Suranta®
UkrainaSpis treści
INSTRUKCJA dot. stosowania leku Suranta® (SURVANTA®)
Skład:
substancja czynna:
1 ml zawiesiny zawiera beraktant, który zawiera fosfolipidy ogółem – 25 mg/ml (w tym fosfatydylocholinę nasyconą – 11-15,5 mg/ml (standardyzowaną dipalmitoylofosfatydylocholiną), kwasy tłuszczowe wolne – 1,4-3,5 mg/ml (standardyzowane kwasem palmitynowym), trójglicerydy – 0,5-1,75 mg/ml (standardyzowane tripalmityną), białka związane z surfaktantem;
substancje pomocnicze: Natrium chloridum, aqua pro injectione, natrii hydroxidum, acidum hydrochloricum dilutum.
Postać leku. Zawiesina do wstrzykiwