Victoza®
Ukraina
Spis treści
INSTRUKCJA DOTYCZĄCA STOSOWANIA LĘKU VICTOZA®
Skład:
substancja czynna: liraglutydu;
1 ml roztworu zawiera 6 mg liraglutydu – analogu ludzkiego peptydu podobnego do glukagonu-1 (GLP-1), wyprodukowanego przy użyciu technologii rekombinowanego DNA w Saccharomyces cerevisiae. Jedna wstępnie wypełniona strzykawka-pen zawiera 18 mg liraglutydu w 3 ml;
substancje pomocnicze: fosforan sodu dwuwodny, glikol propylenowy, fenol, wodorotlenek sodu, kwas chlorowodorowy, woda do wstrzykiwań.
Postać leku. Roztwór do wstrzykiwań.
Główne właściwości fizykochemiczne: przezroczysty, bezbarwny roztwór izotoniczny, pH = 8,15.
Grupa farmakoterapeutyczna. Leki stosowane w cukrzycy, analogi peptydu podobnego do glukagonu-1 (GLP-1).
Kod ATX A10BJ02.
Właściwości farmakologiczne.
Farmakodynamika.
Mechanizm działania
Liraglutyd jest analogiem GLP-1 o sekwencji aminokwasów w 97 % homologicznej do ludzkiego GLP-1, wiąże się z receptorami GLP-1 i aktywuje je. Receptor GLP-1 jest celem działania endogennie wydzielanego hormonu incretyny GLP-1, który wzmacnia zależną od glukozy sekrecję insuliny przez komórki β trzustki. W przeciwieństwie do naturalnego GLP-1 farmakokinetyka i farmakodynamika liraglutydu u ludzi umożliwiają jego podawanie raz na dobę. Działanie przedłużone po podaniu liraglutydu podskórnie wynika z trzech mechanizmów: samoasocjacji, która spowalnia wchłanianie, wiązania się z albuminą krwi oraz zwiększonej odporności na działanie enzymów dipeptydylopeptydazy-4 (DPP-4) i neutralnej endopeptydazy (NEP), co objawia się długim okresie półtrwania leku w osoczu.
Działanie liraglutydu jest pośrednie i wynika ze specyficznego wiązania się z receptorami GLP-1, prowadzącego do zwiększenia stężenia cyklicznego adenozynomonofosforanu (cAMP). Liraglutyd stymuluje wydzielanie insuliny zależne od glukozy i jednocześnie obniża nieadekwatnie wysoką sekrecję glukagonu również w sposób zależny od stężenia glukozy we krwi. W ten sposób przy wysokim stężeniu glukozy we krwi sekrecja insuliny wzrasta, a sekrecja glukagonu maleje. Natomiast w przypadku hipoglikemii liraglutyd zmniejsza sekrecję insuliny, ale nie wpływa na sekrecję glukagonu. Mechanizm obniżania stężenia glukozy we krwi obejmuje również nieznaczne spowolnienie opróżniania żołądka. Liraglutyd zmniejsza masę ciała i masę tkanki tłuszczowej poprzez mechanizmy zmniejszające uczucie głodu i spożycie energii.
GLP-1 jest fizjologicznym regulatorem apetytu i spożycia pokarmu, jednak dokładny mechanizm jego działania nie został w pełni ustalony. W badaniach na zwierzętach podanie liraglutydu drogą obwodową prowadziło do jego gromadzenia się w określonych obszarach mózgu zaangażowanych w regulację apetytu, gdzie dzięki specyficznemu aktywowaniu receptora GLP-1 (GLP-1R) zwiększał on uczucie sytości i zmniejszał kluczowe sygnały głodu, co prowadziło do spadku masy ciała.
Receptory GLP-1 są również wyrażane w określonych obszarach serca, naczyń krwionośnych, układzie odpornościowym i nerkach. W modelu miażdżycy u myszy liraglutyd zapobiegał postępowi plamy aorty i zmniejszał stan zapalny w plamie. Ponadto liraglutyd wywierał pozytywny wpływ na lipidy osocza. Liraglutyd nie zmniejszał rozmiaru już istniejących blaszek.
Efekty wynikające z farmakodynamiki leku
Liraglutyd działa przez 24 godziny i poprawia kontrolę glikemii poprzez obniżenie stężenia glukozy we krwi na czczo i po posiłku u pacjentów z cukrzycą typu 2.
Skuteczność kliniczna i bezpieczeństwo
Zarówno poprawa kontroli glikemii, jak i zmniejszenie zachorowalności i śmiertelności sercowo-naczyniowej są nieodłącznymi elementami leczenia cukrzycy typu 2.
W celu oceny wpływu liraglutydu na kontrolę poziomu glikemii przeprowadzono pięć podwójnie ślepych, randomizowanych, kontrolowanych badań klinicznych fazy 3a z udziałem dorosłych (tabela 1). Leczenie liraglutydem prowadziło do klinicznie i statystycznie istotnej normalizacji stężenia hemoglobiny glikowanej A1c (HbA1c), stężenia glukozy w osoczu krwi na czczo i po posiłku w porównaniu do placebo.
Badania te przeprowadzono u 3978 pacjentów z cukrzycą typu 2 (2501 pacjentów otrzymywało liraglutyd): 53,7 % mężczyzn, 46,3 % kobiet, 797 pacjentów (508 otrzymywało liraglutyd) było w wieku ≥ 65 lat, a 113 pacjentów (66 otrzymywało liraglutyd) – w wieku ≥ 75 lat.
Dodatkowo przeprowadzono badania dotyczące działania liraglutydu u 1901 pacjentów w czterech otwartych, randomizowanych, kontrolowanych badaniach klinicznych (odpowiednio 464, 658, 323 i 177 pacjentów w każdym badaniu), a także jedno podwójnie ślepe, randomizowane, kontrolowane badanie kliniczne u pacjentów z cukrzycą typu 2 i umiarkowanym zaburzeniem funkcji nerek (279 pacjentów).
Liraglutyd był również stosowany w dużym badaniu kardiowaskularnym (LEADER®) z udziałem 9340 pacjentów z cukrzycą typu 2 z wysokim ryzykiem chorób sercowo-naczyniowych.
Kontrola glikemii
Monoterapia
Monoterapia liraglutydem przez 52 tygodnie spowodowała statystycznie istotne i trwałe obniżenie poziomu HbA1c w porównaniu do glikamidu w dawce 8 mg (–0,84 % dla dawki 1,2 mg, –1,14 % przy dawce 1,8 mg w porównaniu do –0,51 % w grupie porównawczej) u pacjentów, którzy wcześniej stosowali dietę i ćwiczenia fizyczne lub otrzymywali monoterapię doustnym lekiem obniżającym stężenie glukozy w dawce nieprzekraczającej połowy dawki maksymalnej (tabela 1).
Kombinacja z doustnymi lekami obniżającymi stężenie glukozy
Leczenie liraglutydem przez 26 tygodni w połączeniu z metforminą, glikamidem lub kombinacją metforminy i roziglitazonu, albo inhibitory SGLT2 ± metformina pozwoliło osiągnąć statystycznie istotne i stabilne obniżenie poziomu HbA1c w porównaniu do placebo (tabela 1).
Tabela 1. Zastosowanie liraglutydu w badaniach klinicznych fazy 3a w monoterapii (52 tygodnie) oraz w połączeniu z doustnymi lekami obniżającymi stężenie glukozy (26 tygodni)
| N |
Średnie wyjściowe stężenie HbA1c (%) |
Zmiana średniego stężenia HbA1c w porównaniu do wartości wyjściowej (%) |
Pacjenci (%), którzy osiągnęli poziom HbA1c < 7% |
Średnia początkowa masa ciała (kg) |
Zmiana średniej masy ciała w porównaniu do wartości wyjściowej (kg) |
||||||||
| Monoterapia |
|||||||||||||
| Victoza® 1,2 mg Victoza® 1,8 mg Glimepiryd 8 mg/dzień |
251 246 248 |
8,18 8,19 8,23 |
|
|
92,1 92,6 93,3 |
1,12 |
|||||||
| Dodawanie do metforminy (2000 mg/dziennie) |
|||||||||||||
| Liraglutyd 1,2 mg |
240 |
8,3 |
|
35,31, 52,82 |
88,5 |
|
|||||||
| Liraglutyd 1,8 mg |
242 |
8,4 |
|
42,41, 66,32 |
88,0 |
|
|||||||
| Placebo |
121 |
8,4 |
0,09 |
10,81, 22,52 |
91,0 |
|
|||||||
| Glimpiryda 4 mg/dziennie |
242 |
8,4 |
|
36,31, 56,02 |
89,0 |
0,95 |
|||||||
| Dodawanie do glimepirydy (4 mg/dziennie) |
|||||||||||||
| Liraglutyd 1,2 mg |
228 |
8,5 |
|
34,51, 57,42 |
80,0 |
0,32** |
|||||||
| Liraglutyd 1,8 mg |
234 |
8,5 |
|
41,61, 55,92 |
83,0 |
|
|||||||
| Placebo |
114 |
8,4 |
0,23 |
7,51, 11,82 |
81,9 |
|
|||||||
| Rosyglitazon 4 mg/dziennie |
231 |
8,4 |
|
21,91, 36,12 |
80,6 |
2,11 |
|||||||
| Dodawanie do metforminy (2000 mg/dziennie) + rosyglitazonu (4 mg dwa razy dziennie) |
|||||||||||||
| Liraglutyd 1,2 mg |
177 |
8,48 |
|
57,51 |
95,3 |
|
|||||||
| Liraglutyd 1,8 mg |
178 |
8,56 |
|
53,71 |
94,9 |
|
|||||||
| Placebo |
175 |
8,42 |
|
28,11 |
98,5 |
0,60 |
|||||||
| Dodawanie do metforminy (2,000 mg/dziennie) + glimepirydy (4 mg/dziennie) |
|||||||||||||
| Liraglutyd 1,8 mg |
230 |
8,3 |
|
53,11 |
85,8 |
|
|||||||
| Placebo |
114 |
8,3 |
|
15,31 |
85,4 |
|
|||||||
| Insulina glargina4 |
232 |
8,1 |
|
45,81 |
85,2 |
1,62 |
|||||||
| Dodawanie do inhibitora SGLT25 ± metforminy (≥1500 mg/dziennie) |
|||||||||||||
| Liraglutyd 1,8 mg Placebo |
203 100 |
8,00 7,96 |
-1,02*** -0,28 |
54,8*** 13,9 |
91,0 91,4 |
-2,92 -2,06 |
|||||||
* Wyższa skuteczność (p < 0,01) w porównaniu z aktywnym lekiem porównawczym.
