Vilate 1000 MO
UkrainaSpis treści
INSTRUKCJA dot. stosowania leczniczego leku VILATE 500 MO VILATE 1000 MO
Skład:
Substancje czynne: czynnik krzepnięcia krwi VIII, czynnik von Willebranda;
1 fiolka zawiera 500 lub 1000 MI czynnika krzepnięcia krwi VIII oraz 500 MI lub 1000 MI czynnika von Willebranda; zawartość całkowitego białka ≤ 7,5 mg (500 MI) lub ≤ 15 mg (1000 MI);
Substancje pomocnicze: glicyna, sacharoza, chlorek sodu, cytrynian sodu, chlorek wapnia.
Roztwórnik: woda do wstrzykiwań z 0,1% polisorbatem 80.
Postać leku. Proszek i roztwórnik do sporządzenia roztworu do wstrzykiwań.
Główne właściwości fizykochemiczne:
Proszyk: proszek lub krucha masa biała lub bladożółta.
Roztwórnik: przejrzysta, bezbarwna ciecz bez zapachu. Bez widocznych cząstek.
Grupa farmakoterapeutyczna. Leki przeciwkrwotoczne. Czynniki krzepnięcia krwi. Czynnik von Willebranda i czynnik krzepnięcia VIII w kombinacji. Kod ATC B02B D06.
Właściwości farmakologiczne.
Farmakodynamika.
Choroba von Willebranda (VWD)
Czynnik von Willebranda (z koncentratu) jest naturalnym składnikiem osocza krwi człowieka i działa tak samo jak czynnik von Willebranda endogenny.
Podanie czynnika von Willebranda pozwala na korekcję zaburzeń hemostazy występujących u pacjentów z niedoborem czynnika von Willebranda na dwóch poziomach:
- czynnik von Willebranda przywraca adhezję płytek krwi do subendotelium naczyniowego w miejscu uszkodzenia naczynia (ponieważ wiąże się zarówno z subendotelium naczyniowym, jak i z błoną płytek krwi), zapewniając w ten sposób hemostazę pierwotną, co potwierdza skrócenie czasu krwawienia. Ten efekt występuje natychmiastowo i, jak wiadomo, w znacznym stopniu zależy od stopnia polimeryzacji białka;
- czynnik von Willebranda wywołuje opóźnioną korekcję niedoboru związanego z czynnikiem VIII. Po podaniu dożylnym czynnik von Willebranda wiąże endogenny czynnik VIII (który jest normalnie wytwarzany u pacjenta), a dzięki jego stabilizacji przeciwdziała szybkiemu jego rozkładowi. W związku z tym podanie czystego czynnika von Willebranda (preparatu czynnika von Willebranda o niskim stężeniu czynnika VIII) przywraca poziom czynnika VIII:C do normy jako efekt wtórny po pierwszej infuzji. Podanie preparatu czynnika von Willebranda zawierającego czynnik VIII przywraca poziom czynnika VIII:C do normy natychmiast po pierwszej infuzji.
Oprócz swojej roli jako białka ochronnego czynnika VIII, czynnik von Willebranda pełni funkcję pośrednika adhezji płytek krwi do uszkodzonych miejsc naczyń i odgrywa rolę w agregacji płytek krwi.
Hemofilia A
Zespół czynnika VIII/czynnika von Willebranda składa się z dwóch cząsteczek (czynnika VIII i czynnika von Willebranda) o różnych funkcjach fizjologicznych. Po podaniu pacjentowi z hemofilią czynnik VIII wiąże się z czynnikiem von Willebranda w krwiobiegu chorego. Aktywowany czynnik VIII (czynnik VIIIa) działa jako kofaktor aktywowanego czynnika IX (czynnik IXa), przyspieszając przekształcanie czynnika X w aktywowany czynnik X (czynnik Xa). Czynnik Xa przekształca protrombinę w trombinę. Następnie trombina przekształca fibrynogen w fibrynę, umożliwiając powstanie skrzepu.
Hemofilia A jest dziedzicznym, sprzężonym z płcią zaburzeniem krzepnięcia krwi, wynikającym ze zmniejszonych poziomów czynnika VIII:C (czynnik koagulacyjny), co prowadzi do silnych krwawień do stawów, mięśni lub narządów wewnętrznych, zarówno spontanicznie, jak i w wyniku urazu przypadkowego lub chirurgicznego. Dzięki terapii zastępczej poziomy czynnika VIII w osoczu krwi wzrastają, co sprzyja tymczasowej korekcji niedoboru czynnika oraz korekcji skłonności do krwawień.
Należy zaznaczyć, że nie można porównywać średniorocznej częstości krwawień (ABR/СЧК) obserwowanej w różnych badaniach klinicznych ani w przypadku stosowania różnych koncentratów czynnika.
Oprócz swojej roli jako białka ochronnego czynnika VIII, czynnik von Willebranda pełni funkcję pośrednika adhezji płytek krwi do uszkodzonych miejsc naczyń i odgrywa rolę w agregacji płytek krwi.
Farmakokinetyka.
Choroba von Willebranda (VWD)
Czynnik von Willebranda (z koncentratu) jest naturalnym składnikiem osocza krwi człowieka i działa jak endogenny czynnik von Willebranda.
