Vabismou
Ukraina
Spis treści
ULOTKA DO ZASTOSOWANIA MEDYCZNEGO LEKU Vabysmo (Vabysmo®)
Skład:
substancja czynna: faricimab;
1 fiolka zawiera 28,8 mg faricimabu w 0,24 ml roztworu, co odpowiada stężeniu 120 mg/ml;
dawka pojedyncza – 6 mg/0,05 ml;
substancje pomocnicze: L-histydyna, kwas octowy 30%, L-metionina, chlorek sodu, sacharoza, polisorbat 20, woda do wstrzykiwań.
Postać farmaceutyczna. Roztwór do wstrzykiwań.
Główne właściwości fizykochemiczne: przejrzysta lub odrobinę mętna ciecz, bezbarwna lub lekko brązowozłota.
Grupa farmakoterapeutyczna. Leki działające na narządy zmysłów. Leki stosowane w okulistyce. Leki stosowane w chorobach naczyniowych oka. Środki antyneowaskularyzacyjne. Faricimab.
Kod ATC S01L A09.
Właściwości farmakologiczne.
Farmakodynamika.
Mechanizm działania
Faricimab to ludzkie przeciwciało dwuspecyficzne klasy IgG1, działające poprzez hamowanie dwóch oddzielnych szlaków sygnalizacyjnych poprzez neutralizację zarówno Ang-2 (angiopoetyny-2), jak i czynnika wzrostu śródbłonka naczyń A (VEGF-A). Ang-2 powoduje niestabilność naczyń krwionośnych poprzez stymulację destabilizacji śródbłonka, utratę pericytów i patologiczny angiogenezę, co zwiększa w ten sposób wyciek płynu przez naczynia i stan zapalny. Ang-2 czyni również naczynia krwionośne bardziej wrażliwe na działanie VEGF-A, co prowadzi do dalszej destabilizacji naczyń. Ang-2 i VEGF-A działają synergistycznie, zwiększając przepuszczalność naczyń i stymulując neowaskularyzację. Poprzez podwójne hamowanie Ang-2 i VEGF-A faricimab zmniejsza przepuszczalność naczyń i stan zapalny, hamuje patologiczny angiogenezę i przywraca stabilność naczyniową.
W sześciu badaniach klinicznych fazy III opisanych poniżej stwierdzono supresję średniego stężenia wolnego ANG-2 i wolnego VEGF-A w narządach wzroku począwszy od dnia 7 w porównaniu z poziomem wyjściowym.
Neowaskularna (eksudatywna) postać zwyrodnienia plamki związanego z wiekiem (nAMD)
Podobne do obserwowanego przy stosowaniu afliberceptu zmniejszenie średniej grubości centralnego pola fowea (CST – centralna grubość siatkówki), począwszy od poziomu wyjściowego do 48 tygodnia podczas leczenia lekiem Vabismo, było obserwowane. Średnie zmniejszenie CST od poziomu wyjściowego do wizyt oceny punktu końcowego pierwotnego (uśrednione na tygodniach 40, 44 i 48) wynosiło -137 μm i -137 μm dla leku Vabismo z interwałem podania co 8 tygodni, 12 tygodni (co 12 tygodni) lub 16 tygodni (co 16 tygodni) w porównaniu do -129 μm i -131 μm przy stosowaniu afliberceptu w badaniach TENAYA i LUCERNE odpowiednio. Takie średnie zmniejszenie CST utrzymywało się przez rok 2. Obserwowano porównywalny efekt leków Vabismo i afliberceptu w zakresie zmniejszenia płynu wewnątrzsiatkórkowego (IRF), płynu podsiatkórkowego (SRF) oraz odwarstwienia nabłonka barwnikowego (PED). Podczas wizyt oceny punktu końcowego pierwotnego w badaniach TENAYA i LUCERNE odsetek pacjentów bez IRF wynosił odpowiednio 76–82 % i 78–85 % przy leczeniu lekiem Vabismo w porównaniu do 74–85 % i 78–84 % przy leczeniu afliberceptem. Odsetek pacjentów bez SRF w obu badaniach wynosił 70–79 % i 66–78 % przy leczeniu lekiem Vabismo w porównaniu do 66–78 % i 62–76 % przy leczeniu afliberceptem. Odsetek pacjentów bez PED w obu badaniach wynosił 3–8 % i 3–6 % przy leczeniu lekiem Vabismo w porównaniu do 8–10 % i 7–9 % przy leczeniu afliberceptem. Takie zmniejszenie IRF, SRF i PED utrzymywało się przez rok 2 (tygodnie 104–108).
W 48 tygodniu obserwowano porównywalne zmiany od poziomu wyjściowego wskaźnika całkowitego obszaru uszkodzenia spowodowanego neowaskularyzacją naczyniówki (CNV) oraz porównywalne zmniejszenie strefy wycieku CNV z ekstrawazacją krwi i płynu w obu badaniach u pacjentów leczonych lekiem Vabismo i afliberceptem.
Cukrzycowy obrzęk plamki (DME)
Zmniejszenie średniej wartości CST od poziomu wyjściowego obserwowane w badaniach YOSEMITE i RHINE było wyższe liczbowo u pacjentów leczonych lekiem Vabismo z interwałem co 8 tygodni oraz z dostosowanym interwałem do co 16 tygodni w porównaniu z pacjentami leczonymi afliberceptem co 8 tygodni w okresie od 4 do 100 tygodnia. W obu badaniach większa część pacjentów w grupach leku Vabismo osiągnęła brak IRF i brak DME (określony jako osiągnięcie CST poniżej 325 μm w badaniu tomografią koherentną optyczną [OCT]) w porównaniu z grupą afliberceptu. Porównywalne zmniejszenie SRF w czasie obserwowano w grupach leczonych lekiem Vabismo i afliberceptem w obu badaniach. Średnie zmniejszenie CST od poziomu wyjściowego do wizyt oceny punktu końcowego pierwotnego (uśrednione na tygodniach 48, 52 i 56) w badaniu YOSEMITE wyniosło 207 μm i 197 μm u pacjentów leczonych lekiem Vabismo co 8 tygodni oraz z dostosowanym interwałem do co 16 tygodni w porównaniu do 170 μm u pacjentów leczonych afliberceptem co 8 tygodni; w badaniu RHINE wyniki wynosiły odpowiednio 196 μm, 188 μm i 170 μm. Takie zmniejszenie średniej wartości CST utrzymywało się do 2 roku. Odsetek pacjentów bez DME podczas wizyt oceny punktu końcowego pierwotnego (wartość minimalna/maksymalna) w badaniu YOSEMITE wyniósł 77–87 % i 80–82 % w grupie leczenia lekiem Vabismo co 8 tygodni i z dostosowanym interwałem do co 16 tygodni odpowiednio, w porównaniu do 64–71 % w grupie leczenia afliberceptem co 8 tygodni; w badaniu RHINE wyniki wyniosły odpowiednio 85–90 %, 83–87 % i 71–77 %. Wyniki te utrzymywały się do 2 roku.
W badaniu YOSEMITE odsetek pacjentów bez IRF podczas wizyt oceny punktu końcowego pierwotnego (uśrednione na tygodniach 48, 52 i 56) wyniósł 42–48 % i 34–43 % w grupie leczenia lekiem Vabismo co 8 tygodni i z dostosowanym interwałem do co 16 tygodni odpowiednio, w porównaniu do 22–25 % w grupie leczenia afliberceptem co 8 tygodni; w badaniu RHINE wyniki wyniosły odpowiednio 39–43 %, 33–41 % i 23–29 %. Wyniki te utrzymywały się do 2 roku.
Zator gałęzi żyły siatkówki (BRVO) i zator środkowej żyły siatkówki (CRVO)
W badaniach fazy III u pacjentów z zatorem gałęzi żyły siatkówki (BRVO; BALATON) oraz środkowym/półsiatkówkowym zatorem żyły siatkówki (CRVO/HRVO; COMINO) obserwowano zmniejszenie średniej wartości CST od poziomu wyjściowego do 24 tygodnia przy stosowaniu Vabismo co 4 tygodnie i było porównywalne z efektem przy stosowaniu afliberceptu co 4 tygodnie. Średnie zmniejszenie CST od poziomu wyjściowego do 24 tygodnia wyniosło 311,4 μm przy stosowaniu Vabismo w porównaniu do 304,4 μm przy stosowaniu afliberceptu oraz 461,6 μm przy stosowaniu Vabismo co 4 tygodnie w porównaniu do 448,8 μm przy stosowaniu afliberceptu co 4 tygodnie w badaniach BALATON i COMINO odpowiednio. Zmniejszenie CST utrzymywało się do 72 tygodnia, kiedy pacjenci przeszli na dostosowany schemat dawkowania Vabismo – co 16 tygodni.
Porównywalna część pacjentów w grupach stosowania Vabismo i afliberceptu osiągnęła brak IRF, brak SRF oraz brak obrzęku plamki (określonego jako osiągnięcie wartości CST poniżej 325 μm) w okresie do 24 tygodnia w obu badaniach. Wyniki te utrzymywały się do 72 tygodnia, kiedy pacjenci przeszli na dostosowany schemat dawkowania Vabismo – co 16 tygodni.
W badaniu BALATON w 24 tygodniu odsetek pacjentów bez obrzęku plamki wyniósł 95,3 % w grupie Vabismo co 4 tygodnie w porównaniu do 93,9 % w grupie afliberceptu co 4 tygodnie; odsetek pacjentów bez IRF wyniósł 72,5 % w grupie Vabismo co 4 tygodnie w porównaniu do 66 % w grupie afliberceptu co 4 tygodnie. Odsetek pacjentów bez SRF wyniósł 91,3 % w grupie Vabismo co 4 tygodnie w porównaniu do 90,3 % w grupie afliberceptu co 4 tygodnie.
W badaniu COMINO w 24 tygodniu odsetek pacjentów bez obrzęku plamki wyniósł 93,7 % w grupie Vabismo co 4 tygodnie w porównaniu do 92 % w grupie afliberceptu co 4 tygodnie. Odsetek pacjentów bez IRF wyniósł 76,2 % w grupie Vabismo co 4 tygodnie w porównaniu do 70,8 % w grupie afliberceptu co 4 tygodnie; odsetek pacjentów bez SRF wyniósł 96,4 % w grupie Vabismo co 4 tygodnie w porównaniu do 93,4 % w grupie afliberceptu co 4 tygodnie.
Skuteczność kliniczna i bezpieczeństwo
Leczenie nAMD
Skuteczność i bezpieczeństwo stosowania faricimabu w porównaniu z leczeniem anty-VEGF badano w dwóch randomizowanych (1:1), wieloośrodkowych, podwójnie ślepych badaniach z dwiema grupami (TENAYA i LUCERNE) z udziałem pacjentów z nAMD. Leczenie (faricimab, 6 mg, lub aflibercept, 2 mg) prowadzono przez iniekcje wewnątrzwitowe, początkowo z 4-tygodniowymi interwałami. W grupie afliberceptu po 3 początkowych iniekcjach interwał między dawkami wynosił 8 tygodni do końca badania (co 8 tygodni). W grupie faricimabu interwał między dawkami był indywidualnie dostosowywany po 4 początkowych dawkach. Końcowy (ustalony) interwał między dawkami wynosił 8 tygodni (co 8 tygodni), 12 tygodni (co 12 tygodni) lub maksymalnie 16 tygodni (co 16 tygodni), w zależności od ustalonej protokołem zmiany CST, określonej na podstawie tomografii koherentnej optycznej spektralnej oraz/lub zmiany BCVA, określonej na podstawie liczby liter w badaniu skuteczności wczesnego leczenia retinopatii cukrzycowej (ETDRS), a także od oceny klinicznej lekarza obecności/braku krwotoku do plamki w 20 i 24 tygodniu. Począwszy od tygodnia 60, pacjenci z grupy leczenia Vabismo byli przenoszeni na elastyczny schemat dawkowania, w którym interwał między dawkami mógł być wydłużany o kroki do 4 tygodni (do co 16 tygodni) lub skracany o kroki do 8 tygodni (do co 8 tygodni) na podstawie zautomatyzowanej obiektywnej oceny wcześniej określonych kryteriów aktywności choroby pod względem wizualnym i anatomicznym. Pacjenci w grupie afliberceptu kontynuowali schemat dawkowania co 8 tygodni przez cały okres badania. Obie studia trwały 112 tygodni.
Badania obejmowały łącznie 1329 wcześniej nieleczone pacjentów, z których 1135 (85 %) ukończyło badanie do tygodnia 112. Łącznie 1326 pacjentów otrzymało co najmniej jedną dawkę (w tym 664 pacjentów z grupy faricimabu). Średni wiek [zakres wieku] populacji badawczej wynosił 75,9 roku [od 50 do 99 lat]. Głównym punktem końcowym skuteczności była średnia zmiana BCVA od poziomu wyjściowego (na podstawie średniej wartości w tygodniach 40, 44 i 48), określona za pomocą tablicy liter ETDRS z odległości 4 metry. W obu badaniach potwierdzono główną hipotezę (nie mniejszą skuteczność): pacjenci leczeni lekiem Vabismo z interwałem do co 16 tygodni oraz pacjenci leczeni afliberceptem co 8 tygodni wykazali porównywalną średnią zmianę BCVA od odpowiedniego poziomu wyjściowego po 1 roku. Istotna poprawa wzroku od stanu wyjściowego była obserwowana do tygodnia 112 w obu grupach leczenia. Szczegółowe wyniki obu badań przedstawiono w tabelach 1 i 2 oraz na rysunku 1.
Odsetek pacjentów w grupach z dostosowanymi interwałami podania leku w 48 tygodniu w badaniach TENAYA i LUCERNE wyniósł odpowiednio: co 16 tygodni – 46 %, 45 %; co 12 tygodni – 34 %, 33 %; co 8 tygodni – 20 %, 22 %.
Odsetek pacjentów w grupach z dostosowanymi interwałami podania leku w 112 tygodniu w badaniach TENAYA i LUCERNE wyniósł odpowiednio: co 16 tygodni – 59 %, 67 %; co 12 tygodni – 15 %, 14 %; co 8 tygodni – 26 %, 19 %.
Tabela 1
Wyniki dotyczące skuteczności podczas wizyt oceny punktu końcowego pierwotnegoa po 2 latachb w badaniu TENAYA
| Wyniki dotyczące skuteczności |
TENAYA |
|||
| Rok 1 |
Rok 2 |
|||
| Vabismou, interwał podawania do co 16 tygodni, N = 334 |
Aflibercept co 8 tygodni, N = 337 |
Vabismou, interwał podawania do co 16 tygodni, N = 334 |
Aflibercept co 8 tygodni, N = 337 |
|
| Średnia zmiana BCVA od wartości wyjściowej wynosząca liczbę liter według ETDRS (95 % CI) |
5,8 (4,6, 7,1) |
5,1 (3,9, 6,4) |
3,7 (2,1, 5,4) |
3,3 (1,7, 4,9) |
| Częstość występowania pacjentów z poprawą rozpoznawania o ≥ 15 liter od wartości wyjściowej (częstość ważona wg CMH, 95 % CI) |
20 % (15,6 %, 24,4 %) |
15,7 % (11,9 %, 19,6 %) |
22,5 % (17,8 %, 27,2 %) |
16,9 % (12,7 %, 21,1 %) |
| Częstość występowania pacjentów, którzy uniknęli pogorszenia rozpoznawania o ≥ 15 liter od wartości wyjściowej (częstość ważona wg CMH, 95 % CI) |
95,4 % (93 %, 97,7 %) |
94,1 % (91,5 %, 96,7 %) |
92,1 % (89,1 %, 95,1 %) |
88,6 % (85,1 %, 92,2 %) |
a Średnia tygodni 40, 44 i 48.
b Średnia tygodni 104, 108, 112.
BVCA – najlepsza skorygowana ostrość wzroku; ETDRS – badanie skuteczności wczesnego leczenia retinopatii cukrzycowej; CI – przedział ufności; CMH – test Cochrana-Mantela-Haenszela, test statystyczny generujący oszacowanie związku z wynikiem binarnym, stosowany do oceny zmiennych kategorycznych.
Tabela 2
Wyniki dotyczące skuteczności podczas wizyt oceny pierwotnego punktu końcowegoa po 2 latachb w badaniu LUCERNE
| Wyniki dotyczące skuteczności |
LUCERNE |
|||
| Rok 1 |
Rok 2 |
|||
| Vabismou, interwał podawania do co 16 tygodni, N = 331 |
Aflibercept co 8 tygodni, N = 327 |
Vabismou, interwał podawania do co 16 tygodni, N = 331 |
Aflibercept co 8 tygodni, N = 327 |
|
| Średnia zmiana BCVA od wartości wyjściowej wynikająca z liczby liter według ETDRS (95 % CI) |
6,6 (5,3, 7,1) |
6,6 (5,3, 7,8) |
5,0 (3,4, 6,6) |
5,2 (3,6, 6,8) |
| Częstość pacjentów ze zwiększeniem widzenia o ≥ 15 liter od wartości wyjściowej (częstość ważona według CMH, 95 % CI) |
20,2 % (15,9 %, 24,6 %) |
22,2 % (17,7 %, 26,8 %) |
22,4 % (17,8 %, 27,1 %) |
21,3 % (16,8 %, 25,9 %) |
| Częstość pacjentów, którzy uniknęli pogorszenia widzenia o ≥ 15 liter od wartości wyjściowej (częstość ważona według CMH, 95 % CI) |
95,8 % (93,6 %, 98,0 %) |
97,3 % (95,5 %, 99,1 %) |
92,9 % (90,1 %, 95,8 %) |
93,2 % (90,2 %, 96,2 %) |
a Średnia tygodni 40, 44 i 48.
b Średnia tygodni 104, 108, 112.
BVCA – najlepsza skorygowana ostrość wzroku; ETDRS – badanie skuteczności wczesnego leczenia retinopatii cukrzycowej; CI – przedział ufności; CMH – test Cochrana-Mantela-Haenszela, test statystyczny generujący oszacowanie związku z wynikiem binarnym, stosowany do oceny zmiennych kategorycznych.
| Skorygowana średnia zmiana od punktu wyjściowego |
|
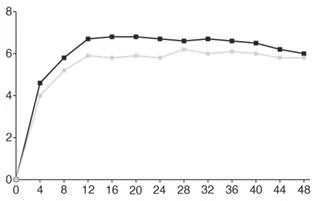
Tygodnie

Faricyklumab, 6 mg, co 16 tygodni (N = 665)

Aflibercept, 2 mg, co 8 tygodni (N = 664)
Rys. 1. Połączone badania III fazy dotyczące AMD związanej z wiekiem (TENAYA i LUCERNE): wykres zmiany BCVA od wartości wyjściowej w badanym oku do 112 tygodnia, metoda MMRM (pierwotna ocena) (populacja ITT)
W obu badaniach TENAYA i LUCERNE poprawa BCVA oraz CST od wartości wyjściowej w 60. tygodniu była porównywalna w obu grupach leczonych i zgodna z obserwacjami w 48. tygodniu.
Wyniki dotyczące skuteczności we wszystkich ocenianych podgrupach pacjentów (np. według wieku, płci, pochodzenia etnicznego, ostrości wzroku na początku, typu zmiany, wielkości zmiany) w każdym badaniu oraz w analizie łącznej były zgodne z wynikami w populacji ogólnej.
W obu badaniach lek Vabismo podawany w odstępach do 16 tygodni wykazał klinicznie istotną poprawę od wartości wyjściowej do 48. tygodnia ogólnego wyniku kwestionariusza oceny funkcji wzroku Narodowego Instytutu Oftalmologii USA (NEI VFQ-25), która była porównywalna z poprawą uzyskaną przy stosowaniu afliberceptu co 8 tygodni. U pacjentów w grupach leczonych lekiem Vabismo w badaniach TENAYA i LUCERNE osiągnięto poprawę ogólnego wyniku NEI VFQ-25 o ≥ 4 punkty od wartości wyjściowej w 48. tygodniu.
Leczenie DMP
Bezpieczeństwo i skuteczność stosowania faricyklumabu w porównaniu z leczeniem anty-VEGF oceniano w dwóch randomizowanych (1:1:1), wieloośrodkowych, podwójnie ślepych badaniach trzech grup (YOSEMITE i RHINE) trwających 2 lata, z udziałem pacjentów z DMP. Pacjenci w trzech badanych grupach otrzymywali wstrzykiwania do ciała szklistego faricyklumabu w dawce 6 mg co 8 tygodni (po 6 miesięcznych wstrzyknięciach na początku leczenia), faricyklumab 6 mg w indywidualnym odstępie między wstrzyknięciami do 16 tygodni (po 4 miesięcznych wstrzyknięciach na początku leczenia) lub aflibercept 2 mg co 8 tygodni (po 5 miesięcznych wstrzyknięciach na początku leczenia).
W grupie faricyklumabu z wydłużonym odstępem między wstrzyknięciami do 16 tygodni dawkowanie leku prowadzono według ujednoliconego schematu leczenia z wydłużaniem odstępów między dawkami. W zależności od zmiany CST w badaniu OCT oraz/lub zmiany BCVA określonej według liczby liter w ETDRS, indywidualny odstęp między wstrzyknięciami w grupie faricyklumabu mógł być wydłużony o 4 tygodnie lub skrócony o 4 lub 8 tygodni podczas każdej wizyty wstrzyknięcia (patrz sekcja „Sposób stosowania i dawki”).
Do badań włączono łącznie 1891 pacjentów (z których około 94 % miało cukrzycę typu 2), 1622 (85,8 %) pacjentów ukończyło udział w badaniu do 100. tygodnia. Łącznie 1887 pacjentów otrzymało co najmniej jedną dawkę do 56. tygodnia (1262 pacjentów otrzymało lek Vabismo). Średni wiek [zakres wieku] badanych pacjentów wynosił 62,2 roku [od 24 do 91 roku]. Populacja badawcza obejmowała pacjentów, którzy wcześniej nie otrzymywali leczenia anty-VEGF (78 %) oraz pacjentów, którzy wcześniej otrzymywali terapię anty-VEGF (22 %).
Pierwotnym punktem końcowym skuteczności była średnia zmiana wyniku BCVA od wartości wyjściowej do końca pierwszego roku (średnio 48., 52. i 56. tydzień), określona za pomocą tablicy liter ETDRS z odległości 4 metry. W obu badaniach potwierdzono pierwotną hipotezę (niegorszą skuteczność) dla obu grup leczonych: pacjenci otrzymujący lek Vabismo co 8 tygodni oraz pacjenci otrzymujący lek Vabismo w trybie wydłużonego odstępu między wstrzyknięciami do 16 tygodni osiągnęli porównywalną średnią zmianę wyniku BCVA od wartości wyjściowej jak pacjenci leczeni afliberceptem co 8 tygodni po 1 roku, a poprawa widzenia utrzymywała się do 2 lat.
Po 4 początkowych miesięcznych wstrzyknięciach pacjenci z grupy leku Vabismo z dostosowanym odstępem wstrzyknięć do 16 tygodni mogli otrzymać łącznie co najmniej 6 i maksymalnie 21 wstrzyknięć do 96. tygodnia. W 52. tygodniu badań YOSEMITE i RHINE odpowiednio 74 % i 71 % pacjentów w odpowiednich grupach leku Vabismo z dostosowanym schematem dawkowania co 16 tygodni osiągnęło odstęp wstrzyknięć co 16 lub co 12 tygodni (53 % i 51 % – co 16 tygodni, 21 % i 20 % – co 12 tygodni). Odpowiednio 75 % i 84 % tych pacjentów w badaniach YOSEMITE i RHINE zachowało odstęp wstrzyknięć co ≥ 12 tygodni bez skrócenia tego odstępu poniżej 12 tygodni do 96. tygodnia; w 52. tygodniu 70 % i 82 % pacjentów leczonych w trybie co 16 tygodni nadal otrzymywało lek co 16 tygodni bez zmniejszania odstępu do 96. tygodnia. W 96. tygodniu w obu badaniach u 78 % pacjentów odpowiedniej grupy leczenia lekiem Vabismo z dostosowanym schematem podawania co 16 tygodni osiągnięto częstotliwość podawania co 16 lub co 12 tygodni (60 % i 65 % pacjentów – co 16 tygodni, 18 % i 14 % pacjentów – co 12 tygodni). U 4 % i 6 % pacjentów w badaniach YOSEMITE i RHINE odpowiednio odstęp wydłużono do 8 tygodni i pacjenci nadal otrzymywali lek w odstępie wstrzyknięć co ≤ 8 tygodni do 96. tygodnia; 3 % i 5 % otrzymywało lek w schemacie podawania co 4 tygodnie do końca 96. tygodnia.
Tabela 3
Wyniki dotyczące skuteczności podczas wizyt oceny pierwotnego punktu końcowego w 1a i 2b roku badania YOSEMITE
| Wyniki dotyczące skuteczności |
YOSEMITE |
|||||
| 1 rok |
2 lata |
|||||
| Vabismou co 8 tygodni, N = 315 |
Vabismou z dostosowanym schematem dawkowania do co 16 tygodni, N = 313 |
Aflibercept co 8 tygodni, N = 312 |
Vabismou co 8 tygodni, N = 315 |
Vabismou z dostosowanym schematem dawkowania do co 16 tygodni, N = 313 |
Aflibercept co 8 tygodni, N = 312 |
|
| Średnia zmiana od wartości wyjściowej wyniku BCVA wyrażona w liczbie liter według ETDRS (97,5 % CI w 1 rok i 95 % w 2 rok) |
10,7 (9,4, 12,0) |
11,6 (10,3, 12,9) |
10,9 (9,6, 12,2) |
10,7 (9,4, 12,1) |
10,7 (9,4, 12,1) |
11,4 (10,0, 12,7) |
| Część pacjentów, u których wartość BCVA wzrosła o co najmniej 15 liter od wartości wyjściowej (częstość ważona wg CMH, 95 % CI w 1 i 2 rok) |
29,2 % (23,9 %, 34,5 %) |
35,5 % (30,1 %, 40,9 %) |
31,8 % (26,6 %, 37,0 %) |
37,2 % (31,4 %, 42,9 %) |
38,2 % (32,8 %, 43,7 %) |
37,4 % (31,7 %, 43,0 %) |
| Część pacjentów, którzy uniknęli spadku wartości BCVA o co najmniej 15 liter od wartości wyjściowej (częstość ważona wg CMH, 95 % CI w 1 i 2 rok) |
98,1 % (96,5 %, 99,7 %) |
98,6 % (97,2 %, 100,0 %) |
98,9 % (97,6 %, 100,0 %) |
97,6 % (95,7 %, 99,5 %) |
97,8 % (96,1 %, 99,5 %) |
98,0 % (96,2 %, 99,7 %) |
a Średnia tygodnie 48, 52, 56.
b Średnia tygodnie 92, 96, 100.
BVCA – najlepsza skorygowana ostrość wzroku; ETDRS – Badanie skuteczności wczesnego leczenia retinopatii cukrzycowej; CI – przedział ufności; CMH – test Cochrana-Mantela-Hanszela, test statystyczny generujący oszacowanie związku z wynikiem binarnym, stosowany do oceny zmiennych kategorycznych.
Uwaga: dane dotyczące CMH-ważonego %, przedstawione dla grupy afliweryceptu, przeznaczone są do porównania dawkowania leku Vabismo co 8 tygodni z afliweryceptem; jednak odpowiedni CMH-ważony % dla porównania skorygowanego schematu leku Vabismo z afliweryceptem jest podobny do powyżej wskazanego.
ETDRS-DRSS: Skala ciężkości retinopatii cukrzycowej z Badania skuteczności wczesnego leczenia retinopatii cukrzycowej (skala oceny retinopatii cukrzycowej z Badania skuteczności wczesnego leczenia retinopatii cukrzycowej).
Tabela 4
Udział pacjentów z poprawą ETDRS-DRSS o co najmniej 2 stopnie w 52. i 96. tygodniu w porównaniu ze stanem wyjściowym w badaniu YOSEMITE (populacja oceniana pod względem retinopatii cukrzycowej)
| Wyniki dotyczące skuteczności |
YOSEMITE |
|||||
| 52 tygodnie |
96 tygodni |
|||||
| Vabismou co 8 tygodni, N = 237 |
Vabismou z dostosowanym trybem dawkowania do co 16 tygodni, N = 242 |
Aflibercept co 8 tygodni, N = 229 |
Vabismou co 8 tygodni, N = 220 |
Vabismou z dostosowanym trybem dawkowania do co 16 tygodni, N = 234 |
Aflibercept co 8 tygodni, N = 221 |
|
| Częstość pacjentów z poprawą o ≥ 2 stopnie od poziomu wyjściowego w skali ETDRS-DRSS (ważona frakcja według CMH) |
46,0 % |
42,5 % |
35,8 % |
51,4 % |
42,8 % |
42,2 % |
ETDRS-DRSS – skala ciężkości retinopatii cukrzycowej według badania oceniającego skuteczność wczesnego leczenia retinopatii cukrzycowej.
DI – przedział ufności; CMH – metoda Cochrana – Mantela – Haenszela, test statystyczny generujący oszacowanie związku z wynikiem binarnym, stosowany do oceny zmiennych kategorycznych.
Uwaga: dane dotyczące % ważonego metodą CMH, przedstawione dla grupy afliberceptu, przeznaczone są do porównania dawkowania leku Vabismo co 8 tygodni z afliberceptem; jednak odpowiedni % ważony metodą CMH do porównania skorygowanego trybu dawkowania leku Vabismo z afliberceptem jest podobny do wskazanego powyżej.
Tabela 5
Wyniki dotyczące skuteczności podczas wizyt oceny pierwotnego punktu końcowego w roku 1a oraz w roku 2b w badaniu RHINE
| Wyniki dotyczące skuteczności |
RHINE |
|||||
| 1 rok |
2 lata |
|||||
| Vabismou co 8 tygodni, N = 317 |
Vabismou z dostosowanym schematem dawkowania do co 16 tygodni, N = 319 |
Aflibercept co 8 tygodni, N = 315 |
Vabismou co 8 tygodni, N = 317 |
Vabismou z dostosowanym schematem dawkowania do co 16 tygodni, N = 319 |
Aflibercept co 8 tygodni, N = 315 |
|
| Średnia zmiana od poziomu wyjściowego w skali BCVA wyrażona w liczbie liter ETDRS (97,5 % CI w 1 roku i 95 % w 2 roku) |
11,8 (10,6, 13,0) |
10,8 (9,6, 11,9) |
10,3 (9,1, 11,4) |
10,9 (9,5, 12,3) |
10,1 (8,7, 11,5) |
9,4 (7,9, 10,8) |
| Częstość pacjentów, u których wartość BCVA wzrosła o co najmniej 15 liter od poziomu wyjściowego (częstość ważona wg CMH, 95 % CI w 1 i 2 roku) |
33,8 % (28,4 %, 39,2 %) |
28,5 % (23,6 %, 33,3 %) |
30,3 % (25,0 %, 35,5 %) |
39,8 % (34,0 %, 45,6 %) |
31,1 % (26,1 %, 36,1 %) |
39,0 % (33,2 %, 44,8 %) |
| Częstość pacjentów, którzy uniknęli spadku BCVA o co najmniej 15 liter od poziomu wyjściowego (częstość ważona wg CMH, 95 % CI w 1 i 2 roku) |
98,9 % (97,6 %, 100,0 %) |
98,7 % (97,4 %, 100,0 %) |
98,6 % (97,2 %, 99,9 %) |
96,6 % (94,4 %, 98,8 %) |
96,8 % (94,8 %, 98,9 %) |
97,6 % (95,7 %, 99,5 %) |
a Średnia tygodnie 48, 52, 56.
b Średnia tygodnie 92, 96, 100.
BVCA – najlepsza skorygowana ostrość wzroku; ETDRS – badanie skuteczności wczesnego leczenia retinopatii cukrzycowej; CI – przedział ufności; CMH – test Cochrana-Mantela-Haenszela, test statystyczny generujący oszacowanie powiązania z wynikiem binarnym, stosowany do oceny zmiennych kategorycznych.
Uwaga: dane dotyczące % ważonego metodą CMH, przedstawione dla grupy aflatyberceptu, przeznaczone są do porównania dawkowania leku Vabismou co 8 tygodni z aflatyberceptem; jednak odpowiedni % ważony metodą CMH dla porównania skorygowanego schematu dawkowania leku Vabismou z aflatyberceptem jest podobny do wskazanego powyżej.
ETDRS-DRSS – skala ciężkości retinopatii cukrzycowej według badania skuteczności wczesnego leczenia retinopatii cukrzycowej (skala oceny retinopatii cukrzycowej z badania skuteczności wczesnego leczenia retinopatii cukrzycowej).
Tabela 6
Udział pacjentów z poprawą wyniku ETDRS-DRSS o co najmniej 2 stopnie w tygodniu 52 i 96 w porównaniu ze stanem wyjściowym w badaniu RHINE (populacja oceniana pod względem retinopatii cukrzycowej)
| Wyniki dotyczące skuteczności |
RHINE |
|||||
| 52 tygodnie |
96 tygodni |
|||||
| Vabismou co 8 tygodni, N = 231 |
Vabismou z dostosowanym schematem dawkowania do co 16 tygodni, N = 251 |
Aflibercept co 8 tygodni, N = 238 |
Vabismou co 8 tygodni, N = 214 |
Vabismou z dostosowanym schematem dawkowania do co 16 tygodni, N = 228 |
Aflibercept co 8 tygodni, N = 203 |
|
| Częstość pacjentów z poprawą o ≥ 2 stopnie w skali ETDRS-DRSS od poziomu wyjściowego (ważona częstość wg CMH) |
44,2 % |
43,7 % |
46,8 % |
53,5 % |
44,3 % |
43,8 % |
ETDRS – badanie oceny skuteczności wczesnego leczenia retinopatii cukrzycowej.
DI – przedział ufności; CMH – metoda Cochrana-Mantela-Haenszela, test statystyczny generujący oszacowanie związku z wynikiem binarnym, stosowany do oceny zmiennych kategorycznych.
Uwaga: dane dotyczące % ważonego metodą CMH, przedstawione dla grupy aflatyberceptu, są przeznaczone do porównania dawki leku Vabismo podawanego co 8 tygodni z aflatyberceptem; jednak odpowiedni % ważony metodą CMH dla porównania skorygowanego schematu leku Vabismo z aflatyberceptem jest podobny do powyższego.
| Skorygowana średnia zmiana od wartości wyjściowej |
|
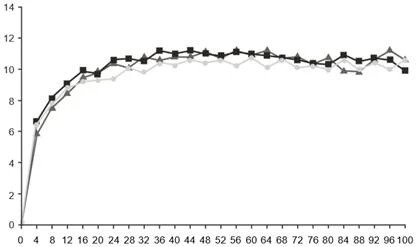
Tygodnie
Faricyklumab, 6 mg, co 16 tygodni dostosowanego dawkowania (N = 632)
Faricyklumab, 6 mg, co 8 tygodni (N = 632)
Aflibercept, 2 mg, co 8 tygodni (N = 627)
Rys. 2. Połączone badania fazy III w DMN (YOSEMITE i RHINE): wykres zmiany BCVA od poziomu wyjściowego do 100. tygodnia badania: metoda MMRM (pierwotna ocena) (populacja ITT)
Wyniki dotyczące skuteczności u pacjentów, którzy przed udziałem w badaniu nie otrzymywali leczenia anty-VEGF, oraz u wszystkich innych ocenianych podgrup pacjentów (w szczególności pod względem wieku, płci, pochodzenia etnicznego, wyjściowego poziomu HbA1c, wyjściowego ostrości widzenia) w każdym badaniu zgadzały się z wynikami w odpowiedniej populacji.
Efekt leczenia nie zależał od kontroli glikemicznej, a porównywalne wyniki osiągano w leczeniu faricyklumabem u pacjentów z poprawą lub pogorszeniem w czasie poziomu HbA1c o > 0,5% lub pozostającym w granicach 0,5% od poziomu wyjściowego.
Leczenie obrzęku plamki spowodowanego OZWS i O/PCWS
Bezpieczeństwo i skuteczność stosowania faricyklumabu badano w dwóch wieloośrodkowych badaniach u pacjentów z obrzękiem plamki spowodowanym OZWS (BALATON) lub O/PCWS (COMINO). Oba badania składały się z początkowej 24-tygodniowej fazy leczenia z randomizacją (1:1) i aktywnym kontrolowaniem (aflibercept). Następnie wszyscy pacjenci (w tym ci, którzy pierwotnie leczono afliberceptem) otrzymywali faricyklumab według indywidualnego schematu dawkowania do 68. tygodnia (ostatnia wizyta w 72. tygodniu). Interwał dawkowania mógł być wydłużony o 4 tygodnie do maksimum – co 16 tygodni – a następnie ponownie skrócony o 4, 8 lub 12 tygodni w razie potrzeby (pogorszenie CST i/lub ostrości widzenia) w zależności od aktywności choroby (ocenianej za pomocą z góry określonej zautomatyzowanej obiektywnej oceny kryteriów wizualnych i anatomicznych). Interwał leczenia nie był ponownie wydłużany po ustabilizowaniu się aktywności choroby u pacjentów, u których konieczne było skrócenie interwału. Z badania wykluczono pacjentów z minimalnym (4-tygodniowym) interwałem między iniekcjami.
Razem 1282 pacjentów (553 w BALATON i 729 w COMINO) zostało włączone do dwóch badań, z czego 1276 pacjentów otrzymało co najmniej jedną dawkę do 24. tygodnia (641 pacjentów – dawkę Vabismou).
Oba badania wykazały skuteczność względem pierwotnego punktu końcowego, zdefiniowanego jako zmiana od poziomu wyjściowego wskaźnika BCVA w 24. tygodniu, mierzonego liczbą liter według skali ETDRS. W obu badaniach pacjenci, którzy otrzymywali Vabismou co cztery tygodnie, osiągali nie gorszą średnią zmianę od poziomu wyjściowego wskaźnika BCVA w 24. tygodniu w porównaniu z pacjentami, którzy otrzymywali aflibercept co cztery tygodnie, a te poprawy wzroku utrzymywały się do 72. tygodnia, kiedy pacjenci przeszli na dostosowany tryb dawkowania Vabismou do interwału co 16 tygodni.
Między 24 a 68 tygodniem 81,5% i 74,0% pacjentów, którzy otrzymywali Vabismou w dawce 6 mg według dostosowanego trybu dawkowania do interwału co 16 tygodni, osiągnęło interwał dawkowania co 16 tygodni lub co 12 tygodni w badaniach BALATON i COMINO odpowiednio. Spośród tych pacjentów 72,1% i 61,6% ukończyło co najmniej jeden cykl co 12 tygodni i utrzymało interwał dawkowania co 16 tygodni lub co 12 tygodni bez skracania interwału poniżej co 12 tygodni do 68. tygodnia w badaniach BALATON i COMINO odpowiednio; 1,2% i 2,5% pacjentów otrzymało dawkę tylko co 4 tygodnie do 68. tygodnia w badaniach BALATON i COMINO odpowiednio.
Szczegółowe wyniki obu badań przedstawiono w tabelach 7 i 8 oraz na rysunkach 3 i 4 poniżej.
Tabela 7
Wyniki dotyczące skuteczności podczas wizyt oceny pierwotnego punktu końcowego w 24. tygodniu oraz na końcu badania BALATON
| Wyniki dotyczące skuteczności |
BALATON |
|||
| 24 tygodnie |
72 tygodniea |
|||
| Vabismou N = 276 |
Aflibercept N = 277 |
Przejście z Vabismou co 4 tygodnie na Vabismou w skorygowanej dawce N = 276 |
Przejście z afliberceptu co 4 tygodnie na Vabismou w skorygowanej dawce N = 277 |
|
| Średnia zmiana od poziomu wyjściowego wskaźnika BCVA w wyniku określenia liczby liter według ETDRS (95 % CI) |
16,9 (15,7; 18,1) |
17,5 (16,3; 18,6) |
18,1 (16,9; 19,4) |
18,8 (17,5; 20,0) |
| Częstość występowania pacjentów ze zwiększeniem ≥ 15 liter w porównaniu z poziomem wyjściowym (ważona częstość według CMH, 95 % CI) |
56,1 % (50,4 %; 61,9 %) |
60,4 % (54,7 %; 66,0 %) |
61,5 % (56,0 %; 67,0 %) |
65,8 % (60,3 %; 71,2 %) |
a Średnia wartość w tygodniach 64, 68, 72.
BCVA – najlepsza skorygowana ostrość wzroku.
ETDRS – badanie skuteczności wczesnego leczenia retinopatii cukrzycowej.
CI – przedział ufności.
CMH – test Cochrana-Mantela-Haenszela, test statystyczny generujący oszacowanie związku z wynikiem binarnym, stosowany do oceny zmiennych kategorycznych.