** Wyższa skuteczność (p < 0,0001) w porównaniu z aktywnym lekiem porównawczym.
*** Wyższa skuteczność (p < 0,001) w porównaniu z aktywnym lekiem porównawczym.
† Nie mniejsza skuteczność (p < 0,0001) w porównaniu z aktywnym lekiem porównawczym.
1 Wszyscy pacjenci.
2 Poprzednia monoterapia doustnym środkiem obniżającym poziom glukozy we krwi.
3 Pacjenci wcześniej stosujący dietę.
4 Badanie z insuliną glarginą było otwarte, a dawkowanie było dostosowywane zgodnie z zaleceniami dotyczącymi tytracji insuliny glarginy.
5Dodawanie leku Victoza® do inhibitora SGLT2 badano we wszystkich zarejestrowanych dawkach inhibitora SGLT2.
Zalecenia dotyczące tytracji insuliny glarginy:
| Samodzielne oznaczanie stężenia glukozy we krwi osoczu na czczo |
Zwiększenie dawki insuliny glarginy (j.m.) |
| ≤ 5,5 mmol/l (≤ 100 mg/dl) Poziom docelowy |
Nie zmieniać dawki |
| > 5,5 i < 6,7 mmol/l (> 100 i < 120 mg/dl) |
0–2 j.m.a |
| ≥ 6,7 mmol/l (≥ 120 mg/dl) |
2 j.m. |
a Zgodnie z indywidualnymi zaleceniami badacza udzielonymi podczas poprzedniej wizyty, np. w zależności od tego, czy u pacjenta wystąpiła hipoglikemia.
Kombinacja z insulina
W badaniu klinicznym trwającym 104 tygodnie 57 % pacjentów z cukrzycą typu 2 leczonych insuliną degludek w połączeniu z metforminą osiągnęło docelowy poziom HbA1c < 7 %, a pozostali pacjenci kontynuowali udział w otwartym badaniu trwającym 26 tygodni, w którym losowo przydzielono im dodatkowe leczenie liraglutydem lub insuliną aspart raz dziennie (podczas największego posiłku). W grupie badawczej stosowanie insuliny degludek z liraglutydem wymagało zmniejszenia dawki insuliny o 20 % w celu zminimalizowania ryzyka hipoglikemii. Dodanie liraglutydu skutkowało istotnie większym statystycznie obniżeniem poziomu HbA1c: – 0,73 % przy stosowaniu liraglutydu i – 0,40 % przy stosowaniu preparatu porównawczego oraz masy ciała – 3,03 vs 0,72 kg odpowiednio. Częstość występowania hipoglikemii (na pacjenta w ciągu roku stosowania leku) była istotnie niższa przy dodaniu liraglutydu w porównaniu z dodaniem insuliny aspart raz dziennie (1,0 vs 8,15; stosunek: 0,13; 95 % CI: od 0,08 do 0,21).
W 52-tygodniowym badaniu klinicznym dodanie insuliny detemiru do liraglutydu u pacjentów, którzy nie osiągnęli docelowego poziomu kontroli glikemii przy leczeniu wyłącznie liraglutydem 1,8 mg i metforminą, skutkowało obniżeniem poziomu HbA1c o 0,54 % od jego początkowego poziomu w porównaniu z pacjentami z grupy kontrolnej, którzy otrzymywali liraglutyd 1,8 mg i metforminę i u których obniżenie wyniosło 0,20 %. Przy tym obserwowano utrzymanie zmniejszenia masy ciała. Zauważono niewielki wzrost liczby przypadków hipoglikemii lekkiej (0,23 vs 0,03 na pacjenta-rok odpowiednio).
W badaniu LEADER® (patrz sekcja „Wpływ na układ sercowo-naczyniowy” poniżej) 873 pacjentów otrzymywało wcześniej zmieszaną insulinę (z doustnymi lekami obniżającymi poziom glukozy we krwi lub bez nich) na początku oraz przez co najmniej kolejne 26 tygodni. Średni poziom HbA1c na początku wynosił 8,7 % przy stosowaniu liraglutydu i placebo. Po 26 tygodniach przybliżona średnia zmiana poziomu HbA1c wynosiła – 1,4 % i – 0,5 % przy stosowaniu liraglutydu i placebo odpowiednio, z oszacowaną różnicą w leczeniu – 0,9 [– 1,00; – 0,70] 95 %. Profil bezpieczeństwa liraglutydu w połączeniu z wcześniej zmieszaną insuliną był ogólnie porównywalny z profilem obserwowanym przy stosowaniu placebo w połączeniu z wcześniej zmieszaną insuliną (patrz sekcja „Reakcje niepożądane”).
Doświadczenie leczenia pacjentów z zaburzeniami funkcji nerek
W podwójnym ślepym badaniu porównującym skuteczność i bezpieczeństwo liraglutydu 1,8 mg i placebo jako dodatku do insuliny i/lub doustnych leków obniżających poziom glukozy we krwi u pacjentów z cukrzycą typu 2 z umiarkowanymi zaburzeniami funkcji nerek liraglutyd wykazał lepsze wyniki niż placebo w zakresie obniżenia poziomu HbA1c po 26 tygodniach leczenia (odpowiednio – 1,05 % vs – 0,38 %). Istotnie większa liczba pacjentów osiągnęła poziom HbA1c poniżej 7 % przy stosowaniu liraglutydu w porównaniu z placebo (52,8 % vs 19,5 %). W obu grupach obserwowano zmniejszenie masy ciała: –2,4 kg przy stosowaniu liraglutydu vs –1,09 z placebo. Względne ryzyko wystąpienia hipoglikemii w obu grupach terapeutycznych było porównywalne. Profil bezpieczeństwa liraglutydu był głównie podobny do profilu obserwowanego w innych badaniach liraglutydu.
Przy monoterapii lekiem Victoza® u pacjentów, u których przed leczeniem poziom HbA1c był wyższy niż 9,5 %, zaobserwowano średnie obniżenie o 2,1 %, a przy leczeniu skojarzonym – o 1,1–2,5 %.
Część pacjentów, u których obniżył się poziom HbA1c
Monoterapia liraglutydem zapewniała istotnie większą część pacjentów, którzy osiągnęli wartość HbA1c ≤ 6,5 % w 52. tygodniu w porównaniu z pacjentami, którzy otrzymywali glikymeperyd (37,6 % przy dawce 1,8 mg i 28,0 % przy dawce 1,2 mg vs 16,2 % przy stosowaniu preparatu porównawczego).
Przy leczeniu przez 26 tygodni liraglutydem w połączeniu z metforminą, glikymeperydem, z metforminą i roziglitazonem lub z inhibitorem SGLT2 ± metforminą zaobserwowano istotnie większy procent pacjentów, u których poziom HbA1c wyniósł ≤ 6,5 % w porównaniu z monoterapią tymi lekami.
Poziom glukozy we krwi na czczo
Leczenie liraglutydem i jego kombinacja z jednym lub dwoma doustnymi lekami przeciwcukrzycowymi prowadziła do obniżenia poziomu glukozy we krwi na czczo o 13 – 43,5 mg/dl (0,72 – 2,42 mmol/l). Takie obniżenie obserwuje się w ciągu pierwszych 2 tygodni leczenia.
