Victoza®
Ucraina
Indice
ISTRUZIONI PER L'USO MEDICINALE DEL MEDICINALE VICTOZA®
Composizione:
Principio attivo: liraglutide;
1 ml di soluzione contiene 6 mg di liraglutide – un analogo del peptide-1 simile al glucagone umano (GLP-1), prodotto mediante tecnologia del DNA ricombinante in Saccharomyces cerevisiae. Una penna preriempita contiene 18 mg di liraglutide in 3 ml;
Eccipienti: fosfato biammidico di sodio diidrato, propilenglicole, fenolo, idrossido di sodio, acido cloridrico, acqua per preparazioni iniettabili.
Forma farmaceutica. Soluzione iniettabile.
Principali proprietà fisico-chimiche: soluzione isotonica trasparente e incolore, pH = 8,15.
Gruppo farmacoterapeutico. Preparati utilizzati nel diabete mellito, analoghi del peptide-1 simile al glucagone (GLP-1).
Codice ATC A10BJ02.
Proprietà farmacologiche.
Farmacodinamica.
Meccanismo d'azione
Liraglutide è un analogo del GLP-1 con una sequenza aminoacidica omologa al GLP-1 umano per il 97%, che si lega ai recettori del GLP-1 e li attiva. Il recettore del GL/1 è un bersaglio per il GLP-1 nativo (ormone incretinico secreto endogenamente), che potenzia la secrezione glucosio-dipendente di insulina da parte delle cellule β del pancreas. A differenza del GLP-1 nativo, la farmacocinetica e la farmacodinamica del liraglutide nell'uomo permettono un'assunzione una volta al giorno. L'azione prolungata del liraglutide somministrato per via sottocutanea è determinata da tre meccanismi: autoassociazione che rallenta l'assorbimento, legame con l'albumina plasmatica e maggiore stabilità all'azione degli enzimi dipeptidil peptidasi-4 (DPP-4) e neprilisina (NEP), che si traduce in un lungo emivita plasmatica del farmaco.
L'effetto del liraglutide è mediato da un'interazione specifica con i recettori del GLP-1, portando ad un aumento del livello di adenosina monofosfato ciclico (cAMP). Il liraglutide stimola la secrezione di insulina in modo glucosio-dipendente e contemporaneamente riduce la secrezione inadeguatamente elevata di glucagone, anch'essa dipendente dal livello di glucosio nel sangue. Pertanto, in caso di alta concentrazione di glucosio nel sangue, la secrezione di insulina aumenta e quella di glucagone diminuisce. Al contrario, in caso di ipoglicemia, il liraglutide riduce la secrezione di insulina, ma non influenza quella di glucagone. Il meccanismo di riduzione del livello di glucosio nel sangue comprende anche un lieve rallentamento dello svuotamento gastrico. Il liraglutide riduce la massa corporea e la massa grassa attraverso meccanismi di riduzione della sensazione di fame e dell'assunzione di energia.
Il GLP-1 è un regolatore fisiologico dell'appetito e dell'assunzione di cibo, ma il meccanismo esatto della sua azione non è completamente chiaro. Negli studi sugli animali, la somministrazione periferica di liraglutide ha portato al suo accumulo in specifiche aree del cervello coinvolte nella regolazione dell'appetito, dove, grazie all'attivazione specifica del recettore del GLP-1 (GLP-1R), aumentava la sensazione di sazietà e riduceva i segnali chiave della fame, causando così una riduzione del peso corporeo.
I recettori del GLP-1 sono espressi anche in determinate aree del cuore, dei vasi sanguigni, del sistema immunitario e dei reni. Nei modelli di aterosclerosi nei topi, il liraglutide ha prevenuto il progresso della placca aortica e ridotto l'infiammazione all'interno della placca. Inoltre, il liraglutide ha mostrato un effetto positivo sui lipidi plasmatici. Il liraglutide non ha ridotto le dimensioni delle placche già esistenti.
Effetti determinati dalla farmacodinamica del farmaco
Il liraglutide agisce per 24 ore e migliora il controllo glicemico riducendo i livelli di glucosio nel sangue a digiuno e dopo i pasti nei pazienti con diabete mellito di tipo 2.
Efficacia clinica e sicurezza
Sia il miglioramento del controllo glicemico che la riduzione della morbilità e mortalità cardiovascolare sono componenti essenziali del trattamento del diabete mellito di tipo 2.
Per valutare l'effetto del liraglutide sul controllo glicemico sono stati condotti cinque studi clinici randomizzati, in doppio cieco e controllati con placebo, di fase 3a su adulti (tabella 1). Il trattamento con liraglutide ha determinato una normalizzazione clinicamente e statisticamente significativa dell'emoglobina glicata A1c (HbA1c), della concentrazione di glucosio nel plasma a digiuno e dopo i pasti, rispetto al placebo.
Questi studi hanno coinvolto 3978 pazienti con diabete mellito di tipo 2 (2501 pazienti hanno ricevuto liraglutide): il 53,7% uomini, il 46,3% donne, 797 pazienti (508 hanno ricevuto liraglutide) avevano un'età ≥ 65 anni e 113 pazienti (66 hanno ricevuto liraglutide) avevano un'età ≥ 75 anni.
Sono stati condotti ulteriori studi sull'effetto del liraglutide su 1901 pazienti in quattro studi clinici randomizzati, aperti e controllati (rispettivamente 464, 658, 323 e 177 pazienti in ciascuno studio), nonché uno studio clinico randomizzato, in doppio cieco e controllato su pazienti con diabete mellito di tipo 2 e compromissione renale di grado moderato (279 pazienti).
Il liraglutide è stato inoltre utilizzato in un ampio studio cardiovascolare (LEADER®) che ha coinvolto 9340 pazienti con diabete mellito di tipo 2 ad alto rischio cardiovascolare.
Controllo glicemico
Monoterapia
La monoterapia con liraglutide per 52 settimane ha determinato una riduzione statisticamente significativa e sostenuta del livello di HbA1c rispetto alla glibenclamide alla dose di 8 mg (-0,84% per la dose di 1,2 mg, -1,14% con la dose di 1,8 mg contro -0,51% con il farmaco di confronto) in pazienti precedentemente trattati con dieta ed esercizio fisico o con monoterapia orale ipoglicemizzante a dosi non superiori alla metà della massima dose consentita (tabella 1).
Combinazione con farmaci ipoglicemizzanti orali
Il trattamento con liraglutide per 26 settimane in combinazione con metformina, glibenclamide o con la combinazione di metformina e rosiglitazone o inibitore del SGLT2 ± metformina ha permesso di ottenere una riduzione statisticamente significativa e stabile del livello di HbA1c rispetto al placebo (tabella 1).