Tabela 8
Wyniki dotyczące skuteczności podczas wizyt oceny pierwotnego punktu końcowego w 24. tygodniu oraz na końcu badania COMINO
| Wyniki dotyczące skuteczności |
COMINO |
|||
| 24 tygodnie |
72 tygodniea |
|||
| Vabismou N = 366 |
Aflibercept N = 363 |
Przejście z Vabismou co 4 tygodnie na Vabismou w dostosowanej dawce N = 366 |
Przejście z afliberceptu co 4 tygodnie na Vabismou w dostosowanej dawce N = 363 |
|
| Średnia zmiana od poziomu wyjściowego wskaźnika BCVA w wyniku oznaczenia liczby liter według ETDRS (95 % CI) |
16,9 (15,4; 18,3) |
17,3 (15,9; 8,8) |
16,9 (15,2; 18,6) |
17,1 (15,4; 18,8) |
| Część pacjentów ze zwiększeniem ≥ 15 liter w porównaniu z poziomem wyjściowym (ważona część według CMH, 95 % CI) |
56,6 % (51,7 %; 61,5 %) |
58,1 % (53,3 %; 62,9 %) |
57,6 % (52,8 %; 62,5 %) |
59,5 % (54,7 %; 64,3 %) |
a Średnia wartość w tygodniach 64, 68, 72.
BCVA – najlepsza skorygowana ostrość wzroku.
ETDRS – badanie skuteczności wczesnego leczenia retinopatii cukrzycowej.
CI – przedział ufności.
CMH – metoda Cochrana-Mantela-Hanszela, test statystyczny generujący oszacowanie związku z wynikiem binarnym, stosowany do oceny zmiennych kategorycznych.
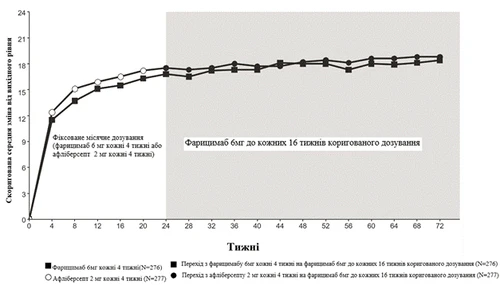
Rys. 3. Badanie fazy III BALATON: wykres zmiany wartości BCVA od wartości wyjściowej w badaniu do tygodnia 72: metoda MMRM (pierwotna ocena) (populacja ITT)
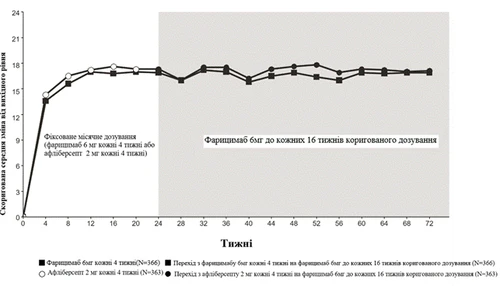
Rys. 4. Badanie fazy III COMINO: wykres zmiany wartości BCVA od wartości wyjściowej w badaniu do tygodnia 72: metoda MMRM (pierwotna ocena) (populacja ITT)
Pacjenci w podeszłym wieku
W sześciu badaniach klinicznych fazy III około 58 % (1496/2571) pacjentów zrandomizowanych do leczenia lekiem Vabismo miało wiek ≥ 65 lat. Populacyjna analiza farmakokinetyczna wykazała wpływ wieku na farmakokinetykę faricymabu w narządzie wzroku, który uznano za klinicznie nieistotny (patrz sekcje „Sposób stosowania i dawka”, „Farmakokinetyka”).
Immunogenność
Istnieje potencjał rozwoju odpowiedzi immunologicznej u pacjentów otrzymujących leczenie lekiem Vabismo (patrz sekcja „Szczególne ostrzeżenia i środki ostrożności przed zastosowaniem”).
Po podaniu leku Vabismo przez okres do 112 (nAMD), 100 (DMN) oraz 72 (OCT i OCVS) tygodni, przeciwciała przeciwko faricymabowi wytworzone podczas leczenia wykryto odpowiednio u około 13,8 %, 9,6 % i 14,4 % pacjentów zrandomizowanych do otrzymywania faricymabu z grupy nAMD, DMN oraz OCT/OCVS. Kliniczne znaczenie przeciwciał przeciwko faricymabowi w odniesieniu do bezpieczeństwa nie jest jeszcze znane. U pacjentów z przeciwciałami przeciwko faricymabowi obserwowano wyższą częstość działań niepożądanych w postaci zapalenia wewnątrzgałkowego. Jednak ogólna częstość występowania przeciwciał przeciwko faricymabowi oraz zapalenia wewnątrzgałkowego w całej populacji badawczej wynosi około 1 %. Przeciwciała przeciwko faricymabowi nie były związane z wpływem na skuteczność kliniczną ani na farmakokinetykę ogólnoustrojową.
Farmakokinetyka.
Absorpcja
Lek Vabismo podaje się do ciała szklistego w celu lokalnego działania na oko. Nie przeprowadzono badań klinicznych innych dróg podania.
Na podstawie populacyjnej analizy farmakokinetycznej (w tym nAMD i DMN, N = 2246), maksymalna stężenie we krwi (Cmax) wolnej (niezwiązanej z VEGF-A i Ang-2) formy faricymabu osiągane jest około 2 dni po podaniu. Średnie (± odchylenie standardowe) Cmax we krwi wolnej formy szacowane jest na poziomie odpowiednio 0,23 (0,07) μg/ml i 0,22 (0,07) μg/ml u pacjentów z nAMD i DMN. Po powtarzanych dawkowaniach średnie minimalne stężenie wolnego faricymabu we krwi przewidywane jest na poziomie 0,002–0,003 μg/ml przy podawaniu co 8 tygodni.
Faricymab wykazał farmakokinetykę proporcjonalną do dawki (na podstawie Cmax i AUC) w zakresie dawek 0,5–6 mg. Nie zaobserwowano kumulacji faricymabu w ciele szklistym ani we krwi po miesięcznym podawaniu, biorąc pod uwagę ekspozycję uzyskaną w modelu populacyjnej farmakokinetyki.
Analiza farmakokinetyczna pacjentów z nAMD, DMN, OCT i OCVS (N = 2977) wykazała porównywalną farmakokinetykę faricymabu u tych pacjentów.
Rozkład
Brak danych.
Metabolizm
Metabolizm faricymabu nie był bezpośrednio badany. Przypuszcza się, że faricymab ulega katabolizmowi do małych peptydów i aminokwasów w lizosomach, podobnie jak endogenne cząsteczki IgG.
Wydalanie
Profil stężenie-czas faricymabu we krwi obniżał się równolegle z profilem stężenie-czas w ciele szklistym i płynie wewnątrzgałkowym. Szacowany średni okres półtrwania wydalenia z oka oraz pozorny ogólnoustrojowy okres półtrwania faricymabu wynosi około 7,5 dnia.
Specjalne grupy pacjentów
Zaburzenia funkcji wątroby
Nie przeprowadzono formalnego badania farmakokinetyki u pacjentów z zaburzeniami funkcji wątroby.
Zaburzenia funkcji nerek
Nie przeprowadzono formalnego badania farmakokinetyki u pacjentów z zaburzeniami funkcji nerek.
Analiza farmakokinetyczna danych z wszystkich badań klinicznych, w tym 1115 pacjentów z łagodnym, 669 pacjentów z umiarkowanym oraz 54 pacjentów z ciężkim zaburzeniem funkcji nerek, nie wykazała różnic w farmakokinetyce ogólnoustrojowej faricymabu po wstrzyknięciu do ciała szklistego leku Vabismo.
Pacjenci w podeszłym wieku
W sześciu badaniach klinicznych fazy III około 58 % (1496/2571) pacjentów zrandomizowanych do leczenia lekiem Vabismo miało wiek ≥ 65 lat. Populacyjna analiza farmakokinetyczna wykazała wpływ wieku na farmakokinetykę faricymabu w narządzie wzroku, który jednak uznano za klinicznie nieistotny.
Inne czynniki demograficzne
Populacyjna analiza farmakokinetyczna wykazała wpływ masy ciała na farmakokinetykę faricymabu w oku oraz na farmakokinetykę ogólnoustrojową faricymabu. Ten efekt uznano za klinicznie nieistotny; z tego względu nie jest wymagana korekta dawki.
Analiza kinetyczna populacyjna wskazuje na brak jakiegokolwiek wpływu pochodzenia etnicznego lub płci pacjenta na farmakokinetykę ogólnoustrojową leku Vabismo.
Dane kliniczne.
Wskazania.
Leczenie nieowaczystej (egsudatycznej) zwyrodnienia plamicy spowodowanego wiekiem (nAMD).
Leczenie obrzęku plamicy spowodowanego cukrzycą (DME).
Leczenie obrzęku plamicy spowodowanego zamknięciem żyły siatkówki (zamknięcie gałęzi żyły siatkówki (BRVO) i zamknięcie środkowej żyły siatkówki (CRVO)).
Przeciwwskazania.
Zakażenia oczu lub zakażenia okołoodbytowe.
Aktywne zapalenie wewnątrzgałkowe.
Ustalona nadwrażliwość na faricizumab lub dowolny składnik pomocniczy leku. Reakcje nadwrażliwościowe mogą objawiać się wysypką, świądem, pokrzywką, rumieniem lub ciężkim zapaleniem wewnątrzgałkowym.
Interakcje z innymi lekami i inne rodzaje interakcji.
Nie przeprowadzono badań interakcji innych leków z lekiem Vabismou.
Szczególne środki ostrożności.
Reakcje związane z iniekcją wewnątrzcieczową
Iniekcje wewnątrzcieczowe, w tym leku Vabismo, były związane z endoftalmią, zapaleniem wewnątrzgałkowym, odwarstwieniem siatkówki spowodowanym rozwarstwieniem, pęknięciem siatkówki oraz zaćmą jatrogenną. Przy podawaniu leku Vabismo należy zawsze przestrzegać odpowiedniej techniki iniekcji aseptycznej. Pacjentów należy poinstruwać o konieczności natychmiastowego zgłaszania wszelkich objawów, takich jak ból, utrata wzroku, światłowstręt, nieostrość widzenia, pływające zacieńczenia lub zaczerwienienie, które mogą wskazywać na endoftalmię lub inne wyżej wymienione zjawiska, w celu zapewnienia szybkiego i odpowiedniego leczenia.
Obserwowano przejściowy wzrost ciśnienia wewnątrzgałkowego (IOP) w ciągu 60 minut po iniekcji wewnątrzcieczowej, w tym po podaniu leku Vabismo. Szczególne środki ostrożności są konieczne u pacjentów z niedobrze kontrolowaną jaskrą (nie podawać leku Vabismo przy IOP ≥ 30 mmHg). We wszystkich przypadkach należy odpowiednio monitorować zarówno IOP, jak i perfuzję tarczy nerwu wzrokowego oraz/lub ostrość wzroku i leczyć w razie potrzeby.
