Vilate 1000 UMI
UcrainaIndice
ISTRUZIONI PER L'USO MEDICINALE DEL MEDICINALE VILATE 500 UMI VILATE 1000 UMI
Composizione:
Principi attivi: fattore della coagulazione del sangue VIII, fattore di von Willebrand;
1 flaconcino contiene 500 oppure 1000 UMI di fattore della coagulazione del sangue VIII e 500 UMI oppure 1000 UMI di fattore di von Willebrand; contenuto di proteine totali ≤ 7,5 mg (500 UMI) oppure ≤ 15 mg (1000 UMI);
Eccipienti: glicina, saccarosio, cloruro di sodio, citrato di sodio, cloruro di calcio.
Solvente: acqua per preparazioni iniettabili con 0,1% di polisorbato 80.
Forma farmaceutica. Polvere e solvente per soluzione iniettabile.
Principali caratteristiche fisico-chimiche:
Polvere: polvere o massa friabile di colore bianco o giallo pallido.
Solvente: liquido limpido incolore, inodore, privo di particelle visibili.
Gruppo farmacoterapeutico. Preparati antiedemorragici. Fattori della coagulazione ematica. Fattore di von Willebrand e fattore della coagulazione VIII in associazione. Codice ATC B02B D06.
Proprietà farmacologiche.
Farmacodinamica.
Malattia di von Willebrand (VWD)
Il fattore di von Willebrand (dal concentrato) è un componente normale del plasma umano ed agisce come il fattore di von Willebrand endogeno.
L'amministrazione del fattore di von Willebrand permette di correggere i disturbi emostatici osservati nei pazienti con deficit del fattore di von Willebrand a due livelli:
- il fattore di von Willebrand ripristina l'adesione delle piastrine all'endotelio subvascolare nel sito di lesione vascolare (poiché si lega sia all'endotelio subvascolare che alla membrana piastrinica), garantendo l'emostasi primaria, come dimostrato dall'accorciamento del tempo di sanguinamento. Questo effetto si manifesta immediatamente e dipende notevolmente dal grado di polimerizzazione della proteina;
- il fattore di von Willebrand induce una correzione ritardata della carenza associata al fattore VIII. Dopo somministrazione endovenosa, il fattore di von Willebrand si lega al fattore VIII endogeno (normalmente prodotto dal paziente), stabilizzandolo e impedendone la rapida degradazione. Di conseguenza, l'infusione di fattore di von Willebrand puro (preparato con basso contenuto di fattore VIII) normalizza i livelli di fattore VIII:C come effetto secondario dopo la prima infusione. L'infusione di un preparato di fattore di von Willebrand contenente fattore VIII normalizza immediatamente i livelli di fattore VIII:C dopo la prima infusione.
Oltre al suo ruolo di proteina di stabilizzazione del fattore VIII, il fattore di von Willebrand media l'adesione piastrinica ai siti di danno vascolare e partecipa all'aggregazione piastrinica.
Emofilia A
Il complesso fattore VIII/fattore di von Willebrand è costituito da due molecole (fattore VIII e fattore di von Willebrand) con diverse funzioni fisiologiche. Dopo somministrazione al paziente emofilico, il fattore VIII si lega al fattore di von Willebrand nella circolazione del paziente. Il fattore VIII attivato (fattore VIIIa) agisce come cofattore del fattore IX attivato (fattore IXa), accelerando la trasformazione del fattore X in fattore X attivato (fattore Xa). Il fattore Xa converte la protrombina in trombina. La trombina a sua volta trasforma il fibrinogeno in fibrina, permettendo la formazione del trombo.
L'emofilia A è una malattia ereditaria legata al sesso, caratterizzata da un disturbo della coagulazione dovuto a livelli ridotti di fattore VIII:C (attività coagulante), che determina emorragie gravi nelle articolazioni, nei muscoli o negli organi interni, spontanee o conseguenti a traumi accidentali o chirurgici. Grazie alla terapia sostitutiva, i livelli plasmatici di fattore VIII aumentano, determinando una correzione temporanea del deficit e riducendo la tendenza all'emorragia.
È importante notare che non è possibile confrontare direttamente la frequenza media annua di emorragie (ABR) osservata in diversi studi clinici o con l'uso di diversi concentrati di fattore.
Oltre al suo ruolo di proteina di stabilizzazione del fattore VIII, il fattore di von Willebrand media l'adesione piastrinica ai siti di danno vascolare e partecipa all'aggregazione piastrinica.
Farmacocinetica.
Malattia di von Willebrand (VWD)
Il fattore di von Willebrand (dal concentrato) è un componente normale del plasma umano ed agisce come il fattore di von Willebrand endogeno.
Tabella 1
Risultati della meta-analisi di tre studi farmacocinetici condotti su 24 pazienti affetti da tutti i tipi di VWD.
| Parametro |
Tutti i tipi di VWD |
VWD di tipo 1 |
VWD di tipo 2 |
VWD di tipo 3 |
|||||||||||||||||
| N |
Media val. |
SD |
Min ∙ |
Max ∙ |
N |
Media val. |
SD |
Min ∙ |
Max ∙ |
N |
Media val. |
SD |
Min ∙ |
Max ∙ |
N |
Media val. |
SD |
Min ∙ |
Max ∙ |
||
| Recupero (%/UI/kg) |
24 |
1,56 |
0,48 |
0,90 |
2,93 |
2 |
1,19 |
0,07 |
1,14 |
1,24 |
5 |
1,83 |
0,86 |
0,98 |
2,93 |
17 |
1,52 |
0,32 |
0,90 |
2,24 |
|
| AUC (0-inf) (g*%) |
23 |
1981 |
960 |
593 |
4831 |
2 |
2062 |
510 |
1701 |
2423 |
5 |
2971 |
1383 |
1511 |
4831 |
16 |
1662 |
622 |
593 |
2606 |
|
| T 1/2 (h) |
24 |
23,3 |
12,6 |
7,4 |
58,4 |
2 |
39,7 |
18,3 |
26,7 |
52,7 |
5 |
34,9 |
16 |
17,5 |
58,4 |
17 |
18 |
6,2 |
7,4 |
30,5 |
|
| MRT (h) |
24 |
33,1 |
19 |
10,1 |
89,7 |
2 |
53,6 |
25,9 |
35,3 |
71,9 |
5 |
53,5 |
24,6 |
27,8 |
89,7 |
17 |
24,7 |
8,5 |
10,1 |
37,7 |
|
| Clearance (ml/h/kg) |
24 |
3,29 |
1,67 |
0,91 |
7,41 |
2 |
2,66 |
0,85 |
2,06 |
3,27 |
5 |
1,95 |
1,02 |
0,91 |
3,31 |
17 |
3,76 |
1,69 |
1,83 |
7,41 |
|
AUC – area sotto la curva; MRT – tempo medio di permanenza del farmaco nell'organismo; SD – deviazione standard.
Emofilia A
Il fattore VIII (da concentrato) è un componente normalmente presente nel plasma umano e agisce come fattore VIII endogeno. Dopo la somministrazione del farmaco, circa da 2/3 a 3/4 del fattore VIII rimane in circolo. Il livello di attività del fattore VIII:C raggiunto nel plasma corrisponde all'80% - 120% dell'attività prevista (attesa) del fattore VIII.
L'attività del fattore VIII diminuisce secondo un decadimento esponenziale bifasico. Nella fase iniziale avviene una distribuzione tra il comparto intravascolare e altri compartimenti (fluidi corporei), con un'emivita plasmatica compresa tra 3 e 6 ore. Nella successiva fase più lenta, l'emivita varia da 8 a 18 ore, con un valore medio di 15 ore. Questo corrisponde all'emivita biologica effettiva.
Tabella 2
Risultati di uno studio clinico condotto su 12 pazienti (analisi cromogenica, determinazione mediante doppia misurazione).
| Parametro |
Visita del livello basale |
Visita a 6 mesi |
||
| Valore medio |
SD |
Valore medio |
SD |
|
| Ritrovamento (%/UML/kg) |
FVIII:C 2,27 |
1,20 |
FVIII:C 2,26 |
1,19 |
| AUCnorm (% × g/UML/kg) |
FVIII:C 31,3 |
7,31 |
FVIII:C 33,8 |
10,9 |
| Emivita (h) |
FVIII:C 11,2 |
2,85 |
FVIII:C 11,8 |
3,37 |
| MRT (h) |
FVIII:C 15,3 |
3,5 |
FVIII:C 16,3 |
4,6 |
| Clearance ml/h/kg |
FVIII:C 3,37 |
0,86 |
FVIII:C 3,24 |
1,04 |
AUC – area sotto la curva; MRT – tempo medio di permanenza del farmaco nell'organismo;
SD – deviazione standard.
Dati dagli studi preclinici di sicurezza
Il VWF e il fattore VIII in Vilate sono componenti normali del plasma sanguigno umano e agiscono come VWF/fattore VIII endogeno.
Uno studio standard di sicurezza di questi componenti negli animali da laboratorio non fornirebbe informazioni aggiuntive rispetto all'esperienza clinica esistente; pertanto, tale studio non è necessario.
Caratteristiche cliniche.
Indicazioni.
Malattia di von Willebrand (VWD)
Prevenzione e trattamento delle emorragie o sanguinamento durante interventi chirurgici nella malattia di von Willebrand (VWD), quando il trattamento con desmopressina (DDAVP) da sola risulta inefficace o è controindicato.
Emofilia A
Trattamento e prevenzione delle emorragie nei pazienti con emofilia A (deficit congenito del fattore di coagulazione del sangue VIII).
Controindicazioni.
Reazioni allergiche ai principi attivi o a qualsiasi degli eccipienti.
Interazioni con altri medicinali e altre forme di interazione.
Non sono note interazioni con altri medicinali.
Caratteristiche di impiego.