Tabela 1
Wyniki metaanalizy trzech badań farmakokinetycznych z udziałem 24 pacjentów ze wszystkimi typami VWD.
| Parametr |
Wszystkie typy VWD |
VWD typu 1 |
VWD typu 2 |
VWD typu 3 |
|||||||||||||||||
| N |
Śr. wsk. |
SD |
Min ∙ |
Maks ∙ |
N |
Śr. wsk. |
SD |
Min ∙ |
Maks ∙ |
N |
Śr. wsk. |
SD |
Min ∙ |
Maks ∙ |
N |
Śr. wsk. |
SD |
Min ∙ |
Maks ∙ |
||
| Odbudowa (%/MO/kg) |
24 |
1,56 |
0,48 |
0,90 |
2,93 |
2 |
1,19 |
0,07 |
1,14 |
1,24 |
5 |
1,83 |
0,86 |
0,98 |
2,93 |
17 |
1,52 |
0,32 |
0,90 |
2,24 |
|
| AUC (0-inf) (g*%) |
23 |
1981 |
960 |
593 |
4831 |
2 |
2062 |
510 |
1701 |
2423 |
5 |
2971 |
1383 |
1511 |
4831 |
16 |
1662 |
622 |
593 |
2606 |
|
| T 1/2 (h) |
24 |
23,3 |
12,6 |
7,4 |
58,4 |
2 |
39,7 |
18,3 |
26,7 |
52,7 |
5 |
34,9 |
16 |
17,5 |
58,4 |
17 |
18 |
6,2 |
7,4 |
30,5 |
|
| MRT (h) |
24 |
33,1 |
19 |
10,1 |
89,7 |
2 |
53,6 |
25,9 |
35,3 |
71,9 |
5 |
53,5 |
24,6 |
27,8 |
89,7 |
17 |
24,7 |
8,5 |
10,1 |
37,7 |
|
| Klirens (ml/h/kg) |
24 |
3,29 |
1,67 |
0,91 |
7,41 |
2 |
2,66 |
0,85 |
2,06 |
3,27 |
5 |
1,95 |
1,02 |
0,91 |
3,31 |
17 |
3,76 |
1,69 |
1,83 |
7,41 |
|
AUC – pole pod krzywą; MRT – średni czas utrzymywania leku w organizmie; SD – odchylenie standardowe.
Hemofilia A
Faktor VIII (z koncentratu) jest naturalnym składnikiem osocza krwi człowieka i działa jako endogenny faktor VIII. Po podaniu leku około od 2/3 do 3/4 faktora VIII pozostaje w krążeniu. Poziom aktywności faktora VIII:C osiągany w osoczu wynosi 80% – 120% przewidywanej (oczekiwanej) aktywności faktora VIII.
Aktywność faktora VIII zmniejsza się w wyniku dwufazowego rozpadu wykładniczego. W wstępnej fazie zachodzi rozdział między środowiskiem wewnątrznaczyniowym a innymi płynami ustrojowymi z okresem półtłumienia z osocza wynoszącym od 3 do 6 godzin. W kolejnej, wolniejszej fazie, okres półtłumienia waha się od 8 do 18 godzin i wynosi średnio 15 godzin. Odpowiada to rzeczywistemu okresowi biologicznego półtłumienia.
Tabela 2
Wyniki jednego badania klinicznego z udziałem 12 pacjentów (analiza chromogenna, oznaczenie metodą podwójnych pomiarów).
| Parametr |
Wizyta wyjściowa |
Wizyta po 6 miesiącach |
||
| Średnia wartość |
SD |
Średnia wartość |
SD |
|
| Odzysk (%/MO/kg) |
FVIII:C 2,27 |
1,20 |
FVIII:C 2,26 |
1,19 |
| AUCnorm (% × g/MO/kg) |
FVIII:C 31,3 |
7,31 |
FVIII:C 33,8 |
10,9 |
| Okres półwyprowadzenia (h) |
FVIII:C 11,2 |
2,85 |
FVIII:C 11,8 |
3,37 |
| MRT (h) |
FVIII:C 15,3 |
3,5 |
FVIII:C 16,3 |
4,6 |
| Klirens ml/h/kg |
FVIII:C 3,37 |
0,86 |
FVIII:C 3,24 |
1,04 |
AUC – pole pod krzywą; MRT – średni czas utrzymywania leku w organizmie;
SD – odchylenie standardowe.
Dane badań przedklinicznych dotyczących bezpieczeństwa
WVF i czynnik VIII w leku Vilate są naturalnymi składnikami ludzkiej osocza krwi i działają jako endogenowy WVF/czynnik VIII.
Zwykłe badania bezpieczeństwa tych składników u zwierząt laboratoryjnych nie dostarczyłyby przydatnych informacji ponad istniejące doświadczenie kliniczne, dlatego takie badanie nie jest konieczne.
Dane kliniczne.
Wskazania.
Choroba von Willebranda (VWD)
Profilaktyka i leczenie krwawień lub krwawień podczas zabiegów chirurgicznych w chorobie von Willebranda (VWD), gdy leczenie wyłącznie desmopresyną (DDAVP) jest nieskuteczne lub jest przeciwwskazane.
Hemofilia A
Leczenie i profilaktyka krwawień u pacjentów z hemofilią A (wrodzony niedobór czynnika krzepnięcia krwi VIII).
Przeciwwskazania.
Reakcje alergiczne na substancje czynne lub na którąkolwiek z substancji pomocniczych.
Interakcje z innymi lekami i inne rodzaje interakcji.
Interakcje z innymi lekami nie są znane.
Szczególne środki ostrożności.
Śledzenie
W celu poprawienia śledzenia produktów biologicznych należy dokładnie odnotowywać nazwę handlową i numer serii wprowadzanego leku.
