTRATION EVERY 2 WEEKS.
Doses of the medicinal product Xolair (mg/dose) administered by subcutaneous injection every 2 weeks
| Baseline IgE (IU/mL) |
Body weight (kg) |
||||||||||
| > 20–25* |
> 25–30* |
> 30–40 |
> 40–50 |
> 50–60 |
> 60–70 |
> 70–80 |
> 80–90 |
> 90–125 |
> 125– 150 |
||
| ≥ 30–100 |
ADMINISTER EVERY 4 WEEKS |
||||||||||
| > 100–200 |
see TABLE 4 |
||||||||||
| > 200–300 |
375 |
||||||||||
| > 300–400 |
450 |
525 |
|||||||||
| > 400–500 |
375 |
375 |
525 |
600 |
|||||||
| > 500–600 |
375 |
450 |
450 |
600 |
|||||||
| > 600–700 |
225 |
375 |
450 |
450 |
525 |
||||||
| > 700–800 |
225 |
225 |
300 |
375 |
450 |
450 |
525 |
600 |
|||
| > 800–900 |
225 |
225 |
300 |
375 |
450 |
525 |
600 |
||||
| > 900–1000 |
225 |
300 |
375 |
450 |
525 |
600 |
|||||
| > 1000–1100 |
225 |
300 |
375 |
450 |
600 |
Insufficient data to recommend a dose. |
|||||
| > 1100–1200 |
300 |
300 |
450 |
525 |
600 |
||||||
| > 1200–1300 |
300 |
375 |
450 |
525 |
|||||||
| > 1300–1500 |
300 |
375 |
525 |
600 |
|||||||
- In the main clinical trials for CRSwNP, use in patients with body weight below 30 kg was not studied.
Administration
For subcutaneous use only. Do not administer intravenously or intramuscularly.
Doses exceeding 150 mg (Table 5) should be divided into two or more injections.
For patients without a history of anaphylaxis, self-administration of Xolair or administration by a caregiver may be considered starting from the 4th dose onward, if deemed appropriate by the physician. The patient or caregiver must be trained in proper injection technique and in recognizing early signs and symptoms of serious allergic reactions.
Patients or caregivers must be instructed to administer the full dose of Xolair according to the instructions in the medical device user manual.
Children aged 6 to 12 years should not self-administer Xolair injections; however, if considered appropriate by the physician, a caregiver may administer the injection to the child after proper training.
Treatment duration, monitoring, and dose adjustment
Allergic asthma
Xolair is intended for long-term treatment. Clinical trials have demonstrated efficacy of Xolair treatment administered for at least 12–16 weeks. At week 16 of Xolair therapy, the patient's response to treatment should be evaluated prior to administering subsequent injections. The decision to continue Xolair therapy should be based on evidence of significant improvement in overall asthma control.
Chronic rhinosinusitis with nasal polyps (CRSwNP)
In clinical trials of CRSwNP, changes in nasal polyp score (NPS) and nasal congestion score (NCS) were observed as early as 4 weeks. The need for continued therapy should be periodically reassessed based on the severity of the patient's disease and level of symptom control.
Allergic asthma and chronic rhinosinusitis with nasal polyps (CRSwNP)
Discontinuation of Xolair treatment in most cases leads to a rebound increase in free IgE levels and development of corresponding symptoms. Total IgE levels remain elevated during treatment and continue to be elevated for up to one year after discontinuation of treatment. Therefore, repeated measurement of IgE levels during Xolair treatment cannot be used to determine the required dose of the drug. Dose determination after a treatment interruption lasting less than one year should be based on the IgE level obtained at initial dose determination. Serum total IgE levels may be re-measured for dose selection if Xolair treatment was interrupted for more than one year.
Dose adjustment is required following significant changes in body weight.
Chronic spontaneous urticaria (CSU), dosing 150 mg/1 mL
The recommended dose is 300 mg administered as subcutaneous injections every 4 weeks.
The need for continued treatment should be periodically re-evaluated.
Clinical trial data regarding treatment duration beyond 6 months for this indication are limited.
Special populations
Elderly (aged 65 years and older)
Limited data are available on the use of Xolair in patients aged over 65 years. However, there are no indications that a different dosing approach is required for elderly patients compared to younger adults.
Patients with renal or hepatic impairment
The effect of renal or hepatic impairment on the pharmacokinetics of Xolair has not been studied. Since omalizumab clearance at clinical doses occurs primarily via the reticuloendothelial system (RES), changes related to renal or hepatic impairment are unlikely. Although there are no specific dose adjustment recommendations, Xolair should be used with caution in these patients.
Paediatric population
The safety and efficacy of Xolair for the treatment of allergic asthma in patients under 6 years of age have not been established. No data are available.