Tabella 1. Utilizzo del liraglutide negli studi clinici di fase 3a in monoterapia (52 settimane) e in combinazione con farmaci ipoglicemizzanti orali (26 settimane)
| N |
Livello medio iniziale di HbA1c (%) |
Variazione del livello medio di HbA1c rispetto al basale (%) |
Pazienti (%) che hanno raggiunto il livello HbA1c < 7% |
Peso medio iniziale (kg) |
Variazione del peso medio rispetto al basale (kg) |
||||||||
| Monoterapia |
|||||||||||||
| Liraglutide 1,2 mg Liraglutide 1,8 mg Glimipiride 8 mg/giorno |
251 246 248 |
8,18 8,19 8,23 |
|
|
92,1 92,6 93,3 |
1,12 |
|||||||
| Aggiunta a metformina (2000 mg/die) |
|||||||||||||
| Liraglutide 1,2 mg |
240 |
8,3 |
|
35,31, 52,82 |
88,5 |
|
|||||||
| Liraglutide 1,8 mg |
242 |
8,4 |
|
42,41, 66,32 |
88,0 |
|
|||||||
| Placebo |
121 |
8,4 |
0,09 |
10,81, 22,52 |
91,0 |
|
|||||||
| Glibenclamide 4 mg/die |
242 |
8,4 |
|
36,31, 56,02 |
89,0 |
0,95 |
|||||||
| Aggiunta a glibenclamide (4 mg/die) |
|||||||||||||
| Liraglutide 1,2 mg |
228 |
8,5 |
|
34,51, 57,42 |
80,0 |
0,32** |
|||||||
| Liraglutide 1,8 mg |
234 |
8,5 |
|
41,61, 55,92 |
83,0 |
|
|||||||
| Placebo |
114 |
8,4 |
0,23 |
7,51, 11,82 |
81,9 |
|
|||||||
| Rosiglitazone 4 mg/die |
231 |
8,4 |
|
21,91, 36,12 |
80,6 |
2,11 |
|||||||
| Aggiunta a metformina (2000 mg/die) + rosiglitazone (4 mg due volte al giorno) |
|||||||||||||
| Liraglutide 1,2 mg |
177 |
8,48 |
|
57,51 |
95,3 |
|
|||||||
| Liraglutide 1,8 mg |
178 |
8,56 |
|
53,71 |
94,9 |
|
|||||||
| Placebo |
175 |
8,42 |
|
28,11 |
98,5 |
0,60 |
|||||||
| Aggiunta a metformina (2.000 mg/die) + glibenclamide (4 mg/die) |
|||||||||||||
| Liraglutide 1,8 mg |
230 |
8,3 |
|
53,11 |
85,8 |
|
|||||||
| Placebo |
114 |
8,3 |
|
15,31 |
85,4 |
|
|||||||
| Insulina glargina4 |
232 |
8,1 |
|
45,81 |
85,2 |
1,62 |
|||||||
| Aggiunta a inibitore SGLT25 ± metformina (≥1500 mg/die) |
|||||||||||||
| Liraglutide 1,8 mg Placebo |
203 100 |
8,00 7,96 |
-1,02*** -0,28 |
54,8*** 13,9 |
91,0 91,4 |
-2,92 -2,06 |
|||||||
* Efficacia superiore (p < 0,01) rispetto al farmaco attivo di confronto.
** Efficacia superiore (p < 0,0001) rispetto al farmaco attivo di confronto.
*** Efficacia superiore (p < 0,001) rispetto al farmaco attivo di confronto.
† Non inferiore (p < 0,0001) rispetto al farmaco attivo di confronto.
1 Tutti i pazienti.
2 Monoterapia precedente con ipoglicemizzante orale.
3 Pazienti precedentemente sottoposti a dieta.
4 Lo studio con insulina glargina era in aperto e il dosaggio era stabilito in base alle raccomandazioni per il titolazione dell'insulina glargina.
5 Aggiunta del medicinale Victoza® a un inibitore SGLT2 studiata in tutte le dosi autorizzate dell'inibitore SGLT2.
Raccomandazioni per la titolazione dell'insulina glargina:
| Autosomministrazione della concentrazione di glucosio nel plasma a digiuno |
Aumento della dose di insulina glargina (UI) |
| ≤ 5,5 mmol/l (≤ 100 mg/dl) Livello target |
Mantenere la dose invariata |
| > 5,5 e < 6,7 mmol/l (> 100 e < 120 mg/dl) |
0–2 UIa |
| ≥ 6,7 mmol/l (≥ 120 mg/dl) |
2 UI |
a Secondo le raccomandazioni individuali fornite dal ricercatore durante la visita precedente, ad esempio, a seconda che il paziente abbia manifestato ipoglicemia.
Combinazione con insulina
In uno studio clinico della durata di 104 settimane, il 57 % dei pazienti con diabete mellito di tipo 2 in trattamento con insulina degludec in associazione a metformina ha raggiunto l’obiettivo di HbA1c < 7 %, mentre i pazienti rimanenti hanno proseguito nello studio aperto della durata di 26 settimane, ricevendo in modo randomizzato un trattamento aggiuntivo con liraglutide o insulina aspart una volta al giorno (con il pasto più abbondante). Nel gruppo di studio, l’insulina degludec è stata associata al liraglutide con una riduzione del 20 % della dose di insulina al fine di minimizzare il rischio di ipoglicemia. L’aggiunta di liraglutide ha determinato una riduzione statisticamente maggiore del livello di HbA1c: – 0,73 % con liraglutide e – 0,40 % con il farmaco di confronto, e del peso corporeo: – 3,03 kg contro – 0,72 kg rispettivamente. L’incidenza di episodi di ipoglicemia (per paziente all’anno di trattamento) è risultata statisticamente significativamente inferiore aggiungendo liraglutide rispetto all’aggiunta di insulina aspart una volta al giorno (1,0 contro 8,15; rapporto: 0,13; IC 95 %: da 0,08 a 0,21).
In uno studio clinico della durata di 52 settimane, l’aggiunta di insulina detemir al liraglutide nei pazienti che non avevano raggiunto un adeguato controllo glicemico con liraglutide 1,8 mg e metformina ha determinato una riduzione dell’HbA1c dello 0,54 % rispetto al valore iniziale, rispetto a una riduzione dello 0,20 % nei pazienti del gruppo di controllo che ricevevano liraglutide 1,8 mg e metformina. In questo caso, la riduzione del peso corporeo si è mantenuta. È stata osservata una lieve aumentata incidenza di ipoglicemia lieve (0,23 contro 0,03 per paziente-anno rispettivamente).
Nello studio LEADER® (vedi paragrafo «Effetto sul sistema cardiovascolare» di seguito) 873 pazienti hanno ricevuto insulina premiscelata (con o senza farmaci orali ipoglicemizzanti) all’inizio e per almeno le successive 26 settimane. Il livello medio di HbA1c all’inizio era dell’8,7 % con liraglutide e placebo. Alla settimana 26, la variazione media approssimativa dell’HbA1c è stata di – 1,4 % e – 0,5 % con liraglutide e placebo rispettivamente, con una differenza stimata nel trattamento di – 0,9 [– 1,00; – 0,70] 95 %. Il profilo di sicurezza del liraglutide in combinazione con insulina premiscelata è stato in generale paragonabile a quello osservato con placebo in combinazione con insulina premiscelata (vedi sezione «Effetti indesiderati»).
Esperienza nel trattamento di pazienti con compromissione renale
In uno studio in doppio cieco volto a confrontare l’efficacia e la sicurezza di liraglutide 1,8 mg e placebo aggiunti all’insulina e/o ai farmaci orali ipoglicemizzanti in pazienti con diabete mellito di tipo 2 e compromissione renale di grado moderato, il liraglutide ha mostrato risultati migliori rispetto al placebo riguardo alla riduzione dell’HbA1c dopo 26 settimane di trattamento (rispettivamente – 1,05 % contro – 0,38 %). Un numero significativamente maggiore di pazienti ha raggiunto un valore di HbA1c inferiore al 7 % con liraglutide rispetto al placebo (52,8 % contro 19,5 %). In entrambi i gruppi si è osservata una riduzione del peso corporeo: –2,4 kg con liraglutide contro –1,09 con placebo. Il rischio relativo di sviluppare ipoglicemia nei due gruppi terapeutici è stato paragonabile. Il profilo di sicurezza del liraglutide è stato sostanzialmente simile a quello osservato negli altri studi con liraglutide.
Con la monoterapia con Victoza®, nei pazienti il cui valore di HbA1c era superiore al 9,5 % prima del trattamento, si è osservata una riduzione media del 2,1 %, mentre con la terapia combinata si è osservata una riduzione del 1,1-2,5 %.
Percentuale di pazienti con riduzione dell’HbA1c
La monoterapia con liraglutide ha determinato una percentuale statisticamente significativa maggiore di pazienti che hanno raggiunto un valore di HbA1c ≤ 6,5 % alla settimana 52 rispetto ai pazienti trattati con glimepiride (37,6 % con dose di 1,8 mg e 28,0 % con dose di 1,2 mg contro 16,2 % con il farmaco di confronto).
Con il trattamento per 26 settimane con liraglutide in combinazione con metformina, glimepiride, metformina e rosiglitazone o inibitore del SGLT2 ± metformina, si è osservata una percentuale statisticamente significativa maggiore di pazienti con valore di HbA1c ≤ 6,5 % rispetto alla monoterapia con questi farmaci.
Livello di glucosio nel plasma a digiuno
Il trattamento con liraglutide e la sua combinazione con uno o due farmaci antidiabetici orali ha determinato una riduzione del livello di glucosio nel plasma a digiuno compresa tra 13 e 43,5 mg/dl (0,72 – 2,42 mmol/l). Tale riduzione si osserva entro le prime 2 settimane di trattamento.