Tracciabilità
Per migliorare il tracciamento dei medicinali biologici, è necessario registrare chiaramente il nome e il numero di lotto del medicinale somministrato.
Ipersensibilità
Nel corso dell'uso del medicinale VILATE possono verificarsi reazioni allergiche. Il medicinale contiene, oltre al fattore VIII, tracce di proteine umane. In caso di comparsa di sintomi di ipersensibilità, ai pazienti deve essere raccomandato di interrompere immediatamente il trattamento e di consultare il medico.
I pazienti devono essere informati sui segni precoci di reazioni allergiche, quali: eruzioni cutanee, orticaria generalizzata, difficoltà respiratorie, dispnea, ipotensione e anafilassi.
In caso di shock, deve essere praticata una terapia antishock standard.
Trasmissione di agenti infettivi
Le misure abituali per prevenire infezioni derivanti dall'uso di medicinali ottenuti dal sangue o dal plasma umano comprendono la selezione dei donatori, lo screening del sangue di singoli donatori e dei lotti di plasma donato per specifici marcatori di infezione e l'inclusione di fasi efficaci nel processo produttivo per l'inattivazione/rimozione dei virus.
Tuttavia, quando si somministrano medicinali ottenuti dal sangue o dal plasma umano, non è possibile escludere completamente la possibilità di trasmissione di agenti infettivi. Ciò vale anche per virus sconosciuti o nuovi e altri agenti patogeni.
Le misure adottate sono considerate efficaci nei confronti di virus dotati di envelope, come il virus dell'immunodeficienza umana (HIV), il virus dell'epatite B (HBV) e il virus dell'epatite C (HCV), nonché nei confronti del virus dell'epatite A (HAV), privo di envelope. Le misure adottate potrebbero avere efficacia limitata nei confronti di virus privi di envelope, come il parvovirus B19.
L'infezione da parvovirus B19 può essere pericolosa per le donne in gravidanza (infezione fetale) e per soggetti con immunodeficienza o eritropoiesi aumentata (ad esempio, anemia emolitica).
Si deve prendere in considerazione la vaccinazione appropriata (epatite A e B) per i pazienti che ricevono ripetutamente o in modo cronico medicinali VWF/fattore VIII ottenuti dal plasma umano.
Si raccomanda vivamente di registrare ogni volta il nome e il numero di lotto del medicinale Vilate somministrato al paziente, al fine di consentire il tracciamento del legame tra lo stato del paziente e il medicinale di un lotto specifico.
Malattia di von Willebrand (VWD)
Complicanze tromboemboliche
Nell'uso di medicinali contenenti VWF e fattore VIII, il medico che effettua il trattamento deve essere consapevole che un trattamento prolungato può portare a un eccessivo aumento del fattore VIII:C (fattore VIII coagulante). Nei pazienti che ricevono medicinali contenenti VWF e fattore VIII, devono essere monitorati i livelli plasmatici di fattore VIII:C per evitare un livello plasmatico costantemente elevato di fattore VIII:C, poiché ciò potrebbe aumentare il rischio di trombosi.
Esiste un rischio di trombosi nell'uso di medicinali contenenti VWF e fattore VIII, specialmente nei pazienti con fattori di rischio clinici o di laboratorio noti. Pertanto, lo stato dei pazienti a rischio deve essere monitorato per la comparsa di segni precoci di trombosi. Deve essere avviata la profilassi delle trombosi venose in conformità con le raccomandazioni vigenti.
Inibitori
Nei pazienti con VWD, specialmente di tipo 3, possono svilupparsi anticorpi neutralizzanti (inibitori) contro il VWF. Se non si raggiungono i livelli attesi di attività VWF:RCo nel plasma o se il sanguinamento non viene controllato con la dose prescritta, si deve effettuare un'analisi appropriata per determinare la presenza di inibitori del VWF.
Nei pazienti con livelli elevati di inibitori, il trattamento con VWF può risultare inefficace; pertanto, si devono considerare altre opzioni terapeutiche. Il trattamento di tali pazienti deve essere effettuato da medici esperti nella gestione di pazienti con alterazioni emostatiche.
Emofilia A
Inibitori
La formazione di anticorpi neutralizzanti (inibitori) contro il fattore VIII è una complicanza nota nel trattamento dei pazienti con emofilia A.
Tali inibitori sono generalmente immunoglobuline di classe G (IgG) dirette contro l'attività procoagulante del fattore VIII, la cui concentrazione viene misurata in unità Bethesda (BU) per 1 ml di plasma mediante un'analisi modificata. Il rischio di sviluppo di inibitori è correlato alla gravità della malattia e all'esposizione al fattore VIII. Questo rischio è massimo nei primi 50 giorni di esposizione e permane per tutta la vita, sebbene sia raro.
La rilevanza clinica dello sviluppo di inibitori dipende dal titolo degli inibitori: con titoli bassi, il rischio di risposta clinica inadeguata è minore rispetto a titoli elevati. In generale, tutti i pazienti trattati con medicinali contenenti fattore VIII devono essere attentamente monitorati per lo sviluppo di inibitori, mediante osservazioni cliniche adeguate e analisi di laboratorio.