Wrażliwość alergiczną
Podczas stosowania leku VILATE mogą wystąpić reakcje alergiczne. Oprócz czynnika VIII, produkt zawiera śladowe ilości białek ludzkich. W przypadku wystąpienia objawów nadwrażliwości pacjentom należy zalecić natychmiastowe zaprzestanie stosowania leku i skontaktowanie się z lekarzem.
Pacjentów należy poinformować o wczesnych objawach reakcji alergicznych, takich jak: wysypka, uogólnione pokrzywki, trudności z oddychaniem, duszność, hipotensja oraz anafilaksja.
W przypadku wstrząsu należy zastosować standardową terapię przeciwwstrząsową.
Przekazywanie patogenów
Zwykłe środki zapobiegające infekcjom w wyniku stosowania leków pochodzących z krwi lub osocza ludzkiego obejmują selekcję dawców, badania przesiewowe krwi poszczególnych dawców i partii osocza dawcy pod kątem specyficznych markerów infekcji oraz włączenie skutecznych etapów procesu produkcyjnego w celu inaktywacji/usunięcia wirusów.
Mimo to, przy stosowaniu leków pochodzących z krwi lub osocza ludzkiego, nie można całkowicie wykluczyć możliwości przekazania patogenów. Dotyczy to również nieznanych lub nowych wirusów oraz innych patogennych mikroorganizmów.
Przyjęte środki uważane są za skuteczne wobec wirusów o powłoce lipidowej, takich jak wirus HIV (ludzkiego zespołu niedoboru odporności), HBV (wirus zapalenia wątroby typu B) i HCV (wirus zapalenia wątroby typu C), a także wobec wirusa zapalenia wątroby typu A (HAV) bez powłoki lipidowej. Przyjęte środki mogą mieć ograniczoną skuteczność wobec wirusów bez powłoki lipidowej, takich jak parwowirus B19.
Zakażenie parwowirusem B19 może być niebezpieczne dla kobiet w ciąży (zakażenie płodu) oraz dla osób z niedoborem odporności lub zwiększonym erytropoezą (np. anemia hemolityczna).
Należy rozważyć odpowiednie szczepienia (zapalenie wątroby typu A i B) u pacjentów, którzy powtarzalnie otrzymują leki VWF/czynnik VIII pochodzące z osocza ludzkiego.
Zaleca się, aby za każdym razem po podaniu leku Vilate do pacjenta odnotowywano nazwę handlową i numer serii produktu, aby możliwe było śledzenie związku pomiędzy stanem pacjenta a podanym lekiem z konkretnej serii.
Choroba von Willebranda (VWD)
Powikłania zakrzepowo-zatorowe
Podczas stosowania leku VWF zawierającego czynnik VIII, lekarz prowadzący terapię powinien wiedzieć, że długotrwałe leczenie może prowadzić do nadmiernego wzrostu czynnika VIII:C (czynnik VIII koagulacyjny). U pacjentów otrzymujących leki VWF zawierające czynnik VIII, należy monitorować poziom czynnika VIII:C we krwi, aby uniknąć trwałego nadmiernego stężenia czynnika VIII:C we krwi, ponieważ może to zwiększyć ryzyko zakrzepicy.
Istnieje ryzyko wystąpienia zakrzepicy przy stosowaniu leków VWF zawierających czynnik VIII, szczególnie u pacjentów z znanymi czynnikami ryzyka klinicznymi lub laboratoryjnymi. Dlatego stan pacjentów z grupy ryzyka należy monitorować pod kątem wczesnych objawów zakrzepicy. Należy rozpocząć profilaktykę zakrzepów żylnych zgodnie z obowiązującymi zaleceniami.
Inhibitory
U pacjentów z VWD, szczególnie typu 3, mogą pojawić się neutralizujące przeciwciała (inhibitory) przeciwko VWF. Jeśli nie zostaną osiągnięte oczekiwane poziomy aktywności VWF:RCo we krwi lub jeśli krwawienie nie będzie kontrolowane podawaną dawką, należy przeprowadzić odpowiednią analizę w celu ustalenia obecności inhibitorów VWF.
U pacjentów z wysokim poziomem inhibitorów leczenie VWF może być nieskuteczne, dlatego należy rozważyć inne opcje terapii. Leczenie takich pacjentów powinno być prowadzone przez lekarzy z doświadczeniem w pracy z pacjentami z zaburzeniami krzepnięcia.
Hemofilia A
Inhibitory
Pojawienie się neutralizujących przeciwciał (inhibitorów) przeciwko czynnikowi VIII jest znanym powikłaniem leczenia pacjentów z hemofilią A.
Takimi inhibitorami są zazwyczaj immunoglobuliny klasy G (IgG) skierowane przeciwko aktywności prokoagulacyjnej czynnika VIII, których stężenie oznacza się w jednostkach Bethesda (BU) na 1 ml osocza za pomocą zmodyfikowanego testu. Ryzyko powstania inhibitorów związane jest z ciężkością choroby oraz ekspozycją na czynnik VIII. To ryzyko jest najwyższe w ciągu pierwszych 50 dni ekspozycji i pozostaje obecne przez całe życie, choć jest rzadkie.
Kliniczne znaczenie rozwoju inhibitorów zależy od ich miana: przy niskim mianie ryzyko niewystarczającej odpowiedzi klinicznej jest mniejsze niż przy wysokim mianie inhibitorów. Ogólnie rzecz biorąc, wszystkich pacjentów, którzy otrzymywali leczenie lekami zawierającymi czynnik VIII, należy dokładnie monitorować pod kątem rozwoju inhibitorów za pomocą odpowiednich obserwacji klinicznych i badań laboratoryjnych.
Jeśli oczekiwany poziom aktywności czynnika VIII we krwi nie zostanie osiągnięty lub jeśli krwawienie nie będzie kontrolowane podawaną dawką, należy przeprowadzić badanie w celu wykrycia obecności inhibitorów przeciwko czynnikowi VIII. U pacjentów z wysokim poziomem inhibitorów leczenie czynnikiem VIII może być nieskuteczne, dlatego należy rozważyć inne opcje terapiwania.
Leczenie takich pacjentów powinno być prowadzone przez lekarzy z doświadczeniem w leczeniu hemofilii i powstawania inhibitorów przeciwko czynnikowi VIII.
Powikłania sercowo-naczyniowe
U pacjentów z istniejącymi czynnikami ryzyka powikłań sercowo-naczyniowych terapia zastępcza FVIII może zwiększyć ryzyko wystąpienia powikłań sercowo-naczyniowych.
Powikłania związane z cewnikiem
Jeśli konieczne jest zastosowanie urządzenia do dostępu do żyły centralnej (CVAD), należy wziąć pod uwagę ryzyko powikłań związanych z CVAD, w tym infekcji miejscowych, bakteriemii i zakrzepicy w miejscu wprowadzenia cewnika.
Ten lek zawiera do 58,7 mg sodu na dawkę 500 MO VWF i czynnika VIII/ampułka oraz do 117,3 mg sodu na dawkę 1000 MO VWF i czynnika VIII/ampułka, co odpowiada odpowiednio 2,94 % i 5,87 % rekomendowanej przez WHO maksymalnej dziennej dawki 2 g sodu dla dorosłego.
Należy zwrócić na to uwagę pacjentom przestrzegającym diety o ograniczonej zawartości soli.
Pacjenci w wieku dziecięcym
Wymienione szczególne wskazówki i środki ostrożności dotyczą zarówno dorosłych, jak i dzieci.
Stosowanie w czasie ciąży lub karmienia piersią.
Nie przeprowadzono badań funkcji rozrodczych na zwierzętach z VWF/czynnikiem VIII.
Choroba von Willebranda (VWD)
Brak jest doświadczenia w leczeniu kobiet w ciąży lub karmiących piersią.
W przypadku niedoboru VWF lek VILATE należy przepisać kobietom w ciąży i karmiącym piersią tylko wtedy, gdy jest to absolutnie wskazane, biorąc pod uwagę zwiększone ryzyko krwawienia u tych pacjentów podczas porodu.
Hemofilia A
Ze względu na rzadkość występowania hemofilii A u kobiet, brak jest doświadczenia w leczeniu w czasie ciąży i karmienia piersią. Dlatego VILATE należy stosować w czasie ciąży i karmienia piersią tylko wtedy, gdy istnieją wyraźne wskazania.
Wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn.
VILATE 500 MO nie wpływa na zdolność prowadzenia pojazdów i obsługiwanie maszyn.
Sposób stosowania i dawki
Leczenie należy rozpoczynać pod nadzorem lekarza posiadającego doświadczenie w leczeniu zaburzeń krzepnięcia krwi. Preparat w fiolce przeznaczony jest do jednorazowego użytku. W przypadku pozostania jakiegokolwiek resztkowego preparatu w fiolce, należy ją zutylizować zgodnie z lokalnymi przepisami.
Choroba von Willebranda (VWF)
Stosunek aktywności VWF:RCo do VWF:Ag (aktywność koofaktora rystocetynowego czynnika von Willebranda do antygenowego czynnika VIII) wynosi około 1:1. Zazwyczaj 1 J/mkg masy ciała VWF:RCo i FVIII:C zwiększa aktywność odpowiedniego białka we krwi o 1,5–2% wartości normalnej. Zwykle wymagana jest dawka od 20 do 50 J Vilate/mkg masy ciała w celu osiągnięcia odpowiedniego hemostazy. Odpowiada to wzrostowi aktywności VWF:RCo i FVIII:C u pacjentów o około 30–100%.
Może być konieczna wyższa dawka początkowa, w zakresie 50–80 J Vilate/mkg masy ciała, szczególnie u pacjentów z typem 3 choroby von Willebranda (VWD), u których utrzymanie odpowiedniego poziomu aktywności we krwi może wymagać wyższych dawek niż u pacjentów z innymi typami VWD.
Pacjenci w wieku dziecięcym
Brakuje wystarczających danych, aby zalecić stosowanie preparatu Vilate dzieciom poniżej 6. roku życia.
Profilaktyka krwawień podczas zabiegów chirurgicznych lub ciężkich urazów
W celu zapobiegania krwawieniom podczas zabiegów chirurgicznych, preparat Vilate należy podawać 1–2 godziny przed rozpoczęciem zabiegu. Należy osiągnąć poziomy VWF:RCo ≥ 60 J/dl (≥ 60%) oraz FVIII:C ≥ 40 J/dl (≥ 40%).
Odpowiednią dawkę należy powtarzać co 12–24 godziny. Dawkę i długość trwania leczenia należy dostosować do stanu klinicznego pacjenta, rodzaju i nasilenia krwawienia oraz poziomów VWF:RCo i FVIII:C.
U pacjentów otrzymujących preparaty VWF zawierające FVIII należy monitorować poziom FVIII:C we krwi w celu uniknięcia trwałego nadmiaru tej aktywności, ponieważ może to zwiększyć ryzyko zakrzepicy, szczególnie u pacjentów z znanymi czynnikami ryzyka klinicznymi lub laboratoryjnymi. W przypadku stwierdzenia nadmiernych poziomów FVIII:C we krwi, należy rozważyć zmniejszenie dawek i/lub wydłużenie odstępu między dawkami lub zastosowanie preparatu VWF o niskim stężeniu FVIII.
Profilaktyka
W celu długoterminowej profilaktyki krwawień u pacjentów z VWD, dawkę 20–40 J/kg masy ciała należy podawać 2 lub 3 razy w tygodniu. W niektórych przypadkach, np. przy krwawieniach przewodu pokarmowego, mogą być wymagane wyższe dawki.
Hemofilia A
Monitorowanie leczenia
Podczas leczenia zaleca się odpowiednie oznaczanie poziomu czynnika VIII w celu ustalenia dawki i częstotliwości powtarzanych infuzji. Indywidualni pacjenci mogą różnie reagować na leczenie czynnikiem VIII, wykazując różne okresy półwydalenia i regeneracji. Pacjentom z niedowagą lub nadwagą może być konieczna korekta dawki zależna od masy ciała. Szczególnie przy dużych zabiegach chirurgicznych konieczne jest dokładne monitorowanie terapii zastępczej poprzez analizę krzepnięcia krwi (aktywność czynnika VIII we krwi).
Dawkowanie
Dawkowanie i długość trwania terapii zastępczej zależą od stopnia nasilenia niedoboru czynnika VIII, lokalizacji i nasilenia krwawienia oraz stanu klinicznego pacjenta.
Ilość jednostek czynnika VIII podawanych pacjentowi wyraża się w jednostkach międzynarodowych (J), zgodnie z obowiązującym międzynarodowym standardem WHO dla preparatów czynnika VIII. Aktywność czynnika VIII we krwi wyrażana jest w procentach (w stosunku do normalnej ludzkiej osocza) lub w jednostkach międzynarodowych (w stosunku do Międzynarodowego Standardu FVIII we krwi).
Jedna jednostka międzynarodowa (J) aktywności czynnika VIII odpowiada ilości czynnika VIII zawartej w 1 ml normalnego osocza ludzkiego.
Leczenie w trybie potrzeby
Dawkę czynnika VIII oblicza się na podstawie danych empirycznych, że 1 J czynnika VIII:C/kg masy ciała zwiększa aktywność czynnika we krwi o 1,5–2% wartości normalnej. Wymaganą dawkę oblicza się według następującego wzoru:
Wymagane J = masa ciała (kg) × pożądane zwiększenie aktywności czynnika VIII (%) (J/dl) × 0,5 J/kg
Ilość i częstotliwość podawania należy zawsze dostosować do skuteczności klinicznej w każdym przypadku. W przypadku wystąpienia krwawień, aktywność czynnika VIII nie powinna spadać poniżej ustalonego poziomu aktywności we krwi (w % wartości normalnej lub J/dl) przez odpowiedni okres czasu.
Poniższa tabela 3 może być wykorzystana do ustalenia dawek w przypadku krwawień i zabiegów chirurgicznych.
Tabela 3
Schemat leczenia przy krwawieniach i zabiegach chirurgicznych
| Stopień krwawienia/ Typ zabiegu chirurgicznego |
Wymagany poziom czynnika VIII (%) (j.m./dL) |
Częstotliwość dawkowania (godziny)/ Czas trwania leczenia (dni) |
| Krwawienie |
||
| Początkowy hemartroza (krwawienie do stawu), krwawienie z mięśni lub z jamy ustnej |
20 – 40 |
Podawanie powtarzać co 12 – 24 godziny przez co najmniej 1 dzień, aż do ustąpienia krwawienia (o czym świadczy ustępujący ból) lub do pełnego wyzdrowienia. |
| Uogólniona hemartroza, krwawienie z mięśni lub hematoma |
30 – 60 |
Podawanie powtarzać co 12 – 24 godziny przez 3 – 4 dni lub dłużej, aż do ustąpienia bólu i ostrych ograniczeń ruchomości. |
| Krwawienia zagrożone dla życia |
60 – 100 |
Podawanie powtarzać co 8 – 24 godziny, aż do przejścia stanu zagrożenia życia. |
| Zabieg chirurgiczny |
||
| Mały zabieg chirurgiczny, w tym usunięcie zęba |
30 – 60 |
Co 24 godziny, co najmniej 1 dzień, aż do pełnego wyzdrowienia. |
| Duży zabieg chirurgiczny |
80 – 100 (przed- i pooperacyjne) |
Podawanie powtarzać co 8 – 24 godziny aż do pełnego gojenia się rany, a następnie kontynuować leczenie przez co najmniej kolejne 7 dni w celu utrzymania aktywności czynnika VIII na poziomie 30–60% (j.m./dL). |
Profilaktyka
W celu długoterminowej profilaktyki krwawień u pacjentów z ciężką hemofilią A należy podawać dawki standardowe w zakresie od 20 do 40 J m. czynnika VIII na 1 kg masy ciała co 2–3 dni. W niektórych przypadkach, szczególnie u młodszych pacjentów, mogą być wymagane krótsze odstępy między podawaniem leku lub wyższe dawki.
Infuzja ciągła
Przed zabiegiem chirurgicznym należy przeprowadzić analizę farmakokinetyczną w celu oszacowania klirensu. Początkową szybkość podawania leku można obliczyć według następującego wzoru:
Szybkość podawania leku (J m./kg/godz) = klirens (ml/kg/godz) × pożądany poziom stężenia w stanie ustalonym (J m./ml)
Po pierwszych 24 godzinach ciągłej infuzji należy codziennie ponownie obliczać klirens, stosując równanie stanu ustalonego, zmierzone stężenie oraz znaną szybkość podawania leku.
Pacjenci w wieku dziecięcym
Brakuje wystarczających danych, aby zalecić stosowanie Vilate 500 MO dzieciom w wieku do 6 lat z hemofilią A.
Sposób stosowania
Podanie dożylnie.
Szybkość wstrzykiwania lub infuzji nie powinna przekraczać 2–3 ml na minutę.
Szczególne środki ostrożności dotyczące utylizacji i dalszego przetwarzania
Należy zapoznać się ze wszystkimi instrukcjami i dokładnie je przestrzegać!
Nie należy stosować leku po upływie daty ważności wskazanej na etykiecie.
Podczas poniżej opisanej procedury należy zachować warunki jałowe!
Przed podaniem należy wizualnie sprawdzić rozcieńczony lek pod kątem obecności widocznych (stałych) cząstek oraz zmiany barwy.
Roztwór powinien być klarowny lub lekko opalescencyjny. Nie wolno stosować roztworów mętnych lub zawierających osad.
Przygotowany roztwór należy stosować natychmiast, aby uniknąć zanieczyszczenia mikroorganizmami.
Należy stosować wyłącznie system wstrzykowy dostarczany wraz z lekiem. Stosowanie innego sprzętu wstrzykowego/infuzyjnego może wiązać się z dodatkowymi ryzykami i prowadzić do nieskuteczności leczenia.
Instrukcja przygotowania roztworu
- Nie należy stosować leku bezpośrednio z lodówki. Roztwórnik i proszek w zamkniętych fiolkach powinny osiągnąć temperaturę pokojową.
- Zdjąć zabezpieczenia z obu fiol i przetrzeć korki gumowe jednym z dostarczonych w opakowaniu tamponów nasączonych alkoholem.
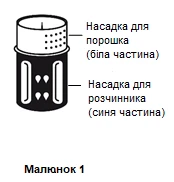
- System do przenoszenia przedstawiono na rysunku 1.
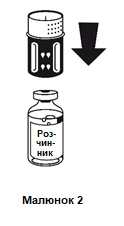
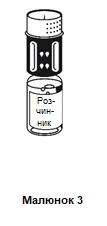
Postawić fiolkę zawierającą rozcieńczalnik na płaskiej, równej powierzchni i trzymać ją mocno. System do przenoszenia należy ustawić niebieską częścią do góry na fiolkę zawierającą rozcieńczalnik i wcisnąć mocno w dół, aż do usłyszenia kliknięcia (patrz rysunki 2 i 3).
Nie należy obracać podczas mocowania.
|
|
|
|
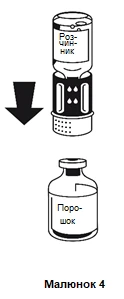
- Postawić fiolkę zawierającą proszek na równej, płaskiej powierzchni i trzymać ją mocno. Wziąć fiolkę z rozpuszczalnikiem razem z zamocowanym systemem do przetaczania i odwrócić ją do góry dnem. Umieścić częścią białą do góry na fiolce zawierającej proszek i nacisnąć mocno w dół, aż usłyszy się kliknięcie (rysunek 4). Nie obracać podczas mocowania. Rozpuszczalnik automatycznie dostanie się do fiolki zawierającej proszek.
- Gdy obie fiolki są nadal połączone, należy delikatnie i ostrożnie mieszać zawartość fiolki z proszkiem, aż lek całkowicie się rozpuści.
Rozpuszczanie odbywa się w mniej niż 10 minut w temperaturze pokojowej. Podczas przygotowywania może pojawić się nieznaczne pienienie. Rozłączyć system do przetaczania na dwie części, rozkręcacając go (rysunek 5). Pienienie zaniknie.
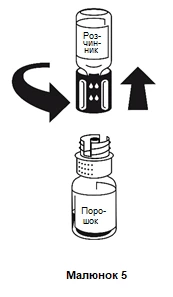
Usuń pustą fiolkę z rozpuszczalnikiem razem z niebieską częścią systemu do przetaczania.
Instrukcja wstrzykiwania
Jako środek ostrożności należy określić częstość pulsowania u pacjenta przed wstrzyknięciem oraz podczas podawania leku. Jeśli stwierdzi się zwiększenie częstości pulsów, należy zmniejszyć tempo wstrzykiwania lub całkowicie przerwać podawanie na pewien czas.
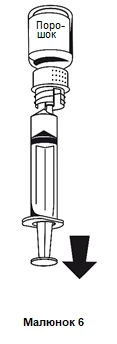
- 1. Zamocować strzykawkę do białej części systemu do przetaczania. Odwrócić fiolkę do góry dnem i nabrać roztwór do strzykawki (rysunek 6).
Roztwór powinien być klarowny lub lekko opalescencyjny.
Natychmiast po napełnieniu strzykawki, trzymając mocno cylinder strzykawki, odłączyć strzykawkę od systemu do przetaczania (rysunek 7).
|
|
|
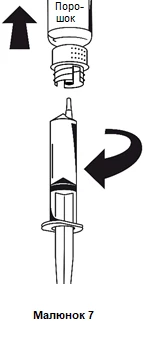
Pojemnik po zużyciu należy zutylizować razem z białą częścią systemu do przenoszenia.