Livello di glucosio nel plasma dopo i pasti
Il liraglutide ha ridotto il livello di glucosio nel plasma dopo tutti e 3 i pasti giornalieri di 31 – 49 mg/dl (1,68 – 2,71 mmol/l).
Funzione delle cellule beta
Sulla base dei dati ottenuti dagli studi clinici con liraglutide mediante la valutazione del modello di omeostasi della funzione delle cellule beta e dai valori del rapporto proinsulina/insulina, si è concluso che la funzione delle cellule beta migliora. Dopo 52 settimane di trattamento con liraglutide in un gruppo di pazienti con diabete mellito di tipo 2 (n = 29) si è osservato un miglioramento della prima e della seconda fase della secrezione di insulina.
Peso corporeo
Con il trattamento con liraglutide in combinazione con metformina, metformina e glimepiride, metformina e rosiglitazone o inibitore del SGLT2, con o senza metformina, i pazienti hanno perso stabilmente da 0,86 a 2,62 kg di peso corporeo rispetto al placebo.
Una riduzione più marcata del peso corporeo è stata osservata nei pazienti con valori più elevati di indice di massa corporea prima dell’inizio del trattamento.
Effetto sul sistema cardiovascolare
Un’analisi retrospettiva di eventi cardiovascolari gravi (mortalità per cause cardiovascolari, infarto miocardico, ictus) durante tutti gli studi di media e lunga durata delle fasi 2 e 3 (della durata da 26 a 100 settimane), in cui hanno partecipato 5607 pazienti (di cui 3651 trattati con liraglutide), ha mostrato l’assenza di un aumento del rischio di eventi cardiovascolari [frequenza degli eventi 0,75 (95 %) IC (intervallo di confidenza) 0,35; 1,63] con liraglutide rispetto a tutti i farmaci di confronto.
Lo studio sull’effetto e l’impatto cardiovascolare del liraglutide nel diabete mellito (LEADER®) è uno studio clinico in doppio cieco, multicentrico, controllato con placebo. 9340 pazienti sono stati randomizzati a ricevere liraglutide (4668) o placebo (4672) come aggiunta al trattamento standard volto a ridurre l’HbA1c e i fattori di rischio cardiovascolari. Lo stato vitale primario o alla fine dello studio era noto nel 99,7 % e nel 99,6 % dei pazienti randomizzati che ricevevano liraglutide o placebo rispettivamente. La durata del follow-up è stata di almeno 3,5 anni e fino a un massimo di 5 anni. Lo studio ha incluso pazienti di età ≥ 65 anni (n = 4329) e ≥ 75 anni (n = 836) e pazienti con compromissione renale lieve (n = 3907), moderata (n = 1934) o grave (n = 224). L’età media dei pazienti era di 64 anni e l’indice di massa corporea medio era di 32,5 kg/m². La durata media della malattia diabetica era di 12,8 anni.
L’endpoint primario era il tempo dalla randomizzazione fino al primo evento di eventi avversi cardiovascolari gravi (MACE): morte cardiovascolare, infarto miocardico non fatale o ictus non fatale. Il liraglutide si è dimostrato superiore al placebo nella prevenzione di eventi avversi cardiovascolari gravi (fig. 1). Il rischio stimato è risultato costantemente inferiore a 1 per tutti e 3 i componenti degli eventi avversi cardiovascolari gravi.
Il liraglutide ha inoltre ridotto significativamente il rischio di sviluppare numerosi eventi avversi cardiovascolari gravi (MACE primari, angina instabile che richiede ospedalizzazione, rivascolarizzazione coronarica o scompenso cardiaco che richiede ospedalizzazione) e altri endpoint secondari (fig. 2).
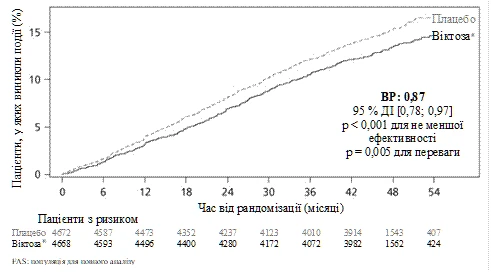
Figura 1.
Curva di Kaplan-Meier. Intervallo di tempo fino al primo evento avverso cardiovascolare grave (MACE) – popolazione FAS
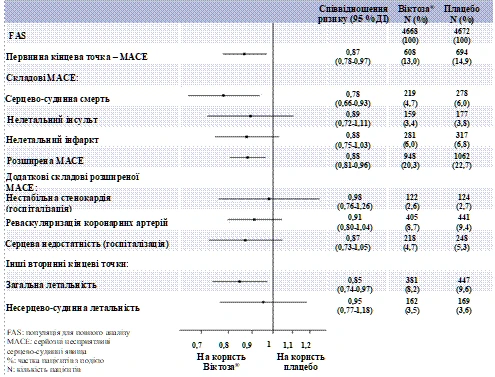
Figura 2. Diagramma foresta dell’analisi degli eventi cardiovascolari individuali – popolazione FAS
Con l’aggiunta di liraglutide al trattamento standard si è osservata una riduzione significativa e sostenuta dell’HbA1c dal basale fino a 36 mesi rispetto al placebo (– 1,16 % contro – 0,77 %; differenza stimata nei metodi di trattamento – 0,40 % [– 0,45; – 0,34]). Con l’uso di liraglutide rispetto al placebo, la necessità di intensificare il trattamento con insulina nei pazienti che precedentemente non avevano usato insulina è diminuita del 48 % (RR 0,52 [0,48; 0,57]).
Pressione arteriosa e frequenza cardiaca
Negli studi di fase 3a, il liraglutide ha ridotto la pressione arteriosa sistolica in media da 2,3 a 6,7 mmHg rispetto al basale rispetto al farmaco attivo di confronto, il cui effetto di riduzione è stato compreso tra 1,9 e 4,5 mmHg.
Negli studi clinici a lungo termine, inclusivo di LEADER®, il liraglutide ha aumentato lievemente la frequenza cardiaca rispetto al basale di 2 – 3 battiti al minuto. Nello studio LEADER® non è stato osservato un impatto clinico a lungo termine dell’aumento della frequenza cardiaca sul rischio di eventi cardiovascolari.
Valutazione delle malattie microvascolari
Nello studio LEADER® gli eventi microvascolari includevano nefropatia e retinopatia. L’analisi dell’intervallo di tempo fino al primo evento microvascolare con liraglutide rispetto a placebo ha mostrato un HR di 0,84 [0,73, 0,97]. L’HR con liraglutide rispetto a placebo è stato di 0,78 [0,67, 0,92] per lo sviluppo del primo episodio di nefropatia e di 1,15 [0,87, 1,52] per lo sviluppo del primo episodio di retinopatia.
Immunogenicità
A causa delle proprietà immunogeniche potenziali dei farmaci contenenti proteine o peptidi, durante il trattamento con liraglutide possono formarsi anticorpi anti-liraglutide nei pazienti. Essi sono stati rilevati in media nell’8,6 % dei pazienti. La formazione di anticorpi non ha portato a una riduzione dell’efficacia del liraglutide.
Pediatria
In uno studio in doppio cieco è stata confrontata l’efficacia e la sicurezza di Victoza® 1,8 mg e placebo aggiunti alla metformina ± insulina in adolescenti e bambini di età ≥ 10 anni con diabete mellito di tipo 2. Dopo 26 settimane di trattamento con Victoza®, il livello di HbA1c è risultato più efficacemente ridotto (−1,06 [−1,65, −0,46]) rispetto al placebo. Dopo ulteriori 26 settimane di trattamento in uno studio aperto aggiuntivo, la differenza nei livelli di HbA1c era di 1,3 %, confermando il controllo glicemico sostenuto con Victoza®.
L’efficacia e il profilo di sicurezza di Victoza® sono risultati analoghi a quelli osservati negli adulti trattati con Victoza®. A seconda del controllo glicemico adeguato o della tollerabilità, il 30 % dei soggetti dello studio ha proseguito il trattamento con dose di 0,6 mg, il 17 % ha aumentato la dose a 1,2 mg e il 53 % a 1,8 mg.