Se non si raggiungono i livelli attesi di attività del fattore VIII nel plasma o se il sanguinamento non viene controllato con la dose prescritta, è necessario effettuare un'analisi per determinare la presenza di inibitori contro il fattore VIII. Nei pazienti con livelli elevati di inibitori, il trattamento con fattore VIII può risultare inefficace; pertanto, si devono considerare altre opzioni terapeutiche.
La gestione di tali pazienti deve essere effettuata da medici esperti nel trattamento dell'emofilia e nella comparsa di inibitori contro il fattore VIII.
Complicanze cardiovascolari
Nei pazienti con fattori di rischio cardiovascolare preesistenti, la terapia sostitutiva con FVIII può aumentare il rischio di complicanze cardiovascolari.
Complicanze legate al catetere
Se è necessario l'uso di un dispositivo di accesso venoso centrale (CVAD), si devono considerare i rischi di complicanze legate al CVAD, inclusi infezioni locali, batteriemia e trombosi nel sito di inserzione del catetere.
Questo medicinale contiene fino a 58,7 mg di sodio per dose di 500 UMI di VWF e fattore VIII/flaconcino e fino a 117,3 mg di sodio per dose di 1000 UMI di VWF e fattore VIII/flaconcino, pari rispettivamente al 2,94% e al 5,87% della dose giornaliera massima raccomandata dall'OMS di 2 g di sodio per l'adulto.
Ciò deve essere tenuto in considerazione nei pazienti sottoposti a dieta controllata priva di sale.
Pazienti pediatrici
Le avvertenze e le precauzioni speciali sopra elencate si applicano sia agli adulti che ai bambini.
Uso durante la gravidanza o l'allattamento.
Non sono stati condotti studi sulla funzione riproduttiva negli animali con VWF/fattore VIII.
Malattia di von Willebrand (VWD)
Non esiste esperienza nel trattamento di donne in gravidanza o che allattano al seno.
Nel caso di deficit di VWF, il medicinale VILATE deve essere somministrato a donne in gravidanza e a quelle che allattano al seno solo se strettamente indicato, considerando che durante il parto esiste un rischio aumentato di sanguinamento in tali pazienti.
Emofilia A
A causa della rarità dei casi di emofilia A nelle donne, non esiste esperienza nel trattamento durante la gravidanza e l'allattamento. Pertanto, VILATE deve essere utilizzato durante la gravidanza e l'allattamento solo in caso di chiare indicazioni.
Capacità di guidare veicoli o di usare macchinari.
VILATE 500 UMI non influenza la capacità di guidare veicoli o di usare macchinari.
Modalità e dosaggio di somministrazione.
La terapia deve essere iniziata sotto la supervisione di un medico esperto nel trattamento dei disturbi della coagulazione. Il contenuto del flaconcino è destinato a un singolo uso. Qualsiasi residuo presente nel flaconcino dopo l'uso deve essere smaltito secondo le normative locali.
Malattia di von Willebrand (VWF)
Il rapporto tra VWF:RCo e FVIII:C (ristocetina/cofattore del fattore von Willebrand e fattore della coagulazione VIII) è approssimativamente 1:1. Generalmente, 1 UI/kg di peso corporeo di VWF:RCo e FVIII:C aumenta l'attività della proteina corrispondente nel plasma di circa il 1,5-2% rispetto al valore normale. Solitamente sono necessarie dosi comprese tra 20 e 50 UI di Vilate/kg di peso corporeo per ottenere un adeguato emostasi, determinando un incremento di VWF:RCo e FVIII:C nei pazienti di circa il 30-100%.
Potrebbe essere necessaria una dose iniziale compresa tra 50 e 80 UI di Vilate/kg di peso corporeo, specialmente nei pazienti con VWD di tipo 3, nei quali il mantenimento di un'adeguata attività plasmatica può richiedere dosi più elevate rispetto ai pazienti con altri tipi di VWD.
Pazienti pediatrici
Non sono disponibili dati sufficienti per raccomandare l'uso di Vilate nei bambini di età inferiore ai 6 anni.
Prevenzione del sanguinamento in caso di intervento chirurgico o trauma grave
Per prevenire il sanguinamento durante un intervento chirurgico, Vilate deve essere somministrato 1-2 ore prima dell'inizio dell'intervento. Devono essere raggiunti livelli di VWF:RCo ≥ 60 UI/dl (≥ 60%) e livelli di FVIII:C ≥ 40 UI/dl (≥ 40%).
La dose appropriata deve essere ripetuta ogni 12-24 ore. La dose e la durata del trattamento dipendono dalle condizioni cliniche del paziente, dal tipo e dalla gravità del sanguinamento, nonché dai livelli di VWF:RCo e FVIII:C.