- 2. Zdezynfekować wybrane miejsce wstrzyknięcia jednym z dostarczonych tamponów nasączonych alkoholem.
- Dołączyć dostarczony system do wstrzykiwania do strzykawki.
- Wprowadzić igłę do wstrzykiwania do odpowiedniej żyły. Jeśli do lepszego zobrazowania żyły stosuje się opaskę uciskową (zawieszającą), należy ją poluzować przed rozpoczęciem wstrzykiwania leku Vilate.
Krew nie powinna dostać się do strzykawki ze względu na ryzyko powstawania skrzeplin fibrynogennych.
- Wstrzykiwać roztwór do żyły bardzo powoli, z prędkością 2–3 ml na minutę.
Jeśli do jednego wstrzyknięcia stosuje się więcej niż jeden fiolę proszku Vilate, można ponownie użyć tej samej igły i strzykawki. System do przenoszenia przeznaczony jest wyłącznie do jednorazowego użytku (używać tylko raz)!
Niezaopatrzone leki lub odpady należy utylizować zgodnie z lokalnymi wymaganiami.
Dzieci.
Choroba von Willebranda (VWD)
Brak wystarczających danych, aby zalecić stosowanie leku Vilate dzieciom poniżej 6. roku życia.
Hemofilia A
Brak wystarczających danych, aby zalecić stosowanie leku Vilate dzieciom poniżej 6. roku życia z hemofilią A.
Przedawkowanie.
Nie odnotowano objawów przedawkowania FVIII ani VWF u ludzi. W przypadku znacznego przedawkowania mogą wystąpić powikłania zakrzepowo-emboliczne.
Niepożądane reakcje.
Skrócony przegląd profilu bezpieczeństwa
Zwiększona wrażliwość lub reakcje alergiczne (które mogą obejmować m.in.: obrzęk naczynioruchowy, uczucie pieczenia i mrowienia w miejscu wstrzyknięcia, dreszcze, zaczerwienienie (prypływy), uogólnione pokrzywienie, rumień, swędzenie, wysypkę, ból głowy, pokrzywienienie, hipotensję, senność, nudności, pobudzenie, tachykardię, duszność, trudności w oddychaniu, uczucie szczypania, wymioty, chrapanie/szumy oddechowe) występują rzadko i w niektórych przypadkach mogą postępować do ciężkiej anafilaksji (w tym wstrząs anafilaktyczny).
Choroba von Willebranda (VWD)
U pacjentów z VWD, szczególnie typu 3, bardzo rzadko mogą rozwijać się neutralizujące przeciwciała przeciwko VWF. Jeśli takie inhibitory wystąpią, będą objawiać się niewystarczającą odpowiedzią kliniczną. Takie przeciwciała mogą pojawiać się równocześnie z reakcjami anafilaktycznymi. W związku z tym pacjentów z reakcją anafilaktyczną należy przebadać pod kątem obecności inhibitorów.
We wszystkich przypadkach reakcji anafilaktycznej zaleca się skonsultowanie się ze specjalistycznym ośrodkiem leczenia hemofilii.
Istnieje ryzyko wystąpienia zakrzepicy, szczególnie u pacjentów z znanymi czynnikami klinicznymi lub laboratoryjnymi zwiększającymi to ryzyko. Należy rozpocząć profilaktykę zakrzepicy żylnej zgodnie z obowiązującymi zaleceniami.
U pacjentów otrzymujących leki zawierające VWF i FVIII, trwałe nadmiarowe stężenia FVIII:C we krwi mogą zwiększać ryzyko powstawania zakrzepów.
Hemofilia A
U pacjentów z hemofilią A, którzy otrzymywali leczenie czynnikiem VIII oraz lekiem Vilate, mogą pojawiać się neutralizujące przeciwciała (inhibitory), patrz sekcja „Właściwości farmakodynamiczne”. W przypadku pojawienia się takich inhibitorów stan będzie się objawiał niewystarczającą odpowiedzią kliniczną. W takich przypadkach zaleca się skonsultowanie się ze specjalistycznym ośrodkiem leczenia hemofilii.
Lista niepożądanych reakcji w formie tabeli
Częstość występowania niepożądanych reakcji szacuje się według następujących umownych oznaczeń: bardzo często (≥ 1/10); często (od ≥ 1/100 do < 1/10); nieczęsto (od ≥ 1/1 000 do < 1/100); rzadko (od ≥ 1/10 000 do < 1/1 000); bardzo rzadko (< 1/10 000); częstość nieznana (nie można oszacować na podstawie dostępnych danych).
W tabeli 4 wymieniono niepożądane reakcje obserwowane w badaniach klinicznych, badaniach pozarejestrowych dotyczących bezpieczeństwa oraz znane z innych pozarejestrowych źródeł. Niepożądane reakcje podano według klas układów narządów (SOC) MedDRA, przy użyciu preferowanych terminów (PT), oraz według częstości.