Skutki systemowe
Zgłaszano niepożądane skutki systemowe, w tym zdarzenia tromboemboliczne tętnicze. Istnieje teoretyczne ryzyko, że te zjawiska mogą być związane z hamowaniem czynnika wzrostu śródbłonka naczyń (VEGF). Niska częstość występowania zdarzeń tętniczych tromboembolicznych była obserwowana w badaniach klinicznych faricymabu u pacjentów z AMD, DR, BRVO, CRVO.
Immunogenność
Substancja czynna leku Vabismo jest białkiem terapeutycznym. Z tego powodu możliwe jest wystąpienie reakcji immunologicznej na lek Vabismo. Pacjentów należy poinstruować o konieczności zgłaszania wszelkich objawów lub objawów zapalenia wewnątrzgałkowego, takich jak utrata wzroku, ból w oku, zwiększona wrażliwość na światło, pływające zacieńczenia lub nasilenie zaczerwienienia oczu, które mogą być objawami klinicznymi związanymi z nadwrażliwością.
Leczenie obu oczu
Bezpieczeństwo i skuteczność leku Vabismo po podaniu do obu oczu nie zostały zbadane.
Jednoczesne stosowanie innych leków przeciwwegf
Brak danych dotyczących jednoczesnego stosowania leku Vabismo z innymi lekami przeciwwegf w jedno oko.
Przerwanie leczenia
Leczenie należy tymczasowo wstrzymać u pacjentów z:
- odwarstwieniem siatkówki spowodowanym rozwarstwieniem, przebiciem siatkówki stopnia 3 lub 4, pęknięciem siatkówki; leczenia nie należy wznowić, dopóki nie nastąpi odpowiednie gojenie;
- spowodowanym leczeniem spadkiem najlepszej skorygowanej ostrości wzroku (BCVA) o ≥ 30 liter w porównaniu z ostatnią oceną ostrości wzroku; leczenia nie należy wznowić wcześniej niż przy następnym zaplanowanym podaniu leku;
- wykonaną lub planowaną operacją wewnątrzgałkową w ciągu poprzednich lub następnych 28 dni; leczenia nie należy wznowić wcześniej niż przy następnym zaplanowanym podaniu leku.
Pęknięcie nabłonka barwnikowego siatkówki
Czynniki ryzyka związane z wystąpieniem pęknięcia nabłonka barwnikowego siatkówki po terapii przeciwwegf z powodu AMD obejmują rozległe i/lub wysokie odwarstwienie nabłonka barwnikowego. Dlatego przy stosowaniu u pacjentów z tymi czynnikami ryzyka należy zachować ostrożność na początku terapii lekiem Vabismo.
Populacje z ograniczonymi danymi
Istnieje jedynie ograniczone doświadczenie w leczeniu pacjentów z DR z poziomem HbA1c powyżej 10%, pacjentów z proliferacyjną retinopatią cukrzycową z wysokim ryzykiem lub pacjentów z AMD, DR, BRVO i CRVO z aktywnymi infekcjami systemowymi. Brak także doświadczenia w leczeniu lekiem Vabismo pacjentów z cukrzycą, BRVO i CRVO z niekontrolowaną nadciśnieniem tętniczym. Lekarz powinien wziąć pod uwagę brak tych informacji przy leczeniu takich pacjentów.
Nadużywanie leków i uzależnienie
Brak danych dotyczących możliwości rozwoju nadużywania i uzależnienia przy stosowaniu leku Vabismo.
Inna informacja
Lek Vabismo, roztwór do wstrzykiwań do stosowania wewnątrzcieczowego, zawiera mniej niż 1 mmol (23 mg)/dawkę sodu, tj. jest praktycznie pozbawiony sodu.
Stosowanie w okresie ciąży lub karmienia piersią.
Kobiety w wieku rozrodczym
Kobietom w wieku rozrodczym należy stosować skuteczną antykoncepcję w trakcie leczenia lekiem Vabismo oraz przez co najmniej 3 miesiące po podaniu ostatniej dawki leku Vabismo.
Ciąża
Brak danych dotyczących stosowania leku Vabismo u kobiet w ciąży.
Nie zaobserwowano skutków ubocznych w badaniu na ciężarnych makakach jawajskich.
Wykazano, że hamowanie VEGF powoduje wady rozwojowe, resorpcję embrionu/łódki oraz zmniejszenie masy ciała płodu. Wykazano również, że hamowanie VEGF wpływa na rozwój pęcherzyków, funkcję ciała żółtego oraz płodność. Brak badań specjalnych dotyczących wpływu hamowania Ang-2 na ciążę. Ze względu na dane przedkliniczne hamowanie Ang-2 może prowadzić do efektów porównywalnych z efektami hamowania VEGF. Ekspozycja systemowa po podaniu leku Vabismo do narządu wzroku jest bardzo niska.
Nie wiadomo, czy faricymab przenika przez łożysko lub powoduje szkodę płodowi przy stosowaniu u kobiet w ciąży. Biorąc pod uwagę mechanizm działania inhibitorów VEGF i Ang-2, istnieje potencjalne ryzyko dla potencjału rozrodczego kobiet oraz dla rozwoju embrionalno-płodowego. Choć ekspozycja systemowa po podaniu do narządu wzroku jest bardzo niska, faricymab nie powinien być stosowany w czasie ciąży, z wyjątkiem przypadków, gdy potrzeba leczenia wynika ze stanu klinicznego kobiety.
Poród
Bezpieczeństwo stosowania leku Vabismo w czasie porodu nie zostało ustalone.
Karmienie piersią
Nie wiadomo, czy lek Vabismo wydostaje się do mleka matki. Nie przeprowadzono badań dotyczących wpływu Vabismo na produkcję mleka ani obecności leku w mleku matki. Ponieważ wiele leków wydostaje się do mleka matki z potencjalnym ryzykiem wchłaniania i szkodliwego wpływu na wzrost i rozwój niemowlęcia, należy zachować ostrożność przy stosowaniu leku Vabismo u kobiet karmiących piersią. Należy porównać korzyści płynące z karmienia piersią dla rozwoju i zdrowia dziecka z kliniczną potrzebą leczenia matki lekiem Vabismo oraz z potencjalnym niepożądanym wpływem leku Vabismo na dziecko spożywające mleko matki.
Płodność
Nie przeprowadzono badań dotyczących wpływu na funkcję rozrodczą lub płodność. Nie zaobserwowano wpływu na narządy rozrodcze w 6-miesięcznym badaniu na makakach jawajskich przy stosowaniu faricymabu w dawkach do 3 mg/oko (ekspozycja 8–10 razy wyższa niż ekspozycja kliniczna, na podstawie parametru AUC). Wykazano, że hamowanie VEGF nie wpływa na rozwój pęcherzyków, funkcję ciała żółtego i płodność. Biorąc pod uwagę mechanizm działania inhibitorów VEGF i Ang-2, istnieje potencjalne ryzyko dla potencjału rozrodczego kobiet i rozwoju embrionalno-płodowego, jednak ryzyko to uważa się za niskie ze względu na niską ekspozycję systemową po podaniu leku do narządu wzroku.
Wpływ na zdolność prowadzenia pojazdów i obsługi mechanizmów.
Lek Vabismo ma nieznaczny wpływ na zdolność prowadzenia pojazdów i obsługi mechanizmów z powodu możliwych tymczasowych zaburzeń widzenia po iniekcji wewnątrzcieczowej i związanego z tym badania oka. Pacjenci nie powinni prowadzić pojazdów ani obsługiwać mechanizmów do czasu wystarczającego odzyskania funkcji wzroku.
Sposób stosowania i dawki.
Ogólna informacja
Wyłącznie do wstrzykiwań do ciała szklistego. Lek Vabismo powinien podawać wykwalifikowany lekarz z doświadczeniem w wykonywaniu wstrzykiwań do ciała szklistego. Zawartość jednego fiolki należy stosować wyłącznie do leczenia jednego oka.
Jednorazowa fiolka szklana z lekiem Vabismo zawiera 28,8 mg faricymabu w 0,24 ml roztworu. Zapewnia to niezbędną ilość roztworu do wstrzyknięcia 0,05 ml, zawierającego 6 mg faricymabu jako dawkę pojedynczą.
Nieowaskularna (egzudatywna) zwyrodnienie plamki starczej (nAMD)
Zalecana dawka leku Vabismo wynosi 6 mg (0,05 ml) wstrzykiwana do ciała szklistego co 4 tygodnie (około co 28 ± 7 dni lub raz w miesiącu) w pierwszych czterech dawkach. Następnie schemat leczenia można indywidualnie dostosować, wydłużając odstępy między dawkami. Na podstawie oceny przez lekarza grubości środkowego pola siatkówki pacjenta (grubość środkowego pola centralnego, CST) oraz/lub ostrości widzenia odstęp między dawkami można wydłużyć maksymalnie do co 16 tygodni (4 miesiące). Odstęp między dawkami należy odpowiednio skrócić w przypadku pogorszenia się wartości CST i/lub ostrości widzenia (patrz sekcja „Farmakodynamika”).
Niektórzy pacjenci mogą wymagać wstrzykiwań co 4 tygodnie (około co 28 ± 7 dni lub raz w miesiącu).
Monitorowanie między wizytami podania leku należy planować w zależności od stanu pacjenta i uznania lekarza.
Światłowidmowy obrzęk plamki spowodowany cukrzycą (DME)
Zalecana dawka leku Vabismo wynosi 6 mg (0,05 ml) wstrzykiwana do ciała szklistego co 4 tygodnie (około co 28 ± 7 dni lub raz w miesiącu) w pierwszych czterech dawkach. Następnie schemat leczenia można indywidualnie dostosować, stosując tryb leczenia z wydłużonymi odstępami między dawkami. Na podstawie oceny przez lekarza CST i/lub ostrości widzenia danego pacjenta odstęp między dawkami można wydłużyć maksymalnie do co 16 tygodni (4 miesiące). Odstęp między dawkami należy odpowiednio skrócić w przypadku pogorszenia się CST i/lub ostrości widzenia (patrz sekcja „Farmakodynamika”).
Monitorowanie między wizytami podania leku należy planować w zależności od stanu pacjenta i uznania lekarza, jednak nie ma wymogu miesięcznego monitorowania między wstrzyknięciami.