Poziom glukozy we krwi po posiłku
Liraglutyd obniżył poziom glukozy we krwi po wszystkich 3 dziennych posiłkach o 31 – 49 mg/dl (1,68 – 2,71 mmol/l).
Funkcja komórek beta
W wyniku badań klinicznych liraglutydu na podstawie danych uzyskanych za pomocą oceny modelu homeostazy funkcji komórek beta i wartości stosunku proinsulina/insulina stwierdzono poprawę funkcjonalnego stanu komórek beta. Po leczeniu liraglutydem przez 52 tygodnie u grupy pacjentów z cukrzycą typu 2 (n = 29) zaobserwowano poprawę sekrecji insuliny w pierwszej i drugiej fazie.
Masa ciała
Przy leczeniu liraglutydem w połączeniu z metforminą, metforminą i glikymeperydem, metforminą i roziglitazonem lub inhibitorem SGLT2, z metforminą i bez niej, pacjenci systematycznie tracili od 0,86 do 2,62 kg masy ciała w porównaniu z placebo.
Bardziej wyraźne zmniejszenie masy ciała obserwowano u pacjentów z wyższymi wartościami wskaźnika masy ciała przed rozpoczęciem leczenia.
Wpływ na układ sercowo-naczyniowy
Retrospektywna analiza poważnych niepożądanych zdarzeń sercowo-naczyniowych (śmierć z przyczyn sercowo-naczyniowych, zawał mięśnia sercowego, udar mózgu) w trakcie wszystkich średnio- i długotrwałych badań fazy 2 i 3 (trwających od 26 do 100 tygodni), w których wzięło udział 5607 pacjentów (z których 3651 otrzymywało liraglutyd), wykazała brak zwiększenia ryzyka zdarzeń sercowo-naczyniowych [częstość występowania 0,75 (95 %) CI (przedział ufności) 0,35; 1,63] przy stosowaniu liraglutydu w porównaniu ze wszystkimi lekami porównawczymi.
Badanie wpływu i działania liraglutydu na układ sercowo-naczyniowy u pacjentów z cukrzycą (LEADER®) to wieloośrodkowe, placebo-kontrolowane, podwójne ślepe badanie kliniczne. 9340 pacjentów losowo otrzymywało liraglutyd (4668) lub placebo (4672) jako dodatek do standardowego leczenia, skierowanego na obniżenie poziomu HbA1c i czynników ryzyka chorób sercowo-naczyniowych. Pierwotny wynik lub status życiowy na koniec badania był znany u 99,7 % i 99,6 % losowo wybranych pacjentów otrzymujących liraglutyd lub placebo odpowiednio. Czas obserwacji wynosił co najmniej 3,5 roku i maksymalnie 5 lat. Badanie obejmowało pacjentów w wieku ≥ 65 lat (n = 4329) i ≥ 75 lat (n = 836) oraz pacjentów z łagodnymi (n = 3907), umiarkowanymi (n = 1934) lub ciężkimi (n = 224) zaburzeniami funkcji nerek. Średni wiek pacjentów wynosił 64 lata, a średni wskaźnik masy ciała wynosił 32,5 kg/m². Średnia długość trwania choroby cukrzycą wynosiła 12,8 roku.
Pierwotnym punktem końcowym był czas od randomizacji do pierwszego wystąpienia poważnych niekorzystnych zdarzeń sercowo-naczyniowych (MACE): śmierci z przyczyn sercowo-naczyniowych, niezakończonego zgonem zawału mięśnia sercowego lub niezakończonego zgonem udaru mózgu. Liraglutyd był lepszy w zapobieganiu poważnym niekorzystnym zdarzeniom sercowo-naczyniowym w porównaniu z placebo (ryc. 1). Szacowane ryzyko było stale niższe 1 dla wszystkich 3 składników poważnych niekorzystnych zdarzeń sercowo-naczyniowych.
Liraglutyd również istotnie zmniejszył ryzyko wystąpienia wielu poważnych niekorzystnych zdarzeń sercowo-naczyniowych (pierwotnych MACE, niestabilnej dławicy piersiowej wymagającej hospitalizacji, rewaskularyzacji wieńcowej lub niewydolności serca wymagającej hospitalizacji) oraz innych wtórnych punktów końcowych (ryc. 2).
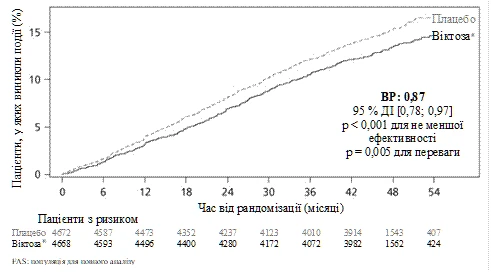
Rycina 1.
Wykres Kaplana-Meiera. Czas do pierwszego wystąpienia poważnego niekorzystnego zdarzenia sercowo-naczyniowego (MACE) – populacja FAS
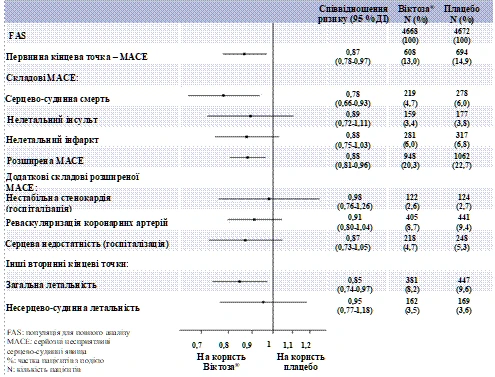
Rycina 2. Diagram lasu analizy indywidualnych zdarzeń sercowo-naczyniowych – populacja FAS
Przy stosowaniu liraglutydu jako dodatku do standardowego leczenia obserwowano istotne i trwałe obniżenie poziomu HbA1c od początku do 36 miesiąca w porównaniu z placebo (– 1,16 % vs – 0,77 %; oszacowana różnica w metodach leczenia – 0,40 % [– 0,45; – 0,34]). Przy stosowaniu liraglutydu w porównaniu z placebo zmniejszyła się o 48 % konieczność intensyfikacji leczenia insuliną u pacjentów, którzy wcześniej nie stosowali insuliny (HR 0,52 [0,48; 0,57]).
Ciśnienie tętnicze i częstość akcji serca
W trakcie badań fazy 3a liraglutyd obniżał średnio ciśnienie tętnicze skurczowe od 2,3 do 6,7 mmHg od poziomu wyjściowego w porównaniu z aktywnym lekiem porównawczym, przy którym obniżenie wyniosło 1,9 – 4,5 mmHg.
W długotrwałych badaniach klinicznych, w tym w badaniu LEADER®, liraglutyd nieznacznie zwiększał częstość akcji serca od poziomu wyjściowego o 2 – 3 uderzenia na minutę. W badaniu LEADER® nie stwierdzono długotrwałego klinicznego wpływu zwiększenia częstości akcji serca na ryzyko wystąpienia zdarzeń sercowo-naczyniowych.
Ocena chorób mikronaczyń
W badaniu LEADER® zdarzenia mikronaczyniowe obejmowały nefropatię i retinopatię. Analiza czasu do pierwszego zdarzenia mikronaczyniowego przy stosowaniu liraglutydu w porównaniu z placebo wykazała HR 0,84 [0,73, 0,97]. HR przy stosowaniu liraglutydu w porównaniu z placebo wyniósł 0,78 [0,67, 0,92] dla pierwszego przypadku nefropatii i 1,15 [0,87, 1,52] dla pierwszego przypadku retinopatii.
Imunogenność
Ze względu na potencjalne właściwości immunogenne leków zawierających białka lub peptydy, przy leczeniu liraglutydem u pacjentów mogą powstawać przeciwciała przeciwko liraglutydowi. Zostały one wykryte średnio u 8,6 % pacjentów. Powstawanie przeciwciał nie prowadziło do zmniejszenia skuteczności liraglutydu.
Dzieci
W podwójnym ślepym badaniu porównano skuteczność i bezpieczeństwo stosowania leku Victoza® 1,8 mg i placebo jako dodatku do metforminy ± insuliny u dzieci i nastolatków w wieku od 10 lat z cukrzycą typu 2. Po 26 tygodniach leczenia lek Victoza® bardziej skutecznie obniżał poziom HbA1c (−1,06 [-1,65, 0,46]) niż placebo. Jeszcze po 26 tygodniach leczenia w otwartym badaniu dodatkowym różnica między poziomami HbA1c wyniosła 1,3 %, co potwierdza trwałą kontrolę glikemii lekiem Victoza®.
Skuteczność i profil bezpieczeństwa leku Victoza® były podobne do obserwowanych u dorosłych otrzymujących lek Victoza®. W zależności od odpowiedniej kontroli glikemii lub tolerancji 30 % uczestników badania kontynuowało leczenie w dawce 0,6 mg, 17 % – dawkę zwiększono do 1,2 mg, a 53 % – dawkę zwiększono do 1,8 mg.