Altri dati clinici
In uno studio aperto è stata confrontata l’efficacia e la sicurezza di liraglutide (1,2 mg e 1,8 mg) e sitagliptin (inibitore della dipeptidil peptidasi-4, 100 mg) nel trattamento di pazienti in cui non era stato raggiunto un adeguato controllo glicemico con metformina (livello medio di HbA1c = 8,5 %). Dopo 26 settimane di trattamento, entrambe le dosi di liraglutide hanno ridotto statisticamente in modo significativo l’HbA1c (−1,24 % e −1,50 %) rispetto al sitagliptin (−0,90 %, p < 0,0001). Nei pazienti trattati con liraglutide si è osservata una riduzione del peso corporeo maggiore (−2,9 kg e −3,4 kg) rispetto al trattamento con sitagliptin (−1,0 kg, p < 0,0001). Nausea transitoria si è verificata più frequentemente nei pazienti che ricevevano liraglutide (20,8 % e 27,1 %) rispetto al sitagliptin (4,6 %). La maggiore riduzione dell’HbA1c osservata dopo 26 settimane di trattamento con liraglutide (1,2 mg e 1,8 mg) si è mantenuta anche dopo 52 settimane (−1,29 % e −1,51 %) rispetto al sitagliptin (−0,88 %, p < 0,0001). La conversione dei pazienti dopo 52 settimane di trattamento con sitagliptin a liraglutide (1,2 mg e 1,8 mg) ha determinato un’ulteriore riduzione statisticamente significativa dell’HbA1c, che alla settimana 78 era di −0,24 % e −0,45 % (IC 95 %: −0,41 % − −0,07 % e −0,67 % − −0,23 %), ma non era presente formalmente un gruppo di controllo.
In uno studio aperto che ha coinvolto pazienti in cui non era stato raggiunto un adeguato controllo glicemico con metformina e/o sulfonilurea (livello medio di HbA1c = 8,3 %), è stata confrontata l’efficacia e la sicurezza di liraglutide 1,8 mg (una volta al giorno) con exenatide 10 mcg (due volte al giorno). Dopo 26 settimane di trattamento, il liraglutide ha ridotto statisticamente in modo significativo l’HbA1C (−1,12 %) rispetto all’exenatide (−0,79 %), con una differenza stimata tra i gruppi di −0,33 % (IC 95 %: −0,47 % − −0,18 %). Il numero di pazienti con HbA1c < 7 % è risultato significativamente maggiore tra quelli che ricevevano liraglutide (54,2 %) rispetto all’exenatide (43,4 %, p = 0,0015). Con entrambi i trattamenti il peso corporeo dei pazienti si è ridotto in media di 3 kg. La conversione dei pazienti dopo 26 settimane di trattamento con exenatide a liraglutide ha determinato un’ulteriore riduzione statisticamente significativa dell’HbA1c, che alla settimana 40 era di −0,32 % (IC 95 %: −0,41 % − −0,24 %), ma non era presente formalmente un gruppo di controllo. Nel corso di 26 settimane di trattamento con liraglutide, 12 complicanze gravi (5,1 %) si sono verificate in 235 pazienti, mentre con exenatide si sono verificate 6 complicanze gravi (2,6 %) in 232 pazienti. Non è stato osservato un chiaro pattern di distribuzione di queste complicanze per sistemi organi.
In uno studio aperto che ha confrontato l’efficacia e la sicurezza di liraglutide 1,8 mg e lixisenatide 20 mcg in 404 pazienti che non avevano raggiunto il controllo glicemico con metformina (livello medio di HbA1c 8,4 %), il liraglutide si è dimostrato più efficace del lixisenatide nel ridurre l’HbA1c dopo 26 settimane di trattamento (– 1,83 % contro – 1,21 %, p < 0,0001). Un numero significativamente maggiore di pazienti ha raggiunto un valore di HbA1c inferiore al 7 % con liraglutide rispetto al lixisenatide (74,2 % contro 45,5 %, p < 0,0001), così come un valore di HbA1c ≤ 6,5 % (54,6 % contro 26,2 %, p < 0,0001). La riduzione del peso corporeo è stata osservata in entrambi i gruppi di pazienti (– 4,3 kg con liraglutide e – 3,7 kg con lixisenatide). Gli effetti indesiderati gastrointestinali si sono verificati più frequentemente nei pazienti che ricevevano liraglutide (43,6 % contro 37,1 %).
Farmacocinetica.
Assorbimento
Dopo somministrazione sottocutanea, il liraglutide viene assorbito lentamente, raggiungendo la concentrazione massima tra le 8 e le 12 ore. Dopo somministrazione sottocutanea singola di 0,6 mg di liraglutide, la concentrazione massima era di 9,4 nmol/l (peso corporeo medio circa 73 kg). Dopo somministrazione di 1,8 mg di liraglutide, la concentrazione media allo steady state (AUCτ/24) raggiungeva circa 34 nmol/l (peso corporeo medio circa 76 kg). L’esposizione al liraglutide diminuisce all’aumentare del peso corporeo. L’esposizione al liraglutide aumenta in modo proporzionale alla dose. In un singolo paziente, il coefficiente di variazione del valore AUC dopo somministrazione singola di liraglutide era dell’11 %.
La biodisponibilità assoluta del liraglutide dopo somministrazione sottocutanea è di circa il 55 %.
Distribuzione
Il volume apparente di distribuzione dopo somministrazione sottocutanea è di 11–17 l. Il volume medio di distribuzione dopo somministrazione endovenosa di liraglutide è di 0,07 l/kg. Il liraglutide si lega estensivamente alle proteine plasmatiche (> 98 %).
Metabolismo
Entro 24 ore dopo somministrazione singola di dose di liraglutide marcato radioattivamente [3H]-liraglutide a volontari sani, il componente principale nel plasma era il liraglutide inalterato. Nel plasma sono stati rilevati in quantità trascurabili due metaboliti (≤ 9 % e ≤ 5 % dell’esposizione totale alla radioattività plasmatica). Il liraglutide viene metabolizzato con gli stessi meccanismi delle grandi proteine. Non è stato identificato un organo specifico responsabile della via principale di eliminazione.
Eliminazione
Dopo somministrazione di dose di [3H]-liraglutide, non è stato rilevato liraglutide inalterato nelle urine e nelle feci. Solo una piccola frazione della radioattività controllata, escretata come metaboliti legati al liraglutide, è stata rilevata nelle urine (6 %) e nelle feci (5 %). La radioattività nelle urine e nelle feci è stata escretata principalmente entro i primi 6 – 8 giorni sotto forma di tre metaboliti in quantità trascurabili rispettivamente.
Dopo somministrazione sottocutanea singola di liraglutide, il valore medio della clearance è di circa 1,2 l/ora, mentre la durata della semivita è di circa 13 ore.
Gruppi particolari di pazienti
Pazienti anziani. Sulla base dei dati di uno studio di farmacocinetica in volontari sani e dell’analisi farmacocinetica in un gruppo di pazienti di età compresa tra 18 e 80 anni, si è concluso che l’età non ha un impatto clinicamente significativo sulla farmacocinetica del liraglutide.
Sesso. Sulla base dei dati dell’analisi farmacocinetica in una popolazione di pazienti di entrambi i sessi e di uno studio farmacocinetico in volontari sani, si è concluso che il sesso non ha un impatto clinicamente rilevante sulla farmacocinetica del liraglutide.
Origine etnica. Sulla base dei dati dell’analisi farmacocinetica in un gruppo di pazienti di razza caucasica, mongoloide e negra, si è concluso che l’origine etnica non ha alcun impatto clinicamente significativo sulla farmacocinetica del liraglutide.
Obesità. Sulla base dei dati dell’analisi farmacocinetica di popolazione, l’indice di massa corporea non ha un impatto clinicamente significativo sui parametri farmacocinetici del liraglutide.
Compromissione epatica. La farmacocinetica del liraglutide è stata studiata in pazienti con diversi gradi di compromissione epatica in uno studio con dose singola. È stato dimostrato che nei pazienti con compromissione epatica lieve e moderata l’esposizione al liraglutide si riduceva del 13 – 23 % rispetto ai volontari sani.
Nei pazienti con compromissione epatica grave (> 9 punti secondo la classificazione Child-Pugh), l’esposizione era significativamente più bassa (44 %).