Nei pazienti che ricevono preparati di VWF contenenti FVIII, è necessario monitorare attentamente i livelli plasmatici di FVIII:C per evitare un'elevazione persistente, poiché ciò potrebbe aumentare il rischio di trombosi, specialmente nei pazienti con fattori di rischio clinici o di laboratorio noti. In caso di rilevazione di livelli eccessivi di FVIII:C nel plasma, si dovrà considerare una riduzione delle dosi e/o un prolungamento dell'intervallo tra le somministrazioni oppure l'uso di un preparato di VWF con basso contenuto di FVIII.
Prevenzione
Per la profilassi a lungo termine del sanguinamento nei pazienti con VWD, si raccomandano dosi di 20-40 UI/kg di peso corporeo da somministrare 2 o 3 volte alla settimana. In alcuni casi, ad esempio nelle emorragie gastrointestinali, potrebbero essere necessarie dosi più elevate.
Emofilia A
Monitoraggio del trattamento
Durante il trattamento è raccomandato un adeguato dosaggio dei livelli del fattore VIII per determinare la dose e la frequenza delle infusioni ripetute. Singoli pazienti possono rispondere in modo diverso al trattamento con fattore VIII, mostrando differenti emivite e tempi di recupero. Pazienti con peso corporeo insufficiente o eccessivo potrebbero necessitare di un aggiustamento della dose basato sul peso corporeo. In particolare, in caso di interventi chirurgici estesi, è obbligatorio un accurato monitoraggio della terapia sostitutiva mediante analisi della coagulazione (attività del fattore VIII nel plasma).
Dosaggio
La dose e la durata della terapia sostitutiva dipendono dal grado di gravità della carenza di fattore VIII, dalla localizzazione e dall'intensità del sanguinamento, nonché dallo stato clinico del paziente.
La quantità di unità di fattore VIII prescritta è espressa in unità internazionali (UI), riferite allo standard internazionale dell'OMS per i prodotti del fattore VIII. L'attività del fattore VIII nel plasma è espressa in percentuale (rispetto al plasma normale umano) oppure in unità internazionali (rispetto allo standard internazionale per il FVIII nel plasma).
Un'unità internazionale (UI) di attività del fattore VIII corrisponde alla quantità di fattore VIII presente in 1 ml di plasma normale umano.
Trattamento su richiesta
Il calcolo della dose necessaria di fattore VIII si basa su dati empirici secondo cui 1 UI di fattore VIII:C/kg di peso corporeo aumenta l'attività del fattore nel plasma di circa il 1,5-2% rispetto al valore normale. La dose richiesta si calcola con la seguente formula:
UI necessarie = peso corporeo (kg) × incremento desiderato del fattore VIII (%) (UI/dl) × 0,5 UI/kg
Quantità e frequenza di somministrazione devono sempre essere adattate in base all'efficacia clinica nel singolo caso. In caso di complicanze emorragiche, l'attività del fattore VIII non deve scendere al di sotto del livello di attività plasmatica stabilito (in % del normale o in UI/dl) per il periodo indicato.
La seguente Tabella 3 può essere utilizzata per determinare le dosi in caso di sanguinamento o intervento chirurgico.
Tabella 3
Schema terapeutico per sanguinamenti e interventi chirurgici
| Grado di emorragia/ Tipo di intervento chirurgico |
Livello necessario di fattore VIII (%) (UI/dl) |
Frequenza delle dosi (ore)/ Durata del trattamento (giorni) |
| Emorragia |
||
| Emartrosi iniziale (emorragia articolare), emorragia muscolare o emorragia orale |
20 – 40 |
Ripetere ogni 12 – 24 ore per almeno 1 giorno, fino alla cessazione dell’emorragia (indicata dal dolore) o fino al recupero. |
| Emartrosi più diffusa, emorragia muscolare o ematoma |
30 – 60 |
Ripetere l’iniezione ogni 12 – 24 ore per 3 – 4 giorni o più, fino alla scomparsa del dolore e del limitato movimento acuto. |
| Emorragie che mettono in pericolo la vita |
60 – 100 |
Ripetere l’iniezione ogni 8 – 24 ore finché il pericolo per la vita non sarà passato. |
| Intervento chirurgico |
||
| Intervento chirurgico minore, inclusa l’estrazione dentale |
30 – 60 |
Ogni 24 ore, per almeno 1 giorno, fino al recupero. |
| Intervento chirurgico maggiore |
80 – 100 (pre- e post-operatorio) |
Ripetere l’iniezione ogni 8 – 24 ore fino alla completa guarigione della ferita, quindi continuare il trattamento per almeno altri 7 giorni per mantenere l’attività del fattore VIII tra il 30% e il 60% (UI/dl). |
Prevenzione
Per la profilassi a lungo termine delle emorragie in pazienti con emofilia A grave, si raccomanda di somministrare dosi abituali di 20-40 UMI di fattore VIII per kg di peso corporeo ogni 2-3 giorni. In alcuni casi, specialmente nei pazienti più giovani, potrebbero essere necessari intervalli più brevi tra le somministrazioni o dosi più elevate.
Infusione continua
Prima di un intervento chirurgico è necessario effettuare un’analisi farmacocinetica per ottenere una stima del clearanc. La velocità iniziale di somministrazione del farmaco può essere calcolata nel seguente modo:
Velocità di somministrazione del farmaco (UMI/kg/ora) = clearanc (ml/kg/ora) × livello desiderato allo stato stazionario (UMI/ml)
Dopo le prime 24 ore di infusione continua, il clearanc deve essere ricalcolato ogni giorno, utilizzando l’equazione dello stato stazionario con il livello misurato e la velocità di somministrazione nota.
Pazienti pediatrici
Non sono disponibili dati sufficienti per raccomandare l’uso di Vilate in bambini di età inferiore a 6 anni affetti da emofilia A.
Modalità di somministrazione
Somministrazione endovenosa.
La velocità di iniezione o infusione non deve superare 2-3 ml al minuto.
Misure precauzionali particolari per lo smaltimento e il riciclaggio
È necessario leggere attentamente tutte le istruzioni e seguirle scrupolosamente!
Non utilizzare il medicinale dopo la data di scadenza indicata sull’etichetta.
Durante la procedura descritta di seguito, è necessario mantenere la sterilità!
Il medicinale ricostituito deve essere ispezionato visivamente per verificare la presenza di particelle visibili (solide) e di variazioni di colore prima della somministrazione.
La soluzione deve essere limpida o leggermente opalescente. Non utilizzare soluzioni torbide o contenenti sedimenti.
Utilizzare immediatamente la soluzione preparata per evitare contaminazioni da microrganismi.
Utilizzare esclusivamente il sistema per iniezione fornito. L’uso di un altro dispositivo per iniezione/infusione può comportare rischi aggiuntivi e portare a un’inefficacia del trattamento.
Istruzioni per la preparazione della soluzione
- Non utilizzare il medicinale direttamente dal frigorifero. Il solvente e la polvere nei flaconcini chiusi devono raggiungere la temperatura ambiente.
- Rimuovere i tappi protettivi da entrambi i flaconcini e disinfettare i tappi di gomma con uno dei tamponi imbevuti di alcol forniti nella confezione.
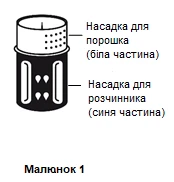
- Il sistema per il trasferimento è illustrato nella figura 1.
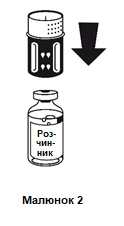
Posizionare il flacone contenente il solvente su una superficie piana e stabile e tenerlo saldamente. Applicare il sistema per il trasferimento con la parte blu verso l’alto sul flacone contenente il solvente, premendo con forza verso il basso finché si sente uno scatto (figure 2 e 3).
Non ruotare durante il fissaggio.
|
|
|
|
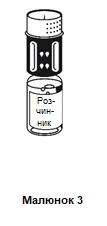
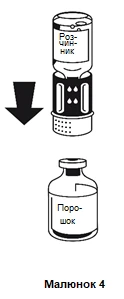
- Posizionare la fiala contenente la polvere su una superficie piana e tenerla saldamente. Prendere la fiala contenente il solvente insieme al sistema di trasferimento fissato e capovolgerla. Posizionare la parte bianca in alto sulla fiala contenente la polvere e premere con forza verso il basso finché non si sente uno scatto (figura 4). Non ruotare durante il fissaggio. Il solvente entrerà automaticamente nella fiala contenente la polvere.
- Quando entrambe le fiale sono ancora unite, ruotare delicatamente e con attenzione la fiala contenente la polvere finché il medicinale non si sarà completamente disciolto.
La dissoluzione avviene in meno di 10 minuti a temperatura ambiente. Durante il processo di preparazione può formarsi una leggera schiuma. Separare il sistema di trasferimento in due parti svitandolo (figura 5). La formazione di schiuma scomparirà.
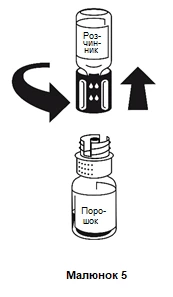
Smaltire la fiala vuota contenente il solvente insieme alla parte blu del sistema di trasferimento.
Istruzioni per l’iniezione
Come misura precauzionale, si raccomanda di controllare la frequenza cardiaca del paziente prima dell’iniezione e durante l’amministrazione del medicinale. Se si osserva un aumento della frequenza cardiaca, ridurre la velocità di somministrazione dell’iniezione o interrompere temporaneamente l’infusione.
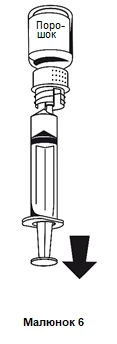
- 1. Fissare la siringa alla parte bianca del sistema di trasferimento. Capovolgere la fiala e aspirare la soluzione nella siringa (figura 6).
La soluzione deve essere limpida o leggermente opalescente.