Inne dane kliniczne
W otwartym badaniu porównano skuteczność i bezpieczeństwo liraglutydu (1,2 mg i 1,8 mg) i sitagliptyny (inhibitor dipeptydylopeptydazy – 4, 100 mg) u pacjentów, u których nie osiągnięto odpowiedniej kontroli glikemii przy stosowaniu metforminy (średni poziom HbA1c = 8,5 %). Po 26 tygodniach leczenia obie dawki liraglutydu istotnie skuteczniej obniżały poziom HbA1c (−1,24 % i −1,50 %) niż sitagliptyna (0,90 %, P < 0,0001). U pacjentów leczonych liraglutydem zaobserwowano większe zmniejszenie masy ciała (−2,9 kg i −3,4 kg) niż przy leczeniu sitagliptyną (−1,0 kg, p <0,0001). Przejściowa nudności częściej obserwowano u pacjentów otrzymujących liraglutyd (20,8 % i 27,1 %) w porównaniu z sitagliptyną (4,6 %). Większe obniżenie poziomu HbA1c, które obserwowano po 26 tygodniach leczenia liraglutydem (1,2 mg i 1,8 mg), utrzymywało się po 52 tygodniach (−1,29 % i −1,51 %) w porównaniu z sitagliptyną (− 0,88 %, p < 0,0001). Przejście pacjentów po 52 tygodniach leczenia sitagliptyną na leczenie liraglutydem (1,2 mg i 1,8 mg) skutkowało dalszym istotnym statystycznie obniżeniem poziomu HbA1c, które po 78 tygodniach wyniosło −0,24 % i −0,45 % (95 % CI: −0,41 % − 0,07 % i − 0,67 % − 0,23 %), ale formalnie brakowało grupy kontrolnej.
W otwartym badaniu z udziałem pacjentów, u których nie osiągnięto odpowiedniej kontroli glikemii przy leczeniu metforminą i/lub sulfonamidami (średni poziom HbA1c = 8,3 %), porównano skuteczność i bezpieczeństwo stosowania liraglutydu 1,8 mg (1 raz dziennie) z zastosowaniem egzynatyd 10 µg (2 razy dziennie). Po 26 tygodniach leczenia liraglutyd istotnie skuteczniej obniżał poziom HbA1C (−1,12 %) niż egzynatyd (− 0,79 %), oszacowana różnica między grupami wyniosła −0,33 % (95 % CI: −0,47 % − 0,18 %). Liczba pacjentów, u których poziom HbA1c spadł poniżej 7 %, była istotnie większa wśród tych, którzy otrzymywali liraglutyd (54,2 %), w porównaniu z egzynatydem (43,4 %, p = 0,0015). Przy obu metodach leczenia masa ciała pacjentów zmniejszyła się średnio o 3 kg. Przejście pacjentów po 26 tygodniach leczenia egzynatydem na leczenie liraglutydem skutkowało dodatkowym i istotnym statystycznie obniżeniem poziomu HbA1c, które po 40 tygodniach wyniosło −0,32 % (95 % CI: − 0,41 % − 0,24 %), ale formalnie brakowało grupy kontrolnej. W ciągu 26 tygodni leczenia liraglutydem u 235 pacjentów wystąpiło 12 poważnych powikłań (5,1 %), a przy leczeniu egzynatydem — 6 poważnych powikłań (2,6 %) u 232 pacjentów. Nie stwierdzono wyraźnego rozłożenia tych powikłań według układów narządów.
W otwartym badaniu, w którym porównywano skuteczność i bezpieczeństwo liraglutydu 1,8 mg i liksisentyd 20 µg u 404 pacjentów, którzy nie osiągnęli kontroli glikemii przy terapii metforminą (średni poziom HbA1c 8,4 %), liraglutyd był bardziej skuteczny niż liksisentyd w zakresie obniżenia poziomu HbA1c po 26 tygodniach leczenia (– 1,83 % vs – 1,21 %, p < 0,0001). Istotnie więcej pacjentów osiągnęło HbA1c poniżej 7 % przy stosowaniu liraglutydu w porównaniu z liksisentydem (74,2 % vs 45,5 %, p < 0,0001), jak również HbA1c nie więcej niż 6,5 % (54,6 % vs 26,2 %, p < 0,0001). Zmniejszenie masy ciała obserwowano w obu grupach pacjentów (– 4,3 kg przy stosowaniu liraglutydu i – 3,7 kg przy stosowaniu liksisentyd). Efekty niepożądane ze strony przewodu pokarmowego częściej występowały u pacjentów otrzymujących liraglutyd (43,6 % vs 37,1 %).
Farmakokinetyka .
Wchłanianie
Po podaniu podskórnie liraglutyd wchłania się powoli, maksymalna stężenie osiąga się po 8–12 godzinach. Po jednorazowym podaniu podskórnie dawki 0,6 mg liraglutydu maksymalne stężenie wyniosło 9,4 nmol/l (średnia masa ciała około
73 kg). Po podaniu 1,8 mg liraglutydu jego średnie stężenie równowagowe (AUCτ/24) wyniosło około 34 nmol/l (średnia masa ciała około 76 kg). Eksponencja na liraglutyd zmniejsza się wraz ze wzrostem masy ciała. Eksponencja na liraglutyd wzrastała proporcjonalnie do dawki. U tego samego pacjenta współczynnik zmienności wartości AUC po jednorazowym podaniu liraglutydu wyniósł 11 %.
Bezwzględna biodostępność liraglutydu po podaniu podskórnie wynosi około 55 %.
Rozkład
Objętość widoczna rozkładu po podaniu podskórnie wynosi 11–17 l. Średni objętość rozkładu po podaniu dożylnym liraglutydu wynosi 0,07 l/kg. Liraglutyd szeroko wiąże się z białkami osocza krwi (> 98 %).
Metabolizm
W ciągu 24 godzin po jednorazowym podaniu dawki radioaktywnie znakowanego [3H]-liraglutydu zdrowym ochotnikom głównym składnikiem w osoczu krwi był niezmieniony liraglutyd. W osoczu krwi wykryto w niewielkiej ilości dwa metabolity (≤ 9 % i ≤ 5 % całkowitej eksponencji radioaktywności osocza). Liraglutyd metabolizuje się tymi samymi drogami, co duże białka. Nie wykryto specjalnego organu, w którym zachodzi główny szlak eliminacji.
Eliminacja
Po podaniu dawki [3H]-liraglutydu w moczu i kaле nie wykryto niezmienionego liraglutydu. Tylko niewielka część kontrolowanej radioaktywności, wydalonej w postaci metabolitów związanych z liraglutydem, została wykryta w moczu (6 %) i kałe (5 %). Radioaktywność z|do| moczu i kału była głównie wydalana w ciągu pierwszych 6 – 8 dni w postaci trzech metabolitów w niewielkiej ilości odpowiednio.
Po jednorazowym podaniu podskórnie liraglutydu średnia wartość klirensu wynosi około 1,2 l/godz, czas półtrwania – około 13 godzin.
Specjalne grupy pacjentów
Pacjenci starsi. Na podstawie danych z badania farmakokinetyki u zdrowych ochotników i analizy farmakokinetycznej grupy pacjentów w wieku od 18 do 80 lat stwierdzono, że wiek nie wywiera klinicznie istotnego wpływu na farmakokinetykę liraglutydu.
Płeć. Na podstawie danych analizy farmakokinetycznej populacji pacjentów męskich i żeńskich, a także badania farmakokinetycznego u zdrowych ochotników stwierdzono, że płeć nie wywiera istotnego klinicznego wpływu na farmakokinetykę liraglutydu.
Pochodzenie etniczne . Na podstawie danych analizy farmakokinetycznej grupy pacjentów pochodzenia europejskiego, mongolskiego i negroidalnego stwierdzono, że pochodzenie etniczne nie wywiera żadnego istotnego klinicznego wpływu na farmakokinetykę liraglutydu.
Otyłość . Na podstawie danych analizy farmakokinetycznej populacji stwierdzono, że wartość wskaźnika masy ciała nie wywiera istotnego wpływu na wartości parametrów farmakokinetyki liraglutydu.
Zaburzenia funkcji wątroby. Farmakokinetykę liraglutydu badano u pacjentów z różnym stopniem zaburzeń funkcji wątroby w trakcie badania z jednorazową dawką. Wykazano, że u pacjentów z łagodnymi i umiarkowanymi zaburzeniami funkcji wątroby eksponencja na liraglutyd zmniejszała się o 13 – 23 % w porównaniu ze zdrowymi ochotnikami.
U pacjentów z ciężkimi zaburzeniami funkcji wątroby (> 9 punktów wg klasyfikacji Childa-Pugh) eksponencja była istotnie niższa (44 %).