The safety and efficacy of Xolair for the treatment of CRSwNP in patients under 18 years of age have not been established.
The safety and efficacy of Xolair for the treatment of CSU in patients under 12 years of age have not been established.
Special instructions for use
The package contains a pre-filled syringe packed in a plastic tray (Fig. 3).
| Needle protective cap |
| Safety flanges of the syringe |
| Needle cap |
| Viewing window Labelling and Expiry date |
| Finger grips |
| Piston |
| Piston head |
Fig. 3. Appearance of Xofigo, solution for injection, 75 mg/0.5 mL or 150 mg/1 mL, in a pre-filled syringe (blue for 75 mg/0.5 mL; purple for 150 mg/1 mL).
- Do not open the sealed outer plastic packaging until you are ready to use Xofigo, solution for injection.
- Do not use the medicinal product if the seal on the outer carton or the seal of the plastic tray is broken, as this may be hazardous.
- Do not shake the syringe.
- Be careful not to touch the safety wings of the syringe before use. If the wings are touched, the safety syringe may activate prematurely.
- Remove the needle cap immediately before injection only.
- The syringe must not be reused. Dispose of the used syringe immediately after use.
- Before use, remove the syringe from the refrigerator and allow it to reach room temperature (25°C) prior to preparing for injection (this will take approximately 20 minutes). Leave the syringe in the carton to protect it from light. The syringe may be returned to the refrigerator if necessary. The total time the syringe is allowed to remain at room temperature (25°C) before use must not exceed 48 hours.
Injection site
If the injection is administered by a caregiver, the outer area of the upper arm may also be used. |
Preparation of Xolar, solution for injection (75 mg/0.5 ml or 150 mg/1 ml) in a pre-filled syringe, prior to injection
Note: Depending on the dose prescribed by your doctor, you may need to prepare one or more pre-filled syringes and inject the contents of all prepared syringes. Table 8 provides examples of how many injections of each dose are required for the prescribed dose.
Table 8
| Number of syringes required for dose administration |
| Dose |
|
- Remove the box containing the syringe from the refrigerator and leave it open for approximately 20 minutes to reach room temperature (leave the syringe in the box to protect it from light).
- When you are ready to use the syringe, wash your hands thoroughly with soap and water.
- Clean the injection site with an alcohol swab.
- Remove the plastic tray from the box and peel off the paper layer. Remove the syringe from the tray by holding the middle of the protective cap.
- Inspect the syringe. The solution should range from clear to slightly cloudy. The color of the solution may vary from colorless to slightly brownish-yellow. You may see an air bubble, which is normal. DO NOT USE if the syringe is damaged or if the liquid is clearly cloudy, distinctly brown, or contains particles.
- Holding the syringe horizontally, check the expiration date printed on the label through the viewing window. Note: You may rotate the inner part of the syringe assembly to view the label in the viewing window. DO NOT USE if the expiration date has passed.
How to use XOLAR, solution for injection (75 mg/0.5 mL or 150 mg/1 mL) in a pre-filled syringe
|
|
Remove the needle cap from the syringe carefully. Discard the needle cap. You may see a drop of liquid at the tip of the needle. This is normal. |
|
|
|
Gently pinch the skin at the injection site and fully insert the needle as shown to ensure complete delivery of the medication. |
|
|
|
Hold the syringe as shown. Slowly push the plunger fully until the plunger head is completely positioned between the safety wings of the syringe. |
|
|
|
Keep the plunger fully depressed while carefully removing the needle from the injection site. |
|
|
|
Slowly release the plunger, after which the syringe's safety shield will automatically cover the needle. A small amount of blood may appear at the injection site. You may apply gentle pressure with a cotton pad or gauze at the injection site for 30 seconds. Do not rub the injection site. If necessary, the injection site may be covered with a small adhesive bandage. |
Any unused medicinal product or waste material must be disposed of in accordance with local requirements.
Children.
Allergic asthma
Xolair can be used in children aged 6 years and older who are already receiving asthma medications, but whose asthma symptoms are not adequately controlled with such medications as high-dose inhaled corticosteroids and beta-agonist inhalers.
Children aged 6 to 11 years should not self-administer Xolair using the pre-filled syringe. However, if considered appropriate by a physician, a caregiver may administer the Xolair injection to the child after proper training.
Chronic rhinosinusitis with nasal polyps (CRwNP)
Xolair should not be used in children (under 18 years of age) for this indication.
Chronic spontaneous urticaria (CSU)
Xolair can be used in children aged 12 years and older who are already receiving antihistamine medications, but whose CSU symptoms are not adequately controlled by these medications. The dose for adolescents aged 12 years and older is the same as that for adults.
Overdose.
The maximum tolerated dose of Xolair has not been determined. Intravenous administration of single doses up to 4,000 mg to patients did not result in dose-limiting toxicity. The highest cumulative dose administered to a patient was 44,000 mg over a 20-week period and did not cause any acute adverse effects.