Obrzęk plamki spowodowany zatorową żylą siatkówki (BRVO i CRVO)
Zalecana dawka leku Vabismo wynosi 6 mg (0,05 ml) i podaje się ją przez wstrzyknięcie do ciała szklistego co 4 tygodnie (około co 28 dni ± 7 dni lub raz w miesiącu); może być wymagane trzy lub więcej kolejnych miesięcznych wstrzyknięć, aż do osiągnięcia maksymalnej ostrości widzenia i/lub braku objawów aktywności choroby. Następnie leczenie może być kontynuowane na podstawie indywidualnego podejścia. W zależności od oceny przez lekarza grubości środkowego pola centralnego (CST) i/lub ostrości widzenia danego pacjenta, odstęp dawkowania może być wydłużony. Odstęp leczenia należy skrócić w przypadku pogorszenia się CST i/lub ostrości widzenia, a każda nowa próba wydłużenia odstępu po ustabilizowaniu stanu powinna być starannie rozważona (patrz sekcja „Właściwości farmakologiczne”). Odstępy leczenia dłuższe niż 4 miesiące między wstrzyknięciami nie były badane.
Monitorowanie między datami podania dawek powinno opierać się na stanie pacjenta i ocenie lekarza, ale nie ma wymogu obowiązkowego miesięcznego monitorowania między wstrzyknięciami.
W celu zapewnienia śledzenia biologicznych leków lepiej jest dokumentować nazwę handlową i numer serii przy każdym podaniu leku.
Czas trwania leczenia
Lek Vabismo przeznaczony jest do długoterminowego leczenia.
Korekta dawki po wystąpieniu niepożądanych skutków działania/współdziałania
Nie zaleca się zmiany dawki leku Vabismo.
Opóźnione podanie
Jeśli wstrzyknięcie zostało odroczone lub pominięte, pacjent powinien wrócić na wizytę kontrolną w najbliższym możliwym terminie i kontynuować podawanie leku zgodnie z decyzją lekarza.
Jeśli wyniki badań wizualnych i/lub anatomicznych wskazują, że pacjent nie czerpie korzyści z kontynuacji leczenia, leczenie lekiem Vabismo należy przerwać.
Specjalne grupy pacjentów
Zaburzenia funkcji wątroby
Nie przeprowadzono specjalnych badań leku Vabismo u pacjentów z zaburzeniami funkcji wątroby (patrz sekcja „Farmakokinetyka”).
Jednak korekta dawki nie jest wymagana u pacjentów z zaburzeniami funkcji wątroby.
Zaburzenia funkcji nerek
Nie przeprowadzono specjalnych badań leku Vabismo u pacjentów z zaburzeniami funkcji nerek (patrz sekcja „Farmakokinetyka”).
Jednak korekta dawki nie jest wymagana u pacjentów z chorobami nerek.
Pacjenci w podeszłym wieku
W sześciu badaniach fazy III około 58% (1496/2571) pacjentów losowanych do leczenia lekiem Vabismo miało wiek ≥ 65 lat. Analiza populacyjna farmakokinetyki wykazała wpływ wieku na farmakokinetykę faricymabu w narządach wzroku. Jednak efekt ten został oceniony jako klinicznie nieistotny. W tych badaniach nie stwierdzono istotnych różnic w skuteczności ani bezpieczeństwie stosowania faricymabu wraz ze wzrostem wieku pacjenta. Korekta dawki u pacjentów w wieku ≥ 65 lat nie jest wymagana (patrz sekcja „Farmakokinetyka”).
Dzieci
Bezpieczeństwo i skuteczność stosowania leku Vabismo u dzieci nie są ustalone.
Specjalne grupy pacjentów
Nie jest wymagana specjalna korekta dawki dla żadnej z badanych populacji pacjentów (w szczególności pod względem wieku (pacjenci w podeszłym wieku), płci i rasy).
Instrukcje dotyczące stosowania
Przed podaniem lek Vabismo należy wizualnie ocenić pod kątem obecności zanieczyszczeń i zmiany barwy.
Bezpośrednio po wstrzyknięciu do ciała szklistego należy obserwować pacjentów pod kątem podwyższonego ciśnienia wewnątrzgałkowego. Odpowiednie monitorowanie może obejmować sprawdzenie perfuzji tarczy nerwu wzrokowego lub tonometrię. Sterylne wyposażenie do paracentesis powinno być dostępne w razie potrzeby.
Po wstrzyknięciu do ciała szklistego pacjentów należy poinstruować o konieczności natychmiastowego zgłaszania wszelkich objawów sugerujących endoftalmię (np. utrata wzroku, ból oka, zaczerwienienie oka, fotofobia, nieostre widzenie).
Przygotowanie leku do podania
Lek Vabismo jest sterylnym, przezroczystym lub opalizującym roztworem bez konserwantów, bezbarwnym lub jasnobrązowym.
Lek Vabismo należy wizualnie ocenić po wyjęciu z lodówki i przed podaniem.
Nie wolno stosować fiolki, jeśli widoczne są zanieczyszczenia, mętnienie lub zmiana barwy.
Zawartość fiolki i igła z filtrem przelewającym są sterylnymi elementami przeznaczonymi wyłącznie do jednorazowego użytku. Nie należy stosować leku, jeśli opakowanie, fiolka i/lub igła z filtrem przelewającym są uszkodzone lub wygasł termin ważności.
Podczas przygotowania do wstrzyknięcia do ciała szklistego należy przestrzegać zasad aseptyki.
Instrukcje dotyczące stosowania leku
Poniższa informacja jest przeznaczona wyłącznie dla personelu medycznego.
Przed rozpoczęciem:
- Uważnie przeczytać wszystkie instrukcje przed zastosowaniem leku Vabismo.
- Opakowanie leku Vabismo zawiera fiolkę szklaną z lekiem i igłę z filtrem przelewającym. Fiolkę szklaną zawiera tylko jedną dawkę. Igła z filtrem przelewającym przeznaczona jest wyłącznie do jednorazowego użytku.
- Lek Vabismo należy przechowywać w lodówce w temperaturze od 2 do 8 °C. Nie mrozić. Nie wstrząsać.
- Przed podaniem pozwolić lekowi Vabismo osiągnąć temperaturę pokojową (od 20 do 25 °C). Przechowywać fiolkę w oryginalnym opakowaniu kartonowym w celu ochrony przed światłem.
- Fiolkę z lekiem Vabismo można przechowywać w temperaturze pokojowej przez 24 godziny.
- Przed podaniem fiolkę z lekiem Vabismo należy wizualnie ocenić. Lek Vabismo jest przezroczystym lub opalizującym roztworem, bezbarwnym lub jasnobrązowym.
Nie należy stosować leku, jeśli widoczne są zanieczyszczenia, mętnienie lub zmiana barwy.
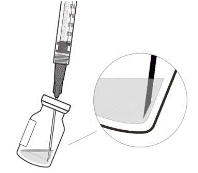
Nie należy stosować leku, jeśli opakowanie, fiolka i/lub igła z filtrem przelewającym są otwarte, uszkodzone lub wygasł termin ważności (patrz rysunek A).
- Podczas przygotowania do wstrzyknięcia do ciała szklistego należy przestrzegać zasad aseptyki.
Rysunek A
Instrukcje dotyczące stosowania fiolki:
- Zebrać następujące materiały:
- Jedna fiolka leku Vabismo (w pudełku kartonowym).
- Jedna sterylna igła z filtrem przelewającym z końcówką tępą, 5 mikron, rozmiar 18G × 11/2 cala lub około 1,2 mm × 40 mm (w pudełku kartonowym).
- Jeden sterylny strzykawka Luer lock o pojemności 1 ml z oznaczeniem dawki 0,05 ml (nie dołączona).
- Jedna sterylna igła do wstrzyknięć, rozmiar 30G × 1/2 cala (nie dołączona).
- Należy zaznaczyć, że igła do wstrzyknięć 30G jest zalecana w celu uniknięcia zwiększonego oporu podczas wstrzykiwania, który może być konieczny przy użyciu igieł o mniejszej średnicy.
- Wacik alkoholowy (nie dołączony).
- Aby cała ciecz opadła na dno fiolki, postawić fiolkę pionowo na płaskiej powierzchni (około 1 minutę) po wyjęciu z opakowania, aby cała ciecz osiadła na dnie fiolki (patrz rysunek B). Ostrożnie stuknąć palcem w fiolkę (patrz rysunek C), ponieważ ciecz może przylegać do górnej części fiolki.
|
|
|
| Rysunek B |
Rysunek C |
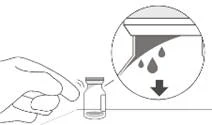
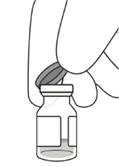
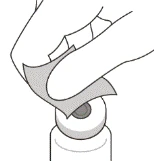
- Zdejmij pokrywkę z fiolki (patrz rysunku D) i przetrzy przegrodę fiolki gazikiem alkoholowym (patrz rysunku E).
|
|
|
| Rysunek D |
Rysunek E |
- Przy zachowaniu zasad aseptyki mocno przymocować dołączoną igłę z filtrem przelewowy 18G × 11/2 cala do strzykawki Luer Lock o pojemności 1 ml (patrz rysunek F).
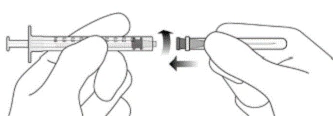
Rysunek F
- Przy zachowaniu zasad aseptyki wprowadzić igłę z filtrem przelewowy do środka przegrody fiolki (patrz rysunek G), wcisnąć ją całkowicie do fiolki, a następnie delikatnie pochylić fiolkę, aby igła dotknęła dolnej krawędzi fiolki (patrz rysunek H).
|
|
|
| Rysunek G |
Rysunek H |
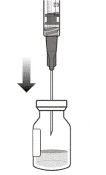
- Trzymać fiolkę lekko pochyloną i powoli wyciągnąć całą ciecz z fiolki (patrz ilustracja I). Trzymać koniec igły z filtrem do transferu zanurzony w cieczy, aby uniknąć dostania się powietrza.
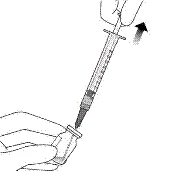
Ilustracja I
- Upewnić się, że trzpień strzykawki jest wystarczająco wyciągnięty do tyłu podczas opróżniania fiolki, aby całkowicie opróżnić igłę z filtrem do transferu (patrz ilustracja I).
- Odłączyć igłę z filtrem do transferu od strzykawki i usunąć ją zgodnie z lokalnymi wymaganiami.