Non appena la soluzione è stata aspirata, tenendo saldamente la siringa per il cilindro, staccare la siringa dal sistema di trasferimento (figura 7).
|
|
|
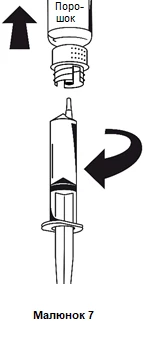
Smaltire il flacone vuoto insieme alla parte bianca del sistema di trasferimento.
- 2. Disinfettare la sede di iniezione scelta con uno dei batuffoli imbevuti di alcol forniti nel confezionamento.
- Applicare al siringa il sistema di somministrazione fornito.
- Inserire l'ago per iniezione nella vena prescelta. Se si utilizza un laccio emostatico (garza emostatica) per rendere più visibile la vena, il laccio deve essere allentato prima di iniziare la somministrazione del medicinale Vilate.
Non deve entrare sangue nella siringa per evitare il rischio di formazione di coaguli di fibrina.
- Somministrare la soluzione per via endovenosa molto lentamente, alla velocità di 2–3 ml al minuto.
Se si utilizzano più di un flacone di polvere Vilate per una singola somministrazione, è possibile riutilizzare lo stesso ago per iniezione e la stessa siringa. Il sistema di trasferimento è destinato all'uso monouso (utilizzare una sola volta)!
Qualsiasi medicinale non utilizzato o rifiuti devono essere smaltiti in conformità con i requisiti locali.
Bambini.
Malattia di von Willebrand (VWD)
Non vi sono dati sufficienti per raccomandare l'uso di Vilate nei bambini di età inferiore ai 6 anni.
Emofilia A
Non vi sono dati sufficienti per raccomandare l'uso di Vilate nei bambini di età inferiore ai 6 anni affetti da emofilia A.
Sovradosaggio.
Non sono stati riportati sintomi di sovradosaggio di FVIII o VWF nell'uomo. In caso di sovradosaggio significativo, possono verificarsi complicanze tromboemboliche.
Effetti indesiderati
Panoramica sintetica del profilo di sicurezza
Reazioni di ipersensibilità o reazioni allergiche (che possono includere: angioedema, sensazione di calore e formicolio nel sito di iniezione, brividi, iperemia (afflusso di sangue), orticaria generalizzata, arrossamento, prurito, eruzione cutanea, cefalea, orticaria, ipotensione, sonnolenza, nausea, eccitazione, tachicardia, difficoltà respiratorie, dispnea, pizzicore, vomito, sibilo/broncospasmo) si verificano raramente e in alcuni casi possono evolvere in una grave anafilassi (incluso shock).
Malattia di von Willebrand (VWD)
Nei pazienti con VWD, in particolare di tipo 3, possono svilupparsi molto raramente anticorpi neutralizzanti verso il FVW. Se tali inibitori compaiono, si manifestano con una risposta clinica inadeguata. Tali anticorpi possono presentarsi contemporaneamente a reazioni anafilattiche. Pertanto, i pazienti con reazioni anafilattiche devono essere sottoposti a screening per la presenza di inibitori.
In tutti i casi di reazione anafilattica si raccomanda di rivolgersi a un centro specializzato nel trattamento dell'emofilia.
Esiste un rischio di trombosi, in particolare nei pazienti con fattori clinici o di laboratorio noti di rischio. È necessario attuare una profilassi antitrombotica in conformità con le raccomandazioni vigenti.
Nei pazienti che ricevono preparati contenenti FVW associato a FVIII, livelli plasmatici costantemente elevati di FVIII:C possono aumentare il rischio di trombosi.
Emofilia A
Nei pazienti affetti da emofilia A trattati con fattore VIII e con il medicinale Vilate 500 UMI, possono svilupparsi anticorpi neutralizzanti (inibitori), vedere il paragrafo «Proprietà farmacodinamiche». In caso di comparsa di tali inibitori, la condizione si manifesterà con una risposta clinica inadeguata. In tali casi si raccomanda di rivolgersi a un centro specializzato nel trattamento dell'emofilia.
Elenco tabulato degli effetti indesiderati
La frequenza degli effetti indesiderati è classificata secondo le seguenti convenzioni: molto comune (≥ 1/10); comune (da ≥ 1/100 a < 1/10); non comune (da ≥ 1/1.000 a < 1/100); raro (da ≥ 1/10.000 a < 1/1.000); molto raro (< 1/10.000); frequenza non nota (non può essere stimata sulla base dei dati disponibili).
Nella Tabella 4 sono riportati gli effetti indesiderati osservati negli studi clinici, negli studi post-marketing sulla sicurezza e noti da altre fonti post-marketing. Gli effetti indesiderati sono elencati per classi di sistemi e organi (SOC) MedDRA, utilizzando i termini preferenziali (PT), e classificati per frequenza.
Tabella 4
| Classe di sistema organo (SOC/MedDRA) |
Reazione avversa |
Frequenza |
| Disturbi del sistema immunitario |
Ipersensibilità Anafilassi |
Non comune Molto raro |
| Patologie generali e condizioni in rapporto alla sede di somministrazione |
Febbre Dolore toracico |
Non comune Frequenza sconosciuta |
| Disturbi del sistema emolinfopoietico |
Inibitori del fattore VIII Inibitori del fattore di von Willebrand |
Non comune (PTPs)* Molto comune (PUPs) Molto raro |
| Disturbi del sistema respiratorio, torace e mediastino |
Tosse |
Frequenza sconosciuta |
| Disturbi del sistema nervoso |
Vertigini |
Frequenza sconosciuta |
| Disturbi gastrointestinali |
Dolore addominale |
Frequenza sconosciuta |
| Disturbi del sistema muscoloscheletrico e del tessuto connettivo |
Dolore alla schiena |
Frequenza sconosciuta |
* La frequenza si basa su studi effettuati con tutti i medicinali contenenti FVIII, condotti su pazienti affetti da emofilia A grave. PTPs – pazienti precedentemente trattati; PUPs – pazienti precedentemente non trattati.
Descrizione delle singole reazioni avverse
Per informazioni sulle singole reazioni avverse, vedere la sezione «Informazioni importanti sull’uso del medicinale».
Segnalazione delle reazioni avverse sospette
La segnalazione delle reazioni avverse dopo l’autorizzazione del medicinale è di fondamentale importanza. Permette di monitorare il rapporto beneficio/rischio del medicinale. I professionisti sanitari e farmaceutici, nonché i pazienti o i loro rappresentanti legali, devono segnalare qualsiasi sospetta reazione avversa o mancanza di efficacia del medicinale attraverso il Sistema informativo automatizzato di farmacovigilanza al seguente indirizzo: https://aisf.dec.gov.ua.
Periodo di validità.
Polvere per soluzione per iniezione – 3 anni.
La stabilità della soluzione ricostituita è mantenuta per 4 ore a temperatura ambiente (non superiore a 25 °C). Tuttavia, per evitare contaminazioni da microrganismi, la soluzione ricostituita deve essere utilizzata immediatamente.
Solvente – conservare per 4 anni.
Il solvente deve essere conservato per 4 anni a una temperatura compresa tra 2 e 8 °C, al riparo dalla luce.
Durante questo periodo, il solvente può essere conservato fino a 6 mesi a una temperatura non superiore a 25 °C; in tal caso, la data di scadenza scadrà dopo 6 mesi.
Condizioni di conservazione.
Conservare a una temperatura compresa tra 2 e 8 °C. Non congelare.
Conservare il flacone nell’imballaggio di cartone per proteggerlo dalla luce.
Tenere fuori dalla portata dei bambini.
Durante il periodo di validità, il medicinale può essere conservato a temperatura ambiente (non superiore a 25 °C) per un massimo di 2 mesi. In questo caso, la data di scadenza scadrà 2 mesi dopo il primo allontanamento del medicinale dal frigorifero.
Il paziente deve indicare la nuova data di scadenza sull’imballaggio esterno di cartone.
La soluzione ricostituita è destinata all’uso singolo. Eventuale soluzione residua nel flacone deve essere smaltita.
Incompatibilità.
In assenza di studi di compatibilità, questo medicinale non deve essere miscelato con altri medicinali né somministrato contemporaneamente ad un altro medicinale per via endovenosa nella stessa linea di infusione. È necessario utilizzare esclusivamente i sistemi per iniezione/infusione forniti, poiché il trattamento potrebbe risultare inefficace a causa dell’adsorbimento (assorbimento) del fattore VIII/fattore di von Willebrand sulla superficie interna di alcuni dispositivi per infusione.
Confezione
Confezione di cartone n. 1: 1 flacone con polvere per soluzione per iniezione (500 UMI) e foglietto illustrativo.
Confezione di cartone n. 2: 1 flacone con solvente (acqua per preparazioni iniettabili con polisorbato 80 allo 0,1 %) da 5 ml, insieme a un kit per somministrazione endovenosa e 2 tamponi alcolici.
Il kit per somministrazione endovenosa comprende: 1 siringa monouso, 1 dispositivo di trasferimento, 1 set per infusione.
La confezione di cartone n. 1 e la confezione di cartone n. 2 sono unite tra loro da un film di plastica.
Confezione di cartone n. 1: 1 flacone con polvere per soluzione per iniezione (1000 UMI) e foglietto illustrativo.
Confezione di cartone n. 2: 1 flacone con solvente (acqua per preparazioni iniettabili con polisorbato 80 allo 0,1 %) da 10 ml, insieme a un kit per somministrazione endovenosa e 2 tamponi alcolici.
Il kit per somministrazione endovenosa comprende: 1 siringa monouso, 1 dispositivo di trasferimento, 1 set per infusione.
La confezione di cartone n. 1 e la confezione di cartone n. 2 sono unite tra loro da un film di plastica.
Categoria di distribuzione. Sotto prescrizione medica.
Produttore. Octapharma Pharmazeutika Produktionsges.m.b.H.
Indirizzo del produttore e sede legale.
Oberlaaer Straße 235, 1100 Vienna, Austria.