Zaburzenia funkcji nerek . Eksponencja na liraglutyd była zmniejszona u pacjentów z zaburzeniami funkcji nerek w porównaniu z osobami z normalną funkcją nerek. U pacjentów z łagodnymi zaburzeniami (klirens kreatyniny 50 – 80 ml/min) zmniejszała się o 33 %, z zaburzeniami umiarkowanymi (klirens kreatyniny 30 – 50 ml/min) – o 14 %, z ciężkimi zaburzeniami (klirens kreatyniny < 30 ml/min) – o 27 %, a w końcowym stadium choroby nerek wymagającej dializy – o 26 %. Analogicznie w 26-tygodniowych badaniach klinicznych u pacjentów z cukrzycą typu 2 z zaburzeniami funkcji nerek (klirens kreatyniny – 30 – 59 ml/min, patrz sekcja „Farmakodynamika”) eksponencja na liraglutyd zmniejszyła się o 26 % w porównaniu z pacjentami z cukrzycą typu 2 z normalną funkcją nerek lub z zaburzeniami łagodnego stopnia.
Dzieci . Własności farmakokinetyczne badano w badaniach klinicznych u dzieci z cukrzycą typu 2 w wieku od 10 lat. Eksponencja na liraglutyd u nastolatków i dzieci odpowiadała eksponencji u dorosłych pacjentów.
Dane przedkliniczne dotyczące bezpieczeństwa
Dane przedkliniczne, uzyskane na podstawie tradycyjnych badań farmakologii bezpieczeństwa, toksyczności powtarzanych dawek lub genotoksyczności, nie wykazały szczególnego ryzyka dla człowieka.
Nieznanej śmiertelności guzy komórek C tarczycy zostały wykryte u szczurów i myszy w trakcie 2-letnich badań kancerogennych. U szczurów nie stwierdzono zwiększenia częstości występowania lub ciężkości niepożądanych efektów. U małp, które otrzymywały leczenie przez 20 miesięcy, takich guzów nie wykryto. Guzy u gryzoni są spowodowane nietoksycznym, specyficznym, zależnym od receptora GLP-1 mechanizmem, do którego częściowo wrażliwe są gryzonie. Znaczenie tego mechanizmu u ludzi jest wystarczająco niskie, ale nie może być całkowicie wykluczone. Nie wykryto rozwoju innych guzów przy leczeniu lekiem Victoza®.
W eksperymentach na zwierzętach nie stwierdzono bezpośredniego szkodliwego wpływu na płodność, jednak przy podaniu najwyższych dawek zaobserwowano niewielkie zwiększenie wcześniejszej śmiertelności zarodków. Podawanie leku Victoza® w okresie połowy ciąży powodowało zmniejszenie masy ciała matki, spowolnienie wzrostu płodu z niejasnym wpływem na rozwój żeber u szczurów i szkieletu u królików. Przy podawaniu leku Victoza® zaobserwowano spowolnienie wzrostu noworodków szczurów, które utrzymuje się w okresie odstawienia od karmienia mlekiem w grupie przyjmującej wysoką dawkę. Pozostaje niejasne, czy spowolnienie wzrostu noworodków szczurów jest spowodowane zmniejszeniem spożycia mleka w wyniku bezpośredniego wpływu GLP-1, czy zmniejszeniem ilości mleka u matki spowodowanym zmniejszeniem kaloryczności spożywanej żywności.
Dane kliniczne.
Wskazania
Lek Victoza® stosuje się w leczeniu nieadekwatnie kontrolowanego cukrzycy typu 2 u dorosłych, młodzieży i dzieci od 10. roku życia jako uzupełnienie diety i aktywności fizycznej:
- w monoterapii, gdy zastosowanie metforminy uznaje się za niemożliwe ze względu na nietolerancję lub przeciwwskazania;
- w kombinacji z innymi lekami stosowanymi w leczeniu cukrzycy.
Wyniki badań dotyczące stosowania w kombinacji z innymi lekami, wpływ na kontrolę glikemii oraz zdarzenia sercowo-naczyniowe, a także badane populacje znajdują się w sekcjach „Szczególne ostrzeżenia i środki ostrożności dotyczące stosowania”, „Interakcje z innymi lekami i inne formy interakcji” oraz „Farmakodynamika”.
Przeciwwskazania
Podwyższona wrażliwość na substancję czynną lub którykolwiek z pozostałych składników leku wymienionych w części „Składniki pomocnicze”.
Interakcje z innymi lekami i inne formy interakcji
In vitro liraglutyd wykazuje bardzo niski potencjał wpływu na farmakokinetykę innych substancji czynnych, których metabolizm wiąże się z cytochromem P450, a także na wiązanie z białkami osocza.
Liraglutyd powoduje nieznaczne opóźnienie opróżniania żołądka, co może wpływać na wchłanianie leków stosowanych jednocześnie doustnie. Badania interakcji nie wykazały żadnego klinicznie istotnego spowolnienia wchłaniania, dlatego korekty dawki nie wymaga. U niektórych pacjentów przyjmujących lek Victoza® odnotowano co najmniej jeden przypadek ciężkiej biegunki. Biegunka może zaburzać wchłanianie leków stosowanych jednocześnie doustnie.
Warakfaryna i inne pochodne kumaryny
Badania interakcji lekowych nie przeprowadzano. Nie można wykluczyć klinicznie istotnej interakcji z substancją czynną o niskiej rozpuszczalności lub wąskim indeksie terapeutycznym, takiej jak warfaryna. Na początku leczenia liraglutydem u pacjentów przyjmujących warfarynę lub inne pochodne kumaryny zaleca się częstsze monitorowanie INR (międzynarodowego znormalizowanego stosunku).
Paracetamol
Liraglutyd nie zmieniał całkowitego narażenia na paracetamol po podaniu pojedynczej dawki 1000 mg. Maksymalne stężenie paracetamolu (Cmax) zmniejszało się o 31%, a czas osiągnięcia maksymalnego stężenia (tmax) wydłużał się do 15 minut. Podczas jednoczesnego stosowania paracetamolu korekta dawki nie jest wymagana.
Atorwastatyna
Liraglutyd nie zmieniał całkowitego narażenia na atorwastatynę na klinicznie istotnym poziomie po pojedynczym podaniu dawki 40 mg. W związku z tym podczas jednoczesnego stosowania z Victozą® korekta dawki atorwastatyny nie jest wymagana. Podczas jednoczesnego podania z liraglutydem Cmax atorwastatyny zmniejszało się o 38%, a tmax wydłużało się z 1 godziny do 3 godzin.
Gryzeofulwina
Liraglutyd nie zmieniał całkowitego narażenia na gryzeofulwinę po pojedynczym podaniu dawki 500 mg. Cmax wzrastało o 37%, natomiast tmax nie zmieniało się. Korekta dawki przy stosowaniu gryzeofulwiny i innych małorozpuszczalnych związków o wysokiej przenikalności nie jest wymagana.
Dygoxyna
Po pojedynczym podaniu 1 mg dygoxyny w połączeniu z liraglutydem zaobserwowano zmniejszenie wartości pola pod krzywą „stężenie-czas” (AUC) dla dygoxyny o 16%, Cmax zmniejszało się o 31%. Średni tmax dygoxyny wydłużał się z 1 godziny do 1,5 godziny. Na podstawie tych wyników korekta dawki dygoxyny nie jest wymagana.
Lizynopryl
Po pojedynczym podaniu 20 mg lizynoprylu zaobserwowano zmniejszenie wartości pola pod krzywą „stężenie-czas” (AUC) dla lizynoprylu o 15%, Cmax zmniejszało się o 27%. Średni tmax lizynoprylu wydłużał się z 6 godzin do 8 godzin. Na podstawie tych wyników korekta dawki lizynoprylu nie jest wymagana.
Środki antykoncepcyjne doustne
Podczas jednoczesnego stosowania pojedynczej dawki środków antykoncepcyjnych doustnych liraglutyd zmniejszał Cmax etynyloestradiolu lub lewonorgestrelu odpowiednio o 12% i 13%, a tmax wydłużał się o 1,5 godziny. Nie wykazano klinicznie istotnego wpływu na całkowite narażenie na etynyloestradiol lub lewonorgestrel, co pozwala stwierdzić, że jednoczesne przyjmowanie liraglutydu nie wpływa na działanie antykoncepcyjne etynyloestradiolu i lewonorgestrelu.
Insulina
U pacjentów ze stabilizowaną cukrzycą typu 2 podczas jednoczesnego podawania insuliny detemiru (5 JEDNOSTEK/kg) i liraglutydu (1,8 mg) nie zaobserwowano objawów interakcji farmakokinetycznej i farmakodynamicznej.
Dzieci
Badania interakcji przeprowadzono wyłącznie na dorosłych.
Szczególne wskazania dotyczące stosowania.
Liraglutyd nie jest stosowany w leczeniu pacjentów z cukrzycą typu I ani z kwasocetem cukrzycowym.
Liraglutyd nie jest substytutem insuliny. W przypadku szybkiego odstawienia lub zmniejszenia dawki insuliny u pacjentów uzależnionych od insuliny opisywano przypadki rozwoju kwasocetu cukrzycowego (patrz dział „Sposób i dawki stosowania”).
Brak doświadczenia terapeutycznego w leczeniu pacjentów z niewydolnością serca klasy IV według klasyfikacji New York Heart Association (NYHA), dlatego liraglutyd nie jest zalecany dla tych pacjentów.