Nie należy używać igły z filtrem do transferu do iniekcji wewnątrzwitrealnej.
- Zgodnie z zasadami aseptyki mocno przymocować igłę do iniekcji 30G × 1 1/2 cala do strzykawki Luer Lock (patrz ilustracja J).
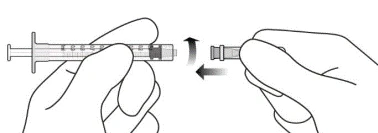
Ilustracja J
- Ostrożnie zdjąć plastikowy kaptur z igły, pociągając za niego.
- Aby sprawdzić obecność pęcherzyków powietrza, trzymać strzykawkę igłą do góry. Jeśli występują pęcherzyki powietrza, delikatnie postukać palcem w strzykawkę, aż pęcherzyki powietrza wypłyną do góry (patrz ilustracja K).
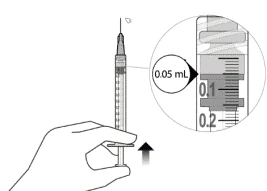
- Delikatnie usunąć powietrze ze strzykawki i igły, powoli naciskając na tłoczek, aż krawędź gumowego uszczelnienia tłoczka wyrówna się z oznaczeniem dawki 0,05 ml. Strzykawka jest teraz gotowa do iniekcji (patrz ilustracja L). Upewnić się, że iniekcja zostanie wykonana natychmiast po przygotowaniu dawki.
|
|
|
| Rysunek K |
Rysunek L |
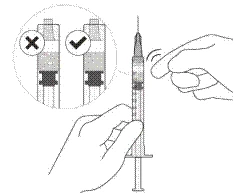
- Wprowadzać powoli, aż uszczelniacz gumowy tłoka osiągnie przednią część strzykawki, aby wprowadzić objętość 0,05 ml. Potwierdzić wprowadzenie pełnej dawki, sprawdzając, czy uszczelniacz gumowy osiągnął przednią część strzykawki po iniekcji.
Każdy niewykorzystany lek lub odpady należy zutylizować zgodnie z lokalnymi wymaganiami.
Dzieci.
Bezpieczeństwo i skuteczność stosowania leku Vabismo u dzieci nie są ustalone.
Przedawkowanie.
Dawek przekraczających zalecane nie badano. Przedawkowanie w wyniku przekroczenia zalecanego objętości iniekcji może prowadzić do podwyższonego ciśnienia wewnątrzgałkowego.
W przypadku przedawkowania należy kontrolować ciśnienie wewnątrzgałkowe i, jeśli lekarz uzna to za stosowne, należy rozpocząć odpowiednie leczenie.
Działania niepożądane.
Podsumowanie profilu bezpieczeństwa na podstawie wyników badań klinicznych
Poniższe dane dotyczące bezpieczeństwa pochodzą z badań fazy III z kontrolą aktywną (aflibercept).
Ogółem 4489 pacjentów weszło w skład populacji oceny bezpieczeństwa w sześciu badaniach klinicznych fazy III (2567 pacjentów otrzymujących leczenie lekiem Vabismou: 664 pacjentów z nAMD, 1262 pacjentów z DMN oraz 641 pacjentów z OGVN i OCVN). Najpoważniejszymi działaniami niepożądanymi były zaćma (0,8%), zapalenie uwey (0,5%), endoftalmia (0,4%), zapalenie ciała szklistego (0,4%), pęknięcia siatkówki (0,2%), odwarstwienie siatkówki o etiologii retymalnej (0,1%) oraz urazowa zaćma (< 0,1%).
Najczęstszymi działaniami niepożądanymi obserwowanymi u pacjentów otrzymujących leczenie lekiem Vabismou były zaćma (10%), krwawienie spojówkowe (7%), odwarstwienie siatkówki (4%), podwyższone ciśnienie wewnątrzgałkowe (4%), pływające zmętnienia ciała szklistego (4%), ból oka (3%) oraz pęknięcie nabłonka barwnikowego siatkówki (tylko u pacjentów z nAMD) (3%).
Poniżej opisane dane dotyczące bezpieczeństwa obejmują wykaz wszystkich zaobserwowanych działań niepożądanych w sześciu badaniach klinicznych fazy III przeprowadzonych u pacjentów z nAMD, DMN, OGVN i OCVN, a także dane z okresu posprzedażowego, z uwzględnieniem podstaw do rozważenia związku przyczynowo-skutkowego z zastrzykiem lub lekiem. Działania niepożądane są wymienione według klasy układów narządów według MedDRA i kategorii częstości: bardzo często (≥ 1/10), często (≥ 1/100 do < 1/10), nieczęsto (≥ 1/1000 do < 1/100), rzadko (≥ 1/10 000 do < 1/1000), częstość nieznana (częstość nie może być oszacowana na podstawie dostępnych danych).
Tabela 9
Podsumowanie działań niepożądanych
Powikłania |
Vabismou, N = 2 567 |
Kategoria częstości |
| Zaburzenia ze strony narządu wzroku |
||
| Katarakta |
9,7 % |
Często |
| Krwawienie do spojówki |
6,7 % |
Często |
| Odwarstwienie siatkówki |
4,2 % |
Często |
| Zwiększone ciśnienie wewnątrzgałkowe |
3,5 % |
Często |
| Unoszące się zmętnienia ciała szklistego |
3,5 % |
Często |
| Odwarstwienie pigmentowego nabłonka siatkówki (tylko nAMD) |
2,9 % |
Często |
| Ból oka |
2,5 % |
Często |
| Erozja rogówki |
0,9 % |
Nieczęsto |
| Zwiększone łzawienie |
0,8 % |
Nieczęsto |
| Irrytacja oczu |
0,8 % |
Nieczęsto |
| Niekomfort w oku |
0,7 % |
Nieczęsto |
| Świąd oczu |
0,7 % |
Nieczęsto |
| Pracenie oczu |
0,7 % |
Nieczęsto |
| Nieostrość widzenia |
0,7 % |
Nieczęsto |
| Spadek ostrości wzroku |
0,6 % |
Nieczęsto |
| Iryt |
0,6 % |
Nieczęsto |
| Uveitis |
0,5 % |
Nieczęsto |
| Endoftalmia |
0,4 % |
Nieczęsto |
| Odczucie ciała obcego |
0,4 % |
Nieczęsto |
| Krwawienie do ciała szklistego |
0,4 % |
Nieczęsto |
| Witryt |
0,4 % |
Nieczęsto |
| Irydocyklit |
0,3 % |
Nieczęsto |
| Pracenie spojówki |
0,2 % |
Nieczęsto |
| Ból podczas procedury |
0,2 % |
Nieczęsto |
| Przerwanie siatkówki |
0,2 % |
Nieczęsto |
| Regmatogenne odwarstwienie siatkówki |
0,1 % |
Nieczęsto |
| Przejściowy spadek ostrości wzroku |
< 0,1 % |
Rzadko |
| Urazowa katarakta |
< 0,1 % |
Rzadko |
| Waskulit siatkówki* |
- |
Częstość nieznana |
| Okludyjny waskulit siatkówki* |
- |
Częstość nieznana |
* Efekty uboczne zgłoszone na podstawie dobrowolnych doniesień pogwarancyjnych. Ponieważ doniesienia te pochodzą z populacji nieokreślonej liczebności, nie można wiarygodnie oszacować częstości występowania tych reakcji.
Opis poszczególnych efektów ubocznych oraz dodatkowe informacje
Po wewnątrzwitrealnej iniekcji inhibitorów VEGF istnieje teoretyczne ryzyko zdarzeń tromboembolii tętniczej, w tym udaru mózgu i zawału mięśnia sercowego. Niska częstość występowania zdarzeń tromboembolii tętniczej była obserwowana w badaniach klinicznych leku Vabismo u pacjentów z zaawansowaną postacią zwyrodnienia plamki, DMD, OZPZ i OZPŚ. Nie zaobserwowano istotnej różnicy między grupami leczonymi lekiem Vabismo a lekiem porównawczym w odniesieniu do różnych wskazań.
Doświadczenie z okresu pozarejestracyjnego
W okresie pozarejestracyjnym rzadko zgłaszano zapalenie naczyń siatkówki i/lub okluzyjne zapalenie naczyń siatkówki. Zapalenie naczyń siatkówki i okluzyjne zapalenie naczyń siatkówki zgłaszano również u pacjentów otrzymujących inną terapię wewnątrzwitrealną.
Zgłaszanie efektów ubocznych po rejestracji leku ma istotne znaczenie. Umożliwia to monitorowanie stosunku korzyści do ryzyka związanego z zastosowaniem tego leku. Osoby medyczne i farmaceutyczne, a także pacjenci lub ich ustawowo upoważnieni przedstawiciele powinni zgłaszać wszystkie przypadki podejrzewanych efektów ubocznych oraz braku skuteczności leku poprzez krajowy system zgłaszania (https://aisf.dec.gov.ua).
Okres ważności.
30 miesięcy.
Warunki przechowywania.
Przechowywać w temperaturze od 2 do 8 °C w oryginalnym opakowaniu w celu ochrony przed światłem.
Nie zamrażać. Nie wstrząsać. Przechowywać w miejscu niedostępnym dla dzieci.
Nieotwarty fiolka leku Vabismo może być przechowywana przed zastosowaniem w temperaturze pokojowej (od 20 do 25 °C) przez okres do 24 godzin.
Należy zapewnić natychmiastowe wykonanie iniekcji po przygotowaniu dawki.
Niezgodność.
W przypadku braku badań dotyczących niezgodności, tego leku nie należy mieszać z innymi lekami.
Opakowanie.
Fiolka
Fiolka o pojemności 2 ml wykonana z bezbarwnego szkła borokrzemowego klasy I, zamknięta gumową septą szarego koloru z butylkauczuku o średnicy 13 mm, warstwowana fluorokauczką, zamknięta aluminiową pokrywką o średnicy 13 mm z plastikowym dyskiem typu „flip-off”.
Igła z filtrem
Igła ze stali nierdzewnej 18 G z filtrem 5 μm wykonanym z akrylowego kopolimeru z pokrywką polipropylenową.
Po 1 fiolce w zestawie z igłą z filtrem, zapakowanym w blister, w pudełku kartonowym.
Kategoria wydawania.
Na receptę.
Producent.
F. Hoffmann-La Roche Ltd
Miejsce położenia producenta oraz adres miejsca prowadzenia działalności.
Wurmisweg, 4303 Kaiseraugst, Szwajcaria