In case of suspected overdose, monitor the patient for any unusual symptoms. Appropriate diagnosis should be established and treatment initiated as needed.
Adverse reactions
Allergic asthma and chronic rhinosinusitis with nasal polyps (CRSwNP)
During clinical trials involving adults and children aged 12 years and older with allergic asthma, the most common adverse reactions were headache and injection site reactions, including injection site pain, swelling, redness, and itching. In clinical trials conducted in children aged 6 to 12 years, the most common drug-related adverse reactions were headache, fever, and upper abdominal pain. The majority of reactions were of mild to moderate severity. In clinical trials in patients aged 18 years and older with CRSwNP, the most common drug-related adverse reactions were headache, dizziness, arthralgia, upper abdominal pain, and injection site reactions.
In Table 9, adverse reactions observed during clinical trials in the overall safety study population for allergic asthma and CRSwNP are listed by MedDRA system organ class and frequency. Within each frequency group, adverse reactions are presented in order of decreasing severity. Frequency is defined as follows: very common (≥ 1/10), common (≥ 1/100 to < 1/10), uncommon (≥ 1/1000 to < 1/100), rare (≥ 1/10,000 to < 1/1000), and very rare (< 1/10,000). The frequency of events reported during the post-marketing period is listed as unknown (cannot be estimated from available data).
Table 9
Adverse reactions observed during clinical trials for allergic asthma and CRSwNP
| Frequency |
Adverse Reactions |
| Infections and infestations |
|
| Uncommon |
Pharyngitis |
| Rare |
Parasitic infestations |
| Blood and lymphatic system disorders |
|
| Frequency unknown |
Idiopathic thrombocytopenia, including severe cases |
| Immune system disorders |
|
| Rare |
Anaphylactic reaction, other serious allergic conditions, development of antibodies to the drug |
| Frequency unknown |
Serum sickness, which may include fever and lymphadenopathy |
| Nervous system disorders |
|
| Common |
Headache* |
| Uncommon |
Syncope, paraesthesia, somnolence, dizziness*** |
| Vascular disorders |
|
| Uncommon |
Postural hypotension, flushing |
| Respiratory, thoracic and mediastinal disorders |
|
| Uncommon |
Allergic bronchospasm, cough |
| Rare |
Laryngeal edema |
| Frequency unknown |
Allergic granulomatous vasculitis (Churg-Strauss syndrome) |
| Gastrointestinal disorders |
|
| Common |
Upper abdominal pain**,*** |
| Uncommon |
Signs or symptoms of dyspepsia, diarrhea, nausea |
| Skin and subcutaneous tissue disorders |
|
| Uncommon |
Photosensitivity, urticaria, rash, pruritus |
| Rare |
Angioneurotic edema |
| Frequency unknown |
Alopecia |
| Musculoskeletal and connective tissue disorders |
|
| Uncommon Rare Frequency unknown |
Arthralgia**** Systemic lupus erythematosus (SLE) Myalgia, joint swelling |
| General disorders and administration site conditions |
|
| Very common |
Pyrexia** |
| Common |
Injection site reactions such as swelling, erythema, pain, pruritus |
| Uncommon |
Influenza-like illness, swelling of extremities, weight increase, fatigue |
* Very common in children aged 6 to 12 years.
** In children aged 6 to 12 years.
*** Common in studies on nasal polyps.
**** Frequency unknown in studies on allergic asthma.
Chronic spontaneous urticaria
The safety and tolerability of omalizumab were evaluated with doses of 75 mg, 150 mg, and 300 mg administered every 4 weeks to 975 patients with CSU, of whom 242 received placebo. Overall, 733 patients received omalizumab for up to 12 weeks and 490 patients for up to 24 weeks. Among them, 412 patients received omalizumab for up to 12 weeks and 333 patients for up to 24 weeks at the dose of 300 mg.
Adverse reactions in CSU occurred following administration of various doses to patients with significantly different risk factors, concomitant diseases, ages, and concomitantly administered medicinal products (e.g., asthma studies included children aged 6 to 12 years); see Table 10.
Table 10 lists adverse reactions (events occurring in ≥1% of patients in any treatment group and at least 2% more frequently in any omalizumab treatment group compared to placebo [after medical evaluation]) observed with the 300 mg dose in three pooled Phase III studies. The listed adverse reactions are divided into two groups: those occurring during the 12-week and 24-week treatment periods.
Adverse reactions are listed by MedDRA system organ class. Within each organ system class, adverse reactions are listed in descending order of frequency, with the most frequent first. Frequency categories are defined as follows: very common (≥1/10); common (≥1/100 to <1/10); uncommon (≥1/1000 to <1/100); rare (≥1/10,000 to <1/1000); very rare (<1/10,000); and not known (frequency cannot be estimated from available data).
| Table 10 |
|||
| Adverse reactions from pooled safety databases in CSU (Day 1 – Week 24) with omalizumab 300 mg |
|||
| 12-week treatment |
Pooled omalizumab studies 1, 2, and 3 |
Frequency category |
|
| Placebo N = 242 |
300 mg N = 412 |
||
| Infections and infestations |
|||
| Sinusitis |
5 (2.1%) |
20 (4.9%) |
Common |
| Nervous system disorders |
|||
| Headache |
7 (2.9%) |
25 (6.1%) |
Common |
| Musculoskeletal and connective tissue disorders |
|||
| Arthralgia |
1 (0.4%) |
12 (2.9%) |
Common |
| General disorders and administration site conditions |
|||
| Injection site reactions* |
2 (0.8%) |
11 (2.7%) |
Common |
| 24-week treatment |
Pooled omalizumab studies 1 and 3 |
Frequency category |
|
| Placebo N = 163 |
300 mg N = 333 |
||
| Infections and infestations |
|||
| Upper respiratory tract infection |
5 (3.1%) |
19 (5.7%) |
Common |
* Although a difference greater than 2% compared to placebo was not observed, injection site reactions were included because in all cases a causal relationship to treatment within the study was established.
Description of selected adverse reactions
Immune system disorders
For additional information, see section "Special warnings and precautions for use".
Anaphylaxis
Anaphylactic reactions were observed very rarely during clinical trials. However, a pooled search of the post-marketing safety database identified a total of 898 cases of anaphylaxis. Based on an estimated exposure of 566,923 patient-years of treatment, this corresponds to a reporting rate of approximately 0.20%.
Arterial thromboembolism (ATE)
A numerical imbalance in ATE events was observed in controlled clinical trials and in the ongoing analysis of observational studies. The composite ATE endpoint included stroke, transient ischemic attack, myocardial infarction, unstable angina, and cardiovascular death (including death from unknown cause). In the final analysis of the observational study, the ATE rate per 1000 patient-years was 7.52 (115/15,286 patient-years) in patients receiving Xolair compared to 5.12 (51/9,963 patient-years) in the control group. In a multivariate analysis adjusted for baseline cardiovascular risk factors, the hazard ratio was 1.32 (95% confidence interval 0.91–1.91). In a separate analysis of pooled clinical trials, which included all randomized, double-blind, placebo-controlled clinical trials of 8 weeks or longer duration, the ATE rate per 1000 patient-years was 2.69 (5/1,856 patient-years) in patients receiving Xolair and 2.38 (4/1,680 patient-years) in the placebo group (hazard ratio 1.13, 95% confidence interval 0.24–5.71).
Platelets
During clinical trials, several patients had platelet counts below the lower limit of the normal laboratory reference range. In none of these cases did this change lead to bleeding or a decrease in hemoglobin levels. Cases of persistent thrombocytopenia, as observed in non-human primates, were not observed in humans (patients aged 6 years and older), although post-marketing surveillance has received isolated reports of idiopathic thrombocytopenia, including severe cases.
Parasitic infections
A placebo-controlled trial revealed a slight increase in the incidence of parasitic infections in patients with a consistently high risk of helminth infestation while receiving omalizumab, which was not statistically significant. The course, severity, and response to treatment of these infestations were unchanged.
Reporting of adverse reactions after drug authorization is of great importance. It allows continuous monitoring of the benefit-risk balance of the drug. Healthcare and pharmaceutical professionals, as well as patients or their legal representatives, should report all suspected adverse reactions and lack of efficacy through the Automated Pharmacovigilance Information System at the following link: https://aisf.dec.gov.ua
Shelf life. 18 months.
The medicinal product may be stored at 25°C for up to 48 hours in total. If necessary, the product may be returned to the refrigerator for further use.
Storage conditions.
Store at 2–8°C in a place inaccessible to children. Do not freeze. Store in the original packaging.
Packaging.
Solution for injection, 75 mg/0.5 ml
0.5 ml of solution for injection in a pre-filled syringe with attached needle and needle cap. One pre-filled syringe in a blister; one blister in a cardboard box.
Solution for injection, 150 mg/1 ml
1 ml of solution for injection in a pre-filled syringe with attached needle and needle cap. One pre-filled syringe in a blister; one blister in a cardboard box.
Prescription status. Prescription only.
Manufacturer.
Novartis Pharma GmbH, Germany / Novartis Pharma GmbH, Germany.
Manufacturer's address and location of operations.
Roonstrasse 25, Gostenhof, Nuremberg, Bavaria, 90429, Germany /
Roonstrasse 25, Gostenhof, Nuremberg, Bavaria, 90429, Germany.