Doświadczenie stosowania liraglutydu u pacjentów z chorobami zapalnymi jelit oraz z gastroparezą cukrzycową jest ograniczone. Stosowanie liraglutydu u tych pacjentów nie jest zalecane, ponieważ wiąże się z tymczasowymi niepożdanymi reakcjami ze strony przewodu pokarmowego, w tym nudnościami, wymiotami i biegunką.
Aspiracja w połączeniu z znieczuleniem ogólnym lub głęboką sedacją
U pacjentów otrzymujących agonisty receptora GLP-1 obserwowano przypadki aspiracji do płuc podczas znieczulenia ogólnego lub głębokiej sedacji. Dlatego przed wykonaniem procedury z wykorzystaniem znieczulenia ogólnego lub głębokiej sedacji należy wziąć pod uwagę zwiększone ryzyko obecności resztkowego zawartości żołądka z powodu opóźnienia opróżniania żołądka (patrz dział „Działania niepożądane”).
Ostry zapalenie trzustki
Ostre zapalenie trzustki obserwowano przy stosowaniu analogów receptora GLP-1.
Pacjentów należy poinformować o charakterystycznych objawach ostrego zapalenia trzustki. W przypadku podejrzenia zapalenia trzustki należy przerwać leczenie liraglutydem. Jeśli zostanie potwierdzone ostre zapalenie trzustki, ponowne stosowanie liraglutydu nie jest zalecane (patrz dział „Działania niepożądane” oraz „Właściwości farmakodynamiczne”).
Choroby tarczycy
W trakcie badań klinicznych obserwowano niepożądane reakcje ze strony tarczycy, takie jak wole, szczególnie u pacjentów z już istniejącymi chorobami tarczycy. Dlatego liraglutyd należy stosować z ostrożnością u tych pacjentów.
Hipoglikemia
U pacjentów otrzymujących liraglutyd jednocześnie z lekami sulfonamidowymi lub insuliną, zwiększa się ryzyko wystąpienia hipoglikemii (patrz dział „Działania niepożądane”). Ryzyko hipoglikemii można zmniejszyć, obniżając dawkę leku sulfonamidowego lub insuliny.
Odewodnienie
U pacjentów leczonych liraglutydem obserwowano objawy odwodnienia, w tym zaburzenia funkcji nerek oraz ostrej niewydolności nerek.
Pacjentów, którym przepisano liraglutyd, należy uprzedzić o możliwości odwodnienia organizmu spowodowanego zaburzeniami układu pokarmowego oraz o konieczności podejmowania środków zapobiegawczych wobec odwodnienia.
Lek Victoza® zawiera mniej niż 1 mmol sodu (23 mg), dlatego można go uznać za lek zawierający niewielkie ilości sodu.
Śledzenie
W celu poprawy śledzenia biologicznych leków należy dokładnie rejestrować nazwę i numer serii stosowanego leku.
Stosowanie w okresie ciąży lub karmienia piersią.
Ciąża
Brak wystarczających danych dotyczących stosowania liraglutydu u kobiet w ciąży. Badania na zwierzętach wykazały toksyczność reprodukcyjną (patrz dział „Dane przedkliniczne dotyczące bezpieczeństwa”). Potencjalne ryzyko dla ludzi jest nieznane.
Liraglutyd nie powinien być stosowany w czasie ciąży – zamiast niego zaleca się stosowanie insuliny. Jeśli pacjentka planuje zajść w ciążę lub jest w ciąży, należy przerwać przyjmowanie leku Victoza®.
Okres karmienia piersią
Nie wiadomo, czy liraglutyd wydzielany jest w mleku matki. Badania na zwierzętach wykazały, że do mleka dostaje się niewielka ilość liraglutydu i jego strukturalnie pokrewnych metabolitów. Badania przedkliniczne wykazały powiązane z lekiem zmniejszenie tempa wzrostu noworodków szczurów (patrz dział „Dane przedkliniczne dotyczące bezpieczeństwa stosowania”). Z uwagi na niewystarczające doświadczenie w okresie karmienia piersią nie należy stosować leku Victoza®.
Plodność
Oprócz nieznacznego zmniejszenia liczby żywych zaimplantowanych embrionów, badania na zwierzętach nie wykazały szkodliwego wpływu leku na płodność.
Wpływ na zdolność prowadzenia pojazdów i obsługiwanie maszyn.
Lek Victoza® nie wpływa lub wpływa nieznacznie na zdolność prowadzenia pojazdów i obsługiwania maszyn. Pacjentom należy zalecić podjęcie środków zapobiegających wystąpieniu hipoglikemii w czasie prowadzenia pojazdów lub innych urządzeń, szczególnie przy jednoczesnym stosowaniu leku Victoza® z lekami sulfonamidowymi lub insuliną.
Sposób stosowania i dawki.
Dawkowanie
Ze względu na poprawę tolerancji ze strony przewodu pokarmowego dawkę początkową stanowi 0,6 mg liraglutynidy na dobę. Po co najmniej 1 tygodniu dawkę należy zwiększyć do 1,2 mg. U niektórych pacjentów po oczekiwaniu poprawy skuteczności po zwiększeniu dawki z 1,2 mg do 1,8 mg oraz w oparciu o odpowiedź na leczenie, po co najmniej 1 tygodniu terapii dawkę można zwiększyć do 1,8 mg w celu dalszego poprawienia kontroli glikemii. Nie zaleca się dawki dobowej wyższej niż 1,8 mg.
W przypadku jednoczesnego stosowania Victoza® oraz leków sulfonilomocznikowych lub insuliny, dawkę leku sulfonilomocznikowego lub insuliny należy zmniejszyć w celu zmniejszenia ryzyka wystąpienia hipoglikemii (patrz sekcja „Szczególne ostrzeżenia i środki ostrożności dotyczące stosowania”). Terapia w połączeniu z lekami sulfonilomocznikowymi może być stosowana wyłącznie u dorosłych pacjentów.
Do doboru dawki leku Victoza® nie jest wymagany samokontrola poziomu glukozy we krwi. Samokontrola poziomu glukozy we krwi jest konieczna do korekty dawki leków sulfonilomocznikowych i insuliny, w szczególności na początku leczenia lekiem Victoza® oraz podczas zmniejszania dawki insuliny. Zaleca się stopniowe zmniejszanie dawki insuliny.
Grupy pacjentów specjalnych
Pacjenci w wieku podeszłym (> 65 lat). Korekty dawki w związku z wiekiem nie wymaga się (patrz sekcja „Farmakokinetyka”).
Naruszenie funkcji nerek. Pacjentom z łagodnym, umiarkowanym lub ciężkim naruszeniem funkcji nerek nie wymaga się korekty dawki. Brak doświadczenia w leczeniu pacjentów w końcowym stadium choroby nerek, dlatego lek Victoza® nie jest zalecany do stosowania u tych pacjentów (patrz sekcje „Farmakodynamika” i „Farmakokinetyka”).
Naruszenie funkcji wątroby. Nie zaleca się korekty dawki u pacjentów z łagodnym lub umiarkowanym naruszeniem funkcji wątroby. Lek Victoza® nie jest zalecany pacjentom z ciężkim naruszeniem funkcji wątroby (patrz sekcja „Farmakokinetyka”).
Sposób podania
Leku Victoza® nie wolno podawać dożylnie ani domięśniowo.
Lek Victoza® podaje się 1 raz na dobę o dowolnej porze niezależnie od przyjmowania pokarmu. Można go podać podskórnie w okolicy przedniej ściany brzucha, uda lub ramienia. Miejsce i czas wstrzyknięć można zmieniać bez konieczności korekty dawki. Należy jednak podawać lek Victoza® mniej więcej o tej samej najbardziej wygodnej porze dnia. Dodatkowe zalecenia dotyczące podania zawarte są w instrukcji obsługi dawaczki do leku Victoza®.
W celu zmniejszenia ryzyka odkładania się amyloidu w miejscu wstrzyknięcia, należy zawsze zmieniać miejsce wstrzyknięcia (patrz sekcja „Efekty niepożądane”).
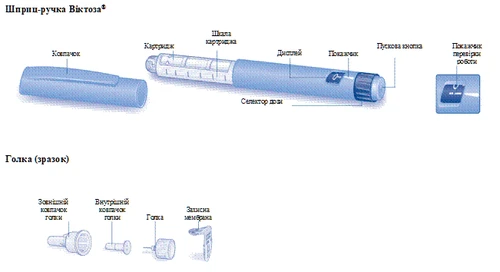
Instrukcja obsługi dawaczki Victoza®
Przed pierwszym użyciem dawaczki Victoza® prosimy uważnie zapoznać się z niniejszą instrukcją.
Dawaczka Victoza® zawiera 18 mg liraglutynidy. Można wybrać następujące dawki: 0,6 mg, 1,2 mg i 1,8 mg.