Tabela 4
| Klasa systemu organów (SOC/КСО) MedDRA |
Reakcja niepożądana |
Częstotliwość |
| Zaburzenia ze strony układu odpornościowego |
Zwiększona wrażliwość Szok anafilaktyczny |
Nieczeście Bardzo rzadko |
| Ogólne zaburzenia i zaburzenia w miejscu podania |
Gorączka Ból w klatce piersiowej |
Nieczeście Częstotliwość nieznana |
| Zaburzenia ze strony układu krwi i chłonnego |
Inhibitory czynnika VIII Inhibitory czynnika von Willebranda |
Nieczeście (PTPs)* Bardzo często (PUPs) Bardzo rzadko |
| Zaburzenia ze strony układu oddechowego, narządów klatki piersiowej i jamy śródpiersia |
Kaszel |
Częstotliwość nieznana |
| Zaburzenia ze strony układu nerwowego |
Zawroty głowy |
Częstotliwość nieznana |
| Zaburzenia ze strony przewodu pokarmowego |
Ból brzucha |
Częstotliwość nieznana |
| Zaburzenia ze strony tkanki mięśniowej i tkanki łącznej |
Ból pleców |
Częstotliwość nieznana |
* Częstotliwość oparta na badaniach z udziałem wszystkich leków zawierających FVIII przeprowadzonych u pacjentów z ciężką hemofilią typu A. PTPs – pacjenci wcześniej leczeni; PUPs – pacjenci wcześniej nieleczeni.
Opis poszczególnych działań niepożądanych
Informacje o poszczególnych działaniach niepożądanych znajdują się w sekcji „Szczególne ostrzeżenia i środki ostrożności podczas stosowania”.
Zgłaszanie podejrzewanych działań niepożądanych
Zgłaszanie działań niepożądanych po rejestracji leku ma istotne znaczenie. Umożliwia to monitorowanie stosunku korzyści do ryzyka związanego z zastosowaniem tego leku. Osoby medyczne i farmaceutyczne, a także pacjenci lub ich ustawowo upoważnieni przedstawiciele, powinni zgłaszać wszystkie przypadki podejrzewanych działań niepożądanych oraz braku skuteczności leku poprzez Automatyczny System Informacyjny Nadzoru Farmakologicznego pod adresem: https://aisf.dec.gov.ua.
Okres ważności.
Proszek do sporządzenia roztworu do wstrzykiwań – 3 lata.
Stabilność rozcieńczonego roztworu utrzymuje się przez 4 godziny w temperaturze pokojowej (nie wyższej niż 25°C). Jednakże, aby uniknąć zanieczyszczenia mikroorganizmami, rozcieńczony roztwór należy użyć natychmiast.
Rozpuszczalnik przechowywać przez 4 lata.
Rozpuszczalnik przechowywać przez 4 lata w temperaturze od 2 do 8°C, w miejscu zabezpieczonym przed światłem.
W trakcie tego okresu rozpuszczalnik można przechowywać do 6 miesięcy w temperaturze do 25°C. W takim przypadku okres ważności kończy się po 6 miesiącach.
Warunki przechowywania.
Przechowywać w temperaturze od 2 do 8°C. Nie zamarzać.
Przechowywać fiolkę w tekturowym opakowaniu w celu ochrony przed światłem.
Przechowywać w miejscu niedostępnym dla dzieci.
W okresie ważności lek może być przechowywany w temperaturze pokojowej (nie wyższej niż 25°C) przez okres do 2 miesięcy. W takim przypadku okres ważności kończy się 2 miesiące po pierwszym wyjęciu leku z lodówki.
Pacjent powinien wpisać nowy termin ważności na zewnętrznym tekturowym opakowaniu.
Rozcieńczony roztwór przeznaczony jest do jednorazowego użycia. Każdy roztwór pozostał w fiolce należy zutylizować.
Niezgodność.
W przypadku braku badań zgodności, tego leku nie należy mieszać z innymi lekami ani podawać jednocześnie z innym lekiem dożylno w tym samym (jednym) systemie infuzyjnym. Należy stosować wyłącznie systemy wstrzykawcze/infuzyjne dostarczane w zestawie, ponieważ leczenie może okazać się nieskuteczne z powodu adsorpcji (wchłaniania) czynnika VIII/czynnika von Willebranda przez wewnętrzną powierzchnię niektórych urządzeń infuzyjnych.
Opakowanie
Tekturowe pudełko nr 1: 1 fiolka z proszkiem do sporządzenia roztworu do wstrzykiwań (500 MO) oraz instrukcja stosowania.
Tekturowe pudełko nr 2: 1 fiolka z rozpuszczalnikiem (woda do wstrzykiwań z 0,1% polisorbata 80) o pojemności 5 ml wraz z zestawem do podania dożylnego oraz 2 alkoholem nasączone tampony.
Zestaw do podania dożylnego składa się z: 1 jednorazowej strzykawki, 1 zestawu do przetaczania, 1 zestawu infuzyjnego.
Tekturowe pudełko nr 1 i tekturowe pudełko nr 2 są połączone ze sobą folią plastikową.
Tekturowe pudełko nr 1: 1 fiolka z proszkiem do sporządzenia roztworu do wstrzykiwań (1000 MO) oraz instrukcja stosowania.
Tekturowe pudełko nr 2: 1 fiolka z rozpuszczalnikiem (woda do wstrzykiwań z 0,1% polisorbata 80) o pojemności 10 ml wraz z zestawem do podania dożylnego oraz 2 alkoholem nasączone tampony.
Zestaw do podania dożylnego składa się z: 1 jednorazowej strzykawki, 1 zestawu do przetaczania, 1 zestawu infuzyjnego.
Tekturowe pudełko nr 1 i tekturowe pudełko nr 2 są połączone ze sobą folią plastikową.
Kategoria wydawania. Na receptę.
Producent. Octapharma Pharmazeutika Produktionsges.m.b.H.
Adres producenta i miejsce prowadzenia działalności.
Oberlaaer Straße 235, 1100 Wiedeń, Austria.