Compromissione renale. L’esposizione al liraglutide era ridotta nei pazienti con compromissione renale rispetto a soggetti con funzione renale normale. Nei pazienti con compromissione lieve (clearance della creatinina 50 – 80 ml/min) si riduceva del 33 %, con compromissione moderata (clearance della creatinina 30 – 50 ml/min) del 14 %, con compromissione grave (clearance della creatinina < 30 ml/min) del 27 % e nelle fasi terminali della malattia renale che richiedono dialisi del 26 %. Analogamente, in studi clinici di 26 settimane in pazienti con diabete mellito di tipo 2 e compromissione renale (clearance della creatinina – 30 – 59 ml/min, vedere sezione «Farmacodinamica»), l’esposizione al liraglutide si riduceva del 26 % rispetto ai pazienti con diabete mellito di tipo 2 e funzione renale normale o compromissione lieve.
Pediatria. Le proprietà farmacocinetiche sono state studiate in studi clinici in bambini con diabete mellito di tipo 2 di età ≥ 10 anni. L’esposizione al liraglutide negli adolescenti e nei bambini corrispondeva all’esposizione negli adulti.
Dati preclinici di sicurezza
I dati preclinici ottenuti da studi tradizionali di farmacologia della sicurezza, tossicità da dosi ripetute o genotossicità non hanno evidenziato rischi particolari per l’uomo.
Neoplasie non letali delle cellule C della tiroide sono state osservate in ratti e topi negli studi di carcinogenicità di 2 anni. Nei ratti non è stato osservato un aumento della frequenza o gravità di effetti avversi. In scimmie trattate per 20 mesi non sono state osservate tali neoplasie. Le neoplasie nei roditori sono attribuibili a un meccanismo specifico non genotossico mediato dal recettore GLP-1, a cui i roditori sono parzialmente sensibili. L’importanza di questo meccanismo nell’uomo è considerata sufficientemente bassa, ma non può essere esclusa completamente. Non sono state osservate altre neoplasie con il trattamento con Victoza®.
Negli studi sugli animali non è stato osservato un effetto dannoso diretto sulla fertilità, tuttavia con le dosi più elevate è stata osservata una lieve aumentata mortalità embrionale precoce. La somministrazione di Victoza® durante la metà della gravidanza ha causato una riduzione del peso corporeo della madre, un rallentamento della crescita fetale con effetto non chiaro sullo sviluppo delle costole nei ratti e dello scheletro nei conigli. Con la somministrazione di Victoza® è stato osservato un rallentamento della crescita nei ratti neonati, che persiste nel periodo di svezzamento nel gruppo con dose alta. Rimane incerto se il rallentamento della crescita nei ratti neonati sia dovuto a una riduzione dell’assunzione di latte a causa di un effetto diretto del GLP-1 o a una riduzione del latte materno dovuta alla riduzione delle calorie assunte.
Caratteristiche cliniche.
Indicazioni.
Il medicinale Victoza® è indicato per il trattamento del diabete mellito di tipo 2 insufficientemente controllato negli adulti, negli adolescenti e nei bambini a partire dai 10 anni di età, come complemento alla dieta e all’attività fisica:
- in monoterapia, quando l’uso della metformina è considerato inadeguato a causa di intolleranza o controindicazioni;
- in associazione con altri medicinali per il trattamento del diabete.
Per i risultati degli studi sull’uso in combinazione con altri agenti, l’impatto sul controllo glicemico e gli eventi cardiovascolari, nonché le popolazioni studiate, si rimanda alle sezioni «Informazioni particolari di impiego», «Interazioni con altri medicinali ed altre forme di interazione» e «Farmacodinamica».
Controindicazioni.
Ipersensibilità all’ingrediente attivo o ad uno qualsiasi degli eccipienti elencati nella sezione degli eccipienti.
Interazioni con altri medicinali ed altre forme di interazione.
In vitro, il liraglutide ha mostrato un potenziale molto basso di interferenza sulla farmacocinetica di altre sostanze attive il cui metabolismo è mediato dal citocromo P450, nonché sul legame con le proteine plasmatiche.
Il liraglutide determina un lieve ritardo nello svuotamento gastrico, che potrebbe influenzare l’assorbimento di medicinali assunti contemporaneamente per via orale. Gli studi sull’interazione non hanno evidenziato un rallentamento clinicamente significativo dell’assorbimento e pertanto non è necessaria alcuna correzione della dose. In alcuni pazienti trattati con Victoza® è stato riportato almeno un episodio di diarrea grave. La diarrea può alterare l’assorbimento di medicinali assunti contemporaneamente per via orale.
Warfarin e altri derivati delle cumarine
Non sono stati condotti studi specifici di interazione farmacologica. Non può essere esclusa un’interazione clinicamente significativa con sostanze attive a bassa solubilità o con un indice terapeutico ristretto, come la warfarin. Nei pazienti in trattamento con warfarin o altri derivati delle cumarine, all’inizio della terapia con liraglutide si raccomanda un monitoraggio più frequente dell’INR (Rapporto Normalizzato Internazionale).
Paracetamolo
Il liraglutide non ha modificato l’esposizione totale al paracetamolo dopo somministrazione di una dose singola da 1000 mg. La concentrazione massima di paracetamolo (Cmax) è diminuita del 31% e il tempo per raggiungere la concentrazione massima (tmax) è aumentato di 15 minuti. Non è necessario alcun aggiustamento della dose del paracetamolo quando assunto contemporaneamente.
Atorvastatina
Il liraglutide non ha modificato in modo clinicamente significativo l’esposizione totale all’atorvastatina dopo somministrazione singola di 40 mg. Pertanto, non è necessario alcun aggiustamento della dose di atorvastatina quando assunta contemporaneamente a Victoza®. Quando somministrata contemporaneamente al liraglutide, la Cmax dell’atorvastatina è diminuita del 38% e il tmax è aumentato da 1 a 3 ore.
Griseofulvina
Il liraglutide non ha modificato l’esposizione totale alla griseofulvina dopo somministrazione singola di 500 mg. La Cmax è aumentata del 37%, mentre il tmax non è cambiato. Non è necessario alcun aggiustamento della dose quando la griseofulvina o altre sostanze poco solubili ad alta permeabilità sono assunte contemporaneamente.
Digossina
Dopo somministrazione singola di 1 mg di digossina in associazione con liraglutide, è stata osservata una riduzione dell’area sotto la curva concentrazione-tempo (AUC) per la digossina del 16% e una riduzione della Cmax del 31%. Il tmax medio della digossina è aumentato da 1 a 1,5 ore. Sulla base di questi risultati, non è necessario alcun aggiustamento della dose di digossina.
Lisinopril
Dopo somministrazione singola di 20 mg di lisinopril, è stata osservata una riduzione dell’AUC del lisinopril del 15% e una riduzione della Cmax del 27%. Il tmax medio del lisinopril è aumentato da 6 a 8 ore. Sulla base di questi risultati, non è necessario alcun aggiustamento della dose di lisinopril.
Contraccettivi orali
Quando una dose singola di contraccettivi orali è stata assunta contemporaneamente al liraglutide, la Cmax di etinilestradiolo o levonorgestrel è diminuita rispettivamente del 12% e del 13%, mentre il tmax è aumentato di 1,5 ore. Tali variazioni non hanno avuto effetti clinici sull’esposizione totale a etinilestradiolo o levonorgestrel, suggerendo che l’assunzione contemporanea di liraglutide non influisce sull’effetto contraccettivo di etinilestradiolo e levonorgestrel.
Insulina
In pazienti con diabete mellito di tipo 2 stabilizzato, la somministrazione contemporanea di insulina detemir (5 UI/kg) e liraglutide (1,8 mg) non ha evidenziato segni di interazione farmacocinetica o farmacodinamica.
Bambini
Gli studi di interazione sono stati condotti esclusivamente su adulti.
Caratteristiche d'uso.
Liraglutide non deve essere utilizzato per il trattamento di pazienti con diabete mellito di tipo I o con chetoacidosi diabetica.
Liraglutide non è un sostituto dell'insulina. Sono stati riportati casi di sviluppo di chetoacidosi diabetica in seguito a interruzione rapida o riduzione della dose di insulina in pazienti dipendenti dall'insulina (vedere la sezione «Modalità di somministrazione e posologia»).
Non esiste esperienza terapeutica nel trattamento di pazienti con insufficienza cardiaca congestizia di classe IV secondo la classificazione della New York Heart Association (NYHA); pertanto, liraglutide non è raccomandato per questi pazienti.
L'esperienza nell'uso di liraglutide in pazienti con malattie infiammatorie intestinali e gastroparesi diabetica è limitata. L'uso di liraglutide in questi pazienti non è raccomandato poiché può essere associato a effetti indesiderati transitori a carico del tratto gastrointestinale, inclusi nausea, vomito e diarrea.
Aspirazione in associazione ad anestesia generale o sedazione profonda
In pazienti trattati con agonisti del recettore GLP-1 sono stati osservati casi di aspirazione polmonare durante anestesia generale o in stato di sedazione profonda. Pertanto, prima di eseguire procedure che richiedono anestesia generale o sedazione profonda, si deve considerare il rischio aumentato di contenuto gastrico residuo dovuto al ritardo dello svuotamento gastrico (vedere la sezione «Effetti indesiderati»).
Pancreatite acuta
È stata riportata pancreatite acuta con l'uso di analoghi del recettore GLP-1.
I pazienti devono essere informati sui sintomi tipici di pancreatite acuta. In caso di sospetto di pancreatite, il trattamento con liraglutide deve essere interrotto. Se la pancreatite acuta viene confermata, non è raccomandato il reimpiego di liraglutide (vedere le sezioni «Effetti indesiderati» e «Proprietà farmacodinamiche»).
Malattie della tiroide
Negli studi clinici sono state osservate reazioni avverse a carico della tiroide, come gozzo, specialmente in pazienti con preesistenti malattie della tiroide. Pertanto, liraglutide deve essere utilizzato con cautela in questi pazienti.
Ipopglicemia
Nei pazienti che ricevono liraglutide in associazione con sulfoniluree o insulina, il rischio di ipoglicemia aumenta (vedere la sezione «Effetti indesiderati»). Il rischio di ipoglicemia può essere ridotto riducendo la dose di sulfonilurea o insulina.
Disidratazione
Nei pazienti trattati con liraglutide sono stati osservati sintomi di disidratazione, inclusi alterazioni della funzionalità renale e insufficienza renale acuta.
I pazienti ai quali viene prescritto liraglutide devono essere informati del rischio di disidratazione dovuta a disturbi gastrointestinali e della necessità di adottare misure preventive contro la disidratazione.
Il medicinale Victoza® contiene meno di 1 mmol di sodio (23 mg); pertanto, questo medicinale può considerarsi praticamente privo di sodio.
Tracciabilità
Al fine di migliorare la tracciabilità dei medicinali biologici, è necessario registrare chiaramente il nome e il numero di lotto del medicinale somministrato.
Uso durante la gravidanza o l’allattamento.
Gravidanza
Non esistono dati adeguati sull'uso di liraglutide in donne in gravidanza. Gli studi sugli animali hanno mostrato tossicità riproduttiva (vedere la sezione «Dati preclinici di sicurezza»). Il rischio potenziale nell'uomo non è noto.
Liraglutide non deve essere usato durante la gravidanza; si raccomanda invece l'uso di insulina. Se una paziente desidera una gravidanza o è in stato di gravidanza, il trattamento con il medicinale Victoza® deve essere interrotto.
Allattamento
Non è noto se liraglutide sia escreto nel latte materno umano. Gli studi sugli animali hanno mostrato che una piccola quantità di liraglutide e dei suoi metaboliti strutturalmente correlati passa nel latte. Gli studi preclinici hanno evidenziato una riduzione della crescita dei neonati nei ratti in seguito all'uso del farmaco (vedere la sezione «Dati preclinici sulla sicurezza d’uso»). A causa della mancanza di esperienza durante l’allattamento, il medicinale Victoza® non deve essere utilizzato in questo periodo.
Fertilità
Gli studi sugli animali non hanno evidenziato effetti dannosi di liraglutide sulla fertilità, eccetto una lieve riduzione del numero di embrioni impiantati e vitali.
Capacità di influenzare la velocità di reazione nella guida di veicoli o nell'uso di macchinari.
Il medicinale Victoza® non ha alcun effetto oppure ha un effetto trascurabile sulla capacità di guidare veicoli o usare macchinari. Ai pazienti si raccomanda di adottare misure preventive per evitare l’insorgenza di ipoglicemia durante la guida di veicoli o l’uso di macchinari, in particolare quando il medicinale Victoza® viene utilizzato in associazione con sulfoniluree o insulina.
Modalità e posologia di somministrazione
Posologia
Per migliorare la tollerabilità a livello del tratto gastrointestinale, la dose iniziale è di 0,6 mg di liraglutide al giorno. Dopo almeno 1 settimana, la dose deve essere aumentata a 1,2 mg. In alcuni pazienti si prevede un ulteriore miglioramento aumentando la dose da 1,2 mg a 1,8 mg e, in base alla risposta al trattamento, dopo almeno 1 settimana di terapia la dose può essere aumentata a 1,8 mg per un ulteriore miglioramento del controllo glicemico. Non è raccomandata una dose giornaliera superiore a 1,8 mg.
Quando Victoza® viene utilizzato in associazione con sulfoniluree o insulina, la dose di sulfonilurea o insulina deve essere ridotta al fine di ridurre il rischio di ipoglicemia (vedere il paragrafo «Speciali avvertenze e precauzioni di impiego»). La terapia in associazione con sulfoniluree è applicabile solo negli adulti.
Per l’aggiustamento della dose di Victoza® non è necessario il monitoraggio autonomo della glicemia. Il monitoraggio autonomo della glicemia è invece necessario per aggiustare la dose di sulfonilurea e di insulina, in particolare all’inizio del trattamento con Victoza® e durante la riduzione della dose di insulina. Si raccomanda una riduzione graduale della dose di insulina.
Pazienti particolari
Pazienti anziani (> 65 anni). Non è necessario alcun aggiustamento posologico in base all’età (vedere il paragrafo «Farmacocinetica»).
Compromissione renale. Nei pazienti con compromissione renale lieve, moderata o grave non è necessario alcun aggiustamento posologico. Non vi è esperienza di trattamento nei pazienti con insufficienza renale allo stadio terminale; pertanto, Victoza® non è raccomandato in questi pazienti (vedere i paragrafi «Farmacodinamica» e «Farmacocinetia»).
Compromissione epatica. Non è raccomandato alcun aggiustamento posologico nei pazienti con compromissione epatica lieve o moderata. Victoza® non è raccomandato nei pazienti con grave compromissione epatica (vedere il paragrafo «Farmacocinetica»).
Modalità di somministrazione
Victoza® non deve essere somministrato per via endovenosa né intramuscolare.
Il medicinale Victoza® va somministrato una volta al giorno, in qualsiasi momento, indipendentemente dai pasti. Può essere iniettato per via sottocutanea nell’area dell’addome, della coscia o della spalla. Il sito e l’orario delle iniezioni possono essere modificati senza necessità di aggiustamento della dose. Tuttavia, si raccomanda di somministrare il medicinale Victoza® approssimativamente alla stessa ora del giorno, scegliendo il momento più comodo. Ulteriori raccomandazioni sulla somministrazione sono riportate nelle istruzioni per l’uso della penna preriempita per Victoza®.
Al fine di ridurre il rischio di deposito di amiloide nel sito di iniezione, è necessario cambiare sempre il sito di iniezione (vedere il paragrafo «Effetti indesiderati»).
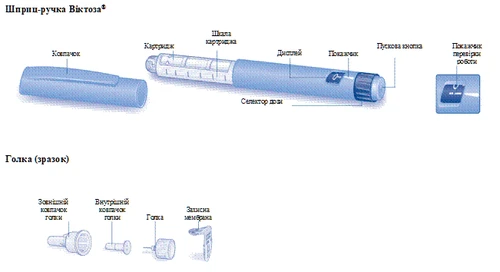
Istruzioni per l’uso della penna preriempita Victoza®
Si prega di leggere attentamente queste istruzioni prima di utilizzare la penna preriempita Victoza®.
La penna preriempita Victoza® contiene 18 mg di liraglutide. È possibile scegliere le seguenti dosi: 0,6 mg, 1,2 mg e 1,8 mg.
La penna preriempita è destinata all’uso con aghi monouso NovoFine® o NovoTwist® con lunghezza fino a 8 mm e calibro fino a 32G.
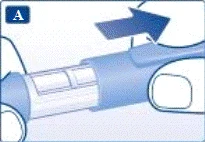
| Preparazione della penna preriempita per l'uso Verificare il nome e l'etichetta colorata della penna preriempita per assicurarsi che contenga liraglutide. L'amministrazione accidentale di un altro medicinale potrebbe causare gravi danni. |
||
|
|
||
|
| ||
|
| |
| |

Per ogni iniezione utilizzare sempre un nuovo ago. Ciò riduce il rischio di contaminazione, infezione, fuoriuscita di liraglutide, otturazione del foro dell'ago e inaccuratezza nella somministrazione della dose.
Fare attenzione: non piegare né danneggiare l'ago.
Per ridurre al minimo il rischio di punture accidentali con l'ago, non rimettere mai il cappuccio interno dell'ago precedentemente rimosso. | | | | Cura della penna preriempita * Non tentare di ripararla né di smontarla. * Evitare il contatto della penna preriempita con polvere, sporcizia e qualsiasi liquido. * Pulire la penna preriempita con un panno inumidito con un detergente delicato. * Non tentare di lavarla, immergerla in acqua né lubrificarla – tutto ciò potrebbe danneggiare la penna preriempita. | | | |
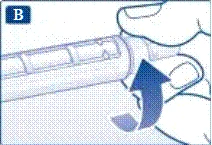
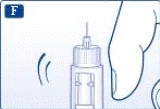
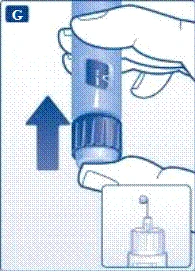
Informazioni importanti * Non dare la propria penna preriempita ad altre persone. * Conservare la penna preriempita Victoza® in un luogo sicuro, fuori dalla portata altrui (soprattutto dei bambini). | | | | Verifica del corretto funzionamento della penna preriempita Prima di utilizzare per la prima volta una nuova penna preriempita, verificare che il farmaco fuoriesca attraverso l'ago. Se la penna preriempita è già stata utilizzata in precedenza, passare alla sezione «Impostazione della dose», punto N. | | | |

| |
| |
| |
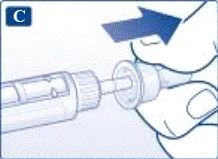
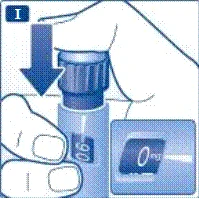
Se si lascia cadere la penna preriempita su una superficie dura o si ritiene che presenti dei malfunzionamenti, collegare ogni volta un nuovo ago monouso e verificare il corretto funzionamento prima della somministrazione della dose. | | | | Impostazione della dose Assicurarsi che il selettore della dose sia impostato sulla posizione «0». | | | |

| |
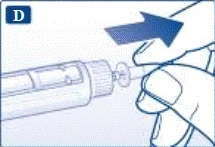
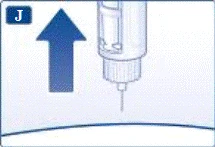
Non tentare di impostare dosi diverse da 0,6 mg, 1,2 mg o 1,8 mg. I numeri sul display devono corrispondere esattamente all'indicatore per garantire la dose corretta. Ruotando il selettio della dose si sentono dei clic. Non fare riferimento al numero di clic durante l’impostazione della dose. Non utilizzare la scala del contenitore per determinare la dose di liraglutide da somministrare, poiché non è sufficientemente precisa. | | | | Somministrazione dell’iniezione Seguendo le istruzioni del medico o dell’infermiere riguardo alla tecnica di iniezione, inserire l’ago nella cute. Procedere quindi come segue. | | | |
| |
| |
| |
| |
Rimuovere sempre l’ago dopo ogni iniezione e conservare la penna preriempita Victoza® senza ago.
Ciò previene contaminazione, infezione o fuoriuscita di liraglutide.
È necessario prestare particolare attenzione durante lo smaltimento degli aghi usati per evitare di ferirsi. | | |
Bambini. Non è necessaria alcuna modifica della dose per adolescenti e bambini di età pari o superiore a 10 anni. Non sono disponibili dati sull’uso nei bambini di età inferiore a 10 anni (vedere le sezioni «Farmacodinamica» e «Farmacocinetica»).
Sovradosaggio.
Negli studi clinici e nelle segnalazioni successive all’immissione in commercio del medicinale Victoza®, sono stati riportati casi di superamento della dose di mantenimento raccomandata fino a 40 volte (72 mg). Le reazioni segnalate comprendevano nausea intensa, vomito, diarrea e grave ipoglicemia.
In caso di sovradosaggio, si deve effettuare un trattamento di supporto in base ai segni e sintomi clinici presenti nel paziente. È necessario monitorare lo stato clinico del paziente per individuare precocemente eventuali segni di disidratazione e controllare il livello di glucosio nel sangue.
Effetti indesiderati.
In cinque lunghi studi clinici di fase 3a, oltre 2500 pazienti adulti hanno ricevuto Victoza® da solo o in combinazione con metformina, gliburide (con o senza metformina), sulfoniluree (con o senza metformina) o metformina + rosiglitazone.
Gli effetti indesiderati più comuni osservati negli studi clinici sono stati disturbi gastrointestinali, tra cui nausea e diarrea molto frequenti, vomito, costipazione, dolore addominale e dispepsia frequenti. I disturbi gastrointestinali si verificano più spesso all’inizio del trattamento, ma generalmente diminuiscono in intensità nel giro di alcuni giorni o settimane con la prosecuzione della terapia. Cefalea e nasofaringite sono stati anch’essi effetti comuni. Inoltre, si è verificata ipoglicemia, molto frequente quando Victoza® è stato utilizzato in combinazione con sulfoniluree. Gli episodi di ipoglicemia grave si sono verificati principalmente durante la terapia combinata con sulfoniluree.
Di seguito è riportato un elenco di effetti indesiderati osservati durante gli studi clinici di lunga durata di fase 3a e nello studio LEADER® (studio cardiovascolare a lungo termine), nonché sulla base di segnalazioni spontanee ricevute dopo l’immissione in commercio del medicinale. La frequenza di tutti gli effetti indesiderati è stata calcolata in base alla frequenza di insorgenza osservata negli studi clinici di fase 3a.
La frequenza degli effetti indesiderati è stata valutata secondo la seguente scala: molto frequente (≥ 1/10), frequente (da ≥ 1/100 a < 1/10), non frequente (da ≥ 1/1000 a < 1/100), raro (da ≥ 1/10000 a < 1/1000), molto raro (< 1/10000), non noto (non può essere stimato sulla base dei dati disponibili). All’interno di ogni gruppo, gli effetti indesiderati sono elencati in ordine decrescente di gravità.
Disturbi del metabolismo e della nutrizione: frequenti – ipoglicemia, anoressia, riduzione dell’appetito; non frequenti – disidratazione*.
Disturbi del sistema nervoso: frequenti – cefalea, capogiri; non frequenti – disgeusia.
Disturbi gastrointestinali: molto frequenti – nausea, diarrea; frequenti – vomito, dispepsia, dolore nell’area superiore dell’addome, costipazione, gastrite, meteorismo, distensione addominale, malattia da reflusso gastroesofageo, disagio gastrico, dolore dentale; non frequenti – rallentamento dello svuotamento gastrico; rari – ostruzione intestinale; molto rari – pancreatite (inclusa pancreatite necrotizzante).
Disturbi del sistema cardiaco: frequenti – aumento della frequenza cardiaca (FC).
Disturbi del sistema immunitario: rari – reazioni anafilattiche.
Infezioni e infestazioni: frequenti – nasofaringite, bronchite.
Disturbi generali e condizioni al sito di iniezione: frequenti – affaticamento, reazioni nel sito di iniezione; non frequenti – malessere.
Disturbi renali e delle vie urinarie: non frequenti – insufficienza renale acuta, alterazione della funzionalità renale.
Disturbi della cute e del tessuto sottocutaneo: frequenti – eruzioni cutanee; non frequenti – orticaria, prurito; non noto – amiloidosi cutanea.
Disturbi epatici e delle vie biliari: non frequenti – calcolosi biliare, colecistite.
Esami di laboratorio: frequenti – aumento del livello di lipasi*, aumento del livello di amilasi*
*Dati da studi clinici di fase 3b e 4, in cui questi parametri sono stati misurati.
Descrizione di specifici effetti indesiderati
Durante lo studio clinico di monoterapia con Victoza®, l’incidenza di ipoglicemia nei pazienti trattati con Victoza® è risultata inferiore rispetto a quella nei pazienti trattati con il farmaco attivo di confronto (glimiperide). Gli effetti indesiderati più comuni sono stati disturbi gastrointestinali, infezioni e infestazioni.
Ipoglicemia
Nella maggior parte dei casi registrati negli studi clinici, l’ipoglicemia confermata è stata di lieve entità. Nessun caso di ipoglicemia grave è stato osservato con la monoterapia a base di liraglutide. L’ipoglicemia grave è rara e si verifica principalmente durante il trattamento combinato di liraglutide e sulfoniluree (0,02 casi per 100 pazienti-anno). Casi molto rari di ipoglicemia (0,001 casi per 100 pazienti-anno) si sono verificati con il trattamento a base di liraglutide in combinazione con altri antidiabetici orali (cioè non con sulfoniluree). Il rischio di ipoglicemia con l’uso combinato di liraglutide e insulina basale è trascurabile (1,0 caso per 100 pazienti-anno, vedere sezione «Proprietà farmacologiche»).
Nello studio LEADER®, gli episodi di ipoglicemia grave sono stati riportati meno frequentemente con liraglutide rispetto al placebo (1,0 vs 1,5 casi per 100 pazienti-anno, rapporto di frequenza stimato 0,69 [0,51-0,93]) (vedere sezione «Farmacodinamica»). Nei pazienti che assumevano insuline miste all’inizio dello studio e per almeno le successive 26 settimane, l’incidenza di episodi di ipoglicemia grave con liraglutide e placebo è stata di 2,2 casi per 100 pazienti-anno.
Disturbi gastrointestinali
Con la terapia combinata di Victoza® e metformina, nausea si è verificata almeno una volta nel 20,7% dei pazienti e diarrea nel 12,6%. Con la terapia combinata di Victoza® e sulfoniluree, nausea si è verificata almeno una volta nel 9,1% dei pazienti e diarrea nel 7,9%. La maggior parte dei casi era di gravità lieve o moderata e mostrava un andamento dose-dipendente. Nei pazienti che hanno manifestato nausea all’inizio del trattamento, la frequenza e la gravità tendevano a diminuire con la prosecuzione della terapia.
Nei pazienti di età pari o superiore a 70 anni, il trattamento con liraglutide può essere associato a una maggiore frequenza di disturbi gastrointestinali.
Nei pazienti con lieve o moderata compromissione della funzionalità renale (clearance della creatinina ≤ 60–90 ml/min e 30–59 ml/min, rispettivamente), il trattamento con liraglutide può essere associato a una maggiore frequenza di disturbi gastrointestinali.
Calcolosi biliare e colecistite
Negli studi clinici controllati di lunga durata di fase 3a con liraglutide, sono stati registrati pochi casi di calcolosi biliare (0,4%) e colecistite (0,1%). Nello studio LEADER®, l’incidenza di calcolosi biliare e colecistite è stata rispettivamente del 1,5% e dell’1,1% con liraglutide e dell’1,1% e dello 0,7% con placebo (vedere sezione «Farmacodinamica»).
Interruzione del trattamento
Negli studi controllati di lunga durata (26 settimane o più), l’incidenza di interruzione del trattamento con liraglutide a causa di effetti indesiderati è stata del 7,8%, rispetto al 3,4% per il farmaco di confronto. Le cause più comuni di interruzione nei pazienti trattati con liraglutide sono state nausea (2,8%) e vomito (1,5%).
Reazioni nel sito di iniezione
Negli studi controllati di lunga durata (26 settimane o più), reazioni nel sito di iniezione di Victoza® sono state osservate in circa il 2% dei pazienti. Tali reazioni erano generalmente lievi.
Pancreatite
Negli studi clinici controllati di lunga durata di fase 3, durante il trattamento con Victoza®, sono stati registrati alcuni casi (< 0,2%) di pancreatite acuta. Casi di pancreatite sono stati riportati anche in segnalazioni spontanee dopo l’immissione in commercio del medicinale. Nello studio LEADER®, l’incidenza di pancreatite acuta confermata da valutazione esperta è stata dello 0,4% con liraglutide e dello 0,5% con placebo, rispettivamente (vedere sezione «Avvertenze particolari e precauzioni d’impiego» e «Farmacodinamica»).
Reazioni allergiche
Dopo l’immissione in commercio di Victoza®, sono state riportate reazioni allergiche, tra cui orticaria, eruzioni cutanee e prurito. Sono stati riportati anche alcuni casi di reazioni anafilattiche con sintomi aggiuntivi come ipotensione, palpitazioni, dispnea e angioedema. Durante l’intero periodo degli studi clinici di lunga durata con Victoza®, è stato osservato raramente (0,05%) angioedema.
Amiloidosi cutanea
L’amiloide cutanea può svilupparsi nel sito di iniezione (vedere sezione «Modalità di somministrazione e dosaggio»).
Bambini
In generale, la frequenza, il tipo e la gravità degli effetti indesiderati nei bambini e adolescenti di età pari o superiore a 10 anni sono simili a quelli negli adulti. L’incidenza di episodi confermati di ipoglicemia è risultata più elevata con liraglutide (0,58 casi/paziente-anno) rispetto al placebo (0,29 casi/paziente-anno). Nei pazienti trattati con insulina prima dell’insorgenza di un episodio confermato di ipoglicemia, l’incidenza è risultata più elevata con liraglutide (1,82 casi/paziente-anno) rispetto al placebo (0,91 casi/paziente-anno). Nel gruppo trattato con liraglutide, non sono stati registrati episodi gravi di ipoglicemia.
Segnalazione di effetti indesiderati e mancata risposta al medicinale
La segnalazione di effetti indesiderati dopo la commercializzazione del medicinale è di fondamentale importanza. Permette un monitoraggio continuo del rapporto beneficio/rischio del farmaco. I professionisti sanitari, i farmacisti, i pazienti o i loro rappresentanti legali devono segnalare tutti i casi sospetti di effetti indesiderati e la mancata efficacia del medicinale attraverso il Sistema Informatizzato Automatizzato per la Farmacovigilanza al seguente indirizzo: https://aisf.dec.gov.ua.
Periodo di validità. 30 mesi.
Dopo il primo utilizzo – 1 mese.
Condizioni di conservazione.
Conservare in un luogo inaccessibile ai bambini. Conservare in frigorifero (2°C – 8°C), lontano dal congelatore. Non congelare.
Dopo il primo utilizzo, conservare a una temperatura inferiore a 30°C o in frigorifero (2°C – 8°C). Non congelare.
Per proteggere dal contatto con la luce, conservare la penna preriempita con il cappuccio chiuso.
Incompatibilità.
L’aggiunta di qualsiasi sostanza a Victoza® può causare degradazione del liraglutide. In assenza di studi di compatibilità, questo medicinale non deve essere miscelato con altri medicinali.
Confezionamento. La penna preriempita multidose monouso contiene una cartuccia da 3 ml realizzata in vetro (tipo 1), chiusa da un lato con un pistone in gomma bromobutile e dall’altro con un disco laminato in gomma bromobutile/polisoprene. La penna è realizzata in poliolefina e poliacetale.
Ogni penna contiene 3 ml di soluzione, sufficiente per 30 dosi da 0,6 mg, 15 dosi da 1,2 mg o 10 dosi da 1,8 mg.
La confezione contiene 1 o 2 penne preriempite.
Categoria di prescrizione. Sotto prescrizione medica.
Produttore. A/S Novo Nordisk, Danimarca / Novo Nordisk A/S, Denmark.
Indirizzo del produttore e sede operativa.
A/S Novo Nordisk, Novo Allé, 2880, Bagsværd, Danimarca. Tel. + 45 4444 8888 /
Novo Nordisk A/S, Novo Alle, 2880, Bagsvaerd, Denmark. Tel. + 45 4444 8888.