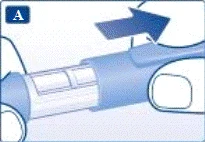
Dawaczka jest przeznaczona do użytku z jednorazowymi igłami NovoFine® lub NovoTwist® o długości do 8 mm i grubości do 32G.
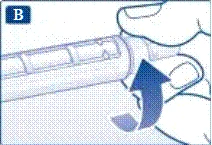
| Przygotowanie do pracy Sprawdź nazwę i kolorowy nalepek swojej ruchomki strzykawki, aby upewnić się, że zawiera ona liraglutyd. Nieumyślne podanie innego leku może poważnie zaszkodzić Twojemu zdrowiu. |
||
|
|
||
|
| ||
|
| |
| |

Zawsze używaj nowej igły do każdego wstrzyknięcia. Zmniejszy to ryzyko zanieczyszczenia, infekcji, wycieku liraglutydu, zatkania się otworu igły oraz niedokładnego dawkowania.
Uważaj: nie zginaj ani nie uszkadzaj igły.
Aby zminimalizować ryzyko przypadkowego ukłucia igłą, nigdy ponownie nie zakładaj wcześniej zdjętego wewnętrznego kapturka igły. | | | | Opieka nad strzykawką-penem * Nie próbuj naprawiać ani rozbierać jej na części. * Unikaj kontaktu strzykawki-pena z kurzem, brudem i jakimikolwiek cieczami. * Czyść strzykawkę-pen za pomocą ściereczki zwilżonej łagodnym środkiem myjącym. * Nie próbuj myć, moczyć ani smarować jej – wszystko to może uszkodzić strzykawkę-pen. | | | |
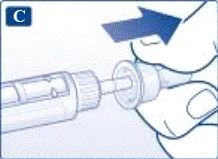
Ważne informacje * Nie przekazuj swojej strzykawki-pena innym osobom. * Przechowuj strzykawkę-pen Victoza® w miejscu niedostępnym dla innych (szczególnie dla dzieci). | | | | Sprawdzenie działania strzykawki-pena Przed pierwszym użyciem nowej strzykawki-pena sprawdź przepływ leku przez igłę. Jeśli strzykawka-pen została już wcześniej używana, przejdź do sekcji „Ustawienie dawki”, krok H. | | | |

| |
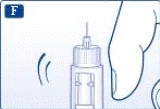
| |
| |
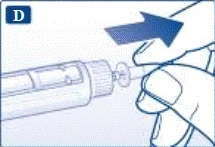
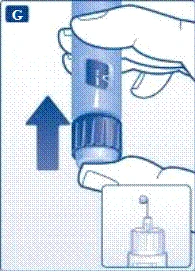
Jeśli upuścisz strzykawkę-pen na twardą powierzchnię lub uważasz, że ma ona jakiekolwiek uszkodzenia, przed każdym wstrzyknięciem podłącz nową jednorazową igłę i sprawdź działanie urządzenia. | | | | Ustawienie dawki Upewnij się, że selektor dawki znajduje się w pozycji „0”. | | | |

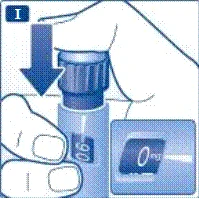
| |
Nie próbuj ustawić dawek innych niż 0,6 mg, 1,2 mg lub 1,8 mg. Cyfry na wyświetlaczu muszą dokładnie pokrywać się z wskaźnikiem, aby zapewnić odpowiednią dawkę. Podczas obracania selektora dawki słychać kliknięcia. Nie należy polegać na liczbie kliknięć podczas ustawiania dawki. Nie używaj skali kartusza do określania dawki liraglutydu do wstrzyknięcia, ponieważ nie jest to wystarczająco dokładne. | | | | Wykonanie wstrzyknięcia Zgodnie z instrukcją lekarza lub pielęgniarki dotyczącą techniki wykonania wstrzyknięcia, wprowadź igłę w skórę. Następnie wykonaj następujące kroki. | | | |
| |
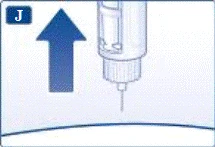
| |
| |
| |
Zawsze po każdym wstrzyknięciu odłącz igłę i przechowuj strzykawkę-pen Victoza® bez igły.
Zapobiegnie to zanieczyszczeniu, zakażeniu lub wyciekowi liraglutydu.
Należy zachować szczególną ostrożność przy usuwaniu używanych igieł, aby nie skaleczyć się. | | |
Dzieci. Korekta dawki u nastolatków i dzieci od 10. roku życia nie jest wymagana. Brak danych dotyczących stosowania u dzieci poniżej 10. roku życia. (patrz sekcje „Farmakodynamika” i „Farmakokinetyka”).
Przedawkowanie .
W badaniach klinicznych oraz w zgłoszeniach po wprowadzeniu leku Victoza® na rynek odnotowano przypadki przekroczenia zalecanej dawki utrzymawczej nawet do 40 razy (72 mg). Opisywane reakcje obejmowały silne nudności, wymioty, biegunkę oraz ciężką hipoglikemię.
W przypadku przedawkowania należy prowadzić leczenie wspomagające zgodnie z objawami klinicznymi występującymi u pacjenta. Należy obserwować stan kliniczny pacjenta w celu wczesnego wykrycia odwodnienia oraz kontrolować poziom glukozy we krwi.
Działania niepożądane.
W pięciu długotrwałych badaniach klinicznych fazy 3a ponad 2500 dorosłych pacjentów otrzymywało lek Victoza® samodzielnie lub w połączeniu z metforminą, glikwidydydą (z metforminą lub bez), sulfonamidem (z metforminą lub bez) lub z metforminą + rosiglitazonem.
Najczęstsze działania niepożądane podczas badań klinicznych to zaburzenia układu pokarmowego, wśród których bardzo często występują nudności i biegunka, często – wymioty, zaparcia, ból brzucha i dyspepsja. Na początku leczenia zaburzenia przewodu pokarmowego pojawiają się częściej, jednak ich nasilenie podczas dalszego leczenia zwykle zmniejsza się w ciągu kilku dni lub tygodni. Często również obserwowano ból głowy i zapalenie gardła i nosa. Ponadto często występowała hipoglikemia, a przy leczeniu lekiem Victoza® w połączeniu z sulfonamidem – bardzo często. Ciężkie przypadki hipoglikemii obserwowano głównie przy leczeniu skojarzonym z sulfonamidem.
Poniżej przedstawiono listę działań niepożądanych zarejestrowanych podczas długotrwałych badań klinicznych fazy 3a, badania LEADER® (długotrwałe badanie kardiowaskularne), a także na podstawie zgłoszeń spontanicznych otrzymanych po wprowadzeniu leku na rynek. Częstość wszystkich działań niepożądanych obliczono na podstawie częstości występowania w badaniach klinicznych fazy 3a.
Ocena częstości występowania działań niepożądanych przeprowadzona została według następującej skali: bardzo często (≥ 1/10), często (od ≥ 1/100 do < 1/10), rzadko (od ≥ 1/1000 do < 1/100), bardzo rzadko (od ≥ 1/10000 do < 1/1000), nieznane (nie można oszacować na podstawie dostępnych danych). W każdej grupie działania niepożądane wymieniono w kolejności zmniejszania się ich nasilenia.
Zaburzenia metabolizmu i odżywiania: często – hipoglikemia, anoreksja, zmniejszony apetyt; rzadko – odwodnienie*.
Zaburzenia układu nerwowego: często – ból głowy, zawroty głowy; rzadko – dysgezja.
Zaburzenia układu pokarmowego: bardzo często – nudności, biegunka; często – wymioty, dyspepsja, ból w górnym odcinku brzucha, zaparcia, zapalenie żołądka, wzdęcia, nadmierne gazy, choroba refluksowa przełyku, dyskomfort w żołądku, ból zęba; rzadko – opóźnienie opróżniania żołądka, niedrożność jelit; bardzo rzadko – zapalenie trzustki (w tym zapalenie trzustki z martwicą).
Zaburzenia układu sercowo-naczyniowego: często – przyspieszenie rytmu serca (HR).
Zaburzenia układu odpornościowego: rzadko – reakcje anafilaktyczne.
Zakażenia i inwazje: często – zapalenie gardła i nosa, zapalenie oskrzeli.
Ogólne zaburzenia i stan w miejscu wstrzyknięcia: często – zmęczenie, reakcje w miejscu wstrzyknięcia; rzadko – niedobór samopoczucia.
Zaburzenia nerek i dróg moczowych: rzadko – ostre niewydolność nerek, zaburzenia funkcji nerek.
Zaburzenia ze strony skóry i tkanek podskórnych: często – wysypka; rzadko – pokrzywka, świąd; nieznane – amyloidoza skóry.
Zaburzenia ze strony wątroby i dróg żółciowych: rzadko – kamica żółciowa, zapalenie pęcherza żółciowego.
Badania laboratoryjne: często – podwyższony poziom lipazy*, podwyższony poziom amylazy*
*Dane z badań klinicznych fazy 3b i 4, w których prowadzono pomiary.
Opis poszczególnych działań niepożądanych
Podczas badania klinicznego monoterapii lekiem Victoza® częstość występowania hipoglikemii u pacjentów przyjmujących lek Victoza® była niższa niż u pacjentów otrzymujących lek porównawczy (glikwidydyd). Najczęstsze działania niepożądane to zaburzenia przewodu pokarmowego, infekcje i inwazje.
Hipoglikemia
W większości przypadków zarejestrowanych podczas badań klinicznych potwierdzona hipoglikemia była niewielka. Przy monoterapii liraglutydem nie odnotowano żadnego przypadku ciężkiej hipoglikemii. Ciężka hipoglikemia występuje rzadko i obserwuje się ją głównie przy leczeniu skojarzonym liraglutydem i sulfonamidem (0,02 przypadku na 100 pacjentów-roku). Bardzo rzadko (0,001 przypadku na 100 pacjentów-roku) występowały przypadki hipoglikemii przy leczeniu liraglutydem w połączeniu z innymi doustnymi lekami przeciwcukrzycowymi (czyli nie z sulfonamidem). Ryzyko rozwoju hipoglikemii przy jednoczesnym stosowaniu liraglutydu i insuliny bazalnej jest niewielkie (1,0 przypadku na 100 pacjentów-roku, patrz sekcja „Właściwości farmakologiczne”).
W badaniu LEADER® o przypadkach ciężkiej hipoglikemii zgłaszano rzadziej przy stosowaniu liraglutydu w porównaniu z placebo (1,0 vs 1,5 przypadku na 100 pacjentów-roku, oszacowane stosunki częstości 0,69 [0,51 do 0,93]) (patrz sekcja „Farmakodynamika”). U pacjentów, którzy otrzymywali wcześniej mieszane insuliny na początku i co najmniej przez kolejne 26 tygodni, częstość przypadków ciężkiej hipoglikemii przy stosowaniu zarówno liraglutydu, jak i placebo wynosiła 2,2 przypadki na 100 pacjentów-roku.
Zaburzenia układu pokarmowego
Przy leczeniu skojarzonym lekiem Victoza® i metforminą nudności występowały co najmniej raz u 20,7% pacjentów, a biegunka – u 12,6%. Przy leczeniu skojarzonym lekiem Victoza® i sulfonamidem nudności występowały co najmniej raz u 9,1% pacjentów, a biegunka – u 7,9%. Większość przypadków miała charakter łagodny lub umiarkowany i była zależna od dawki. U większości pacjentów, którzy na początku leczenia odczuwali nudności, częstość i nasilenie objawów zmniejszały się wraz z przedłużaniem terapii.
U pacjentów w wieku powyżej 70 lat przy leczeniu liraglutydem mogą częściej występować zaburzenia ze strony układu pokarmowego.
U pacjentów z łagodnymi zaburzeniami lub zaburzeniami średniego nasilenia funkcji nerek (klirens kreatyniny ≤ 60 – 90 ml/min i 30 – 59 ml/min odpowiednio) przy leczeniu liraglutydem mogą częściej występować zaburzenia ze strony układu pokarmowego.
Kamica żółciowa i zapalenie pęcherza żółciowego
Podczas długotrwałych kontrolowanych badań klinicznych fazy 3a z liraglutydem odnotowano niewielką liczbę przypadków kamicy żółciowej (0,4%) i zapalenia pęcherza żółciowego (0,1%). W badaniu LEADER® częstość kamicy żółciowej i zapalenia pęcherza żółciowego wynosiła odpowiednio 1,5% i 1,1% przy stosowaniu liraglutydu oraz 1,1% i 0,7% przy stosowaniu placebo (patrz sekcja „Farmakodynamika”).
Przestanie stosowania leku
Podczas długotrwałych kontrolowanych badań (26 tygodni lub dłużej) częstość zaprzestania stosowania liraglutydu z powodu występujących działań niepożądanych wynosiła 7,8%, a zaprzestania leku porównawczego – 3,4%. Najczęstszą przyczyną u pacjentów otrzymujących liraglutyd były nudności (2,8%) i wymioty (1,5%).
Reakcje w miejscu wstrzyknięcia
Podczas długotrwałych kontrolowanych badań (26 tygodni lub dłużej) reakcje w miejscu wstrzyknięcia leku Victoza® odnotowano u około 2% pacjentów. Reakcje te były zazwyczaj łagodne.
Zapalenie trzustki
Podczas długotrwałych kontrolowanych badań klinicznych fazy 3 przy leczeniu lekiem Victoza® odnotowano kilka przypadków (< 0,2%) ostrego zapalenia trzustki. Przypadki zapalenia trzustki odnotowano również w zgłoszeniach spontanicznych po wprowadzeniu leku na rynek. W badaniu LEADER® częstość przypadków ostrego zapalenia trzustki potwierdzonych oceną ekspertów wynosiła 0,4% przy stosowaniu liraglutydu i 0,5% w grupie placebo (patrz sekcja „Szczególne ostrzeżenia i środki ostrożności” oraz „Farmakodynamika”).
Reakcje alergiczne
Po wprowadzeniu leku Victoza® na rynek zgłaszano o wystąpieniu reakcji alergicznych, w tym pokrzywki, wysypki i świądu. Zgłoszono również kilka przypadków reakcji anafilaktycznych z dodatkowymi objawami, takimi jak hipotensja, kołatanie serca, duszność i obrzęk. Podczas całego okresu długotrwałych badań klinicznych przy leczeniu lekiem Victoza® rzadko (0,05%) obserwowano obrzęk naczynioruchowy.
Amyloidoza skóry
Amyloidoza skóry może wystąpić w miejscu wstrzyknięcia (patrz sekcja „Sposób stosowania i dawki”).
Dzieci
Ogólnie częstość występowania, typ i nasilenie działań niepożądanych u nastolatków i dzieci w wieku od 10 lat są takie same jak u dorosłych. Częstość potwierdzonych przypadków hipoglikemii była wyższa przy stosowaniu liraglutydu (0,58 przypadku/pacjent-rok) w porównaniu z placebo (0,29 przypadku/pacjent-rok). U pacjentów, którym podawano insulinę przed wystąpieniem potwierdzonego epizodu hipoglikemii, częstość występowania była wyższa przy stosowaniu liraglutydu (1,82 przypadku/pacjent-rok) w porównaniu z placebo (0,91 przypadku/pacjent-rok). W grupie stosowania liraglutydu nie odnotowano ciężkich epizodów hipoglikemii.
Zgłaszanie działań niepożądanych i braku skuteczności leku
Zgłaszanie działań niepożądanych po rejestracji leku ma istotne znaczenie. Umożliwia monitorowanie stosunku korzyści do ryzyka przy stosowaniu tego leku. Personel medyczny i farmaceutyczny, a także pacjenci lub ich prawni przedstawiciele powinni zgłaszać wszystkie przypadki podejrzewanych działań niepożądanych i braku skuteczności leku poprzez Automatyczny System Informacyjny Nadzoru Farmakologicznego pod adresem: https://aisf.dec.gov.ua.
Okres ważności. 30 miesięcy.
Po pierwszym użyciu – 1 miesiąc.
Warunki przechowywania.
Przechowywać w miejscu niedostępnym dla dzieci. Przechowywać w lodówce (2°C – 8°C), z dala od zamrażarki. Nie zamarzać.
Po pierwszym użyciu przechowywać w temperaturze poniżej 30°C lub w lodówce (2°C – 8°C). Nie zamarzać.
W celu zapobiegania działaniu światła przechowywać długopis strzykawkowy z zamkniętą osłonką.
Niezgodność.
Dodanie jakiejkolwiek substancji do leku Victoza® może spowodować degradację liraglutydu. Bez przeprowadzenia badań na zgodność ten lek nie może być mieszany z innymi lekami.
Opakowanie. Wstępnie napełniony wielodawkowy jednorazowy długopis strzykawkowy zawiera cylinder o pojemności 3 ml wykonany ze szkła (typ 1), zatopiony z jednej strony tłokiem z gumi bromobutylowej, a z drugiej – warstwową wkładką gumową z gumi bromobutylowej/polizoprenowej. Długopis strzykawkowy wykonany jest z poliolefiny i poliacetalu.
Każdy długopis strzykawkowy zawiera 3 ml roztworu, co umożliwia podanie 30 dawek po 0,6 mg, 15 dawek po 1,2 mg lub 10 dawek po 1,8 mg.
Opakowanie zawiera 1 lub 2 wstępnie napełnione długopisy strzykawkowe.
Kategoria wydania. Na receptę.
Producent. A/S Novo Nordisk, Dania / Novo Nordisk A/S, Denmark.
Miejsce produkcji i jego adres.
A/S Novo Nordisk, Novo Allé, 2880, Bagsværd, Dania. Tel. + 45 4444 8888 /
Novo Nordisk A/S, Novo Alle, 2880, Bagsvaerd, Denmark. Tel. + 45 4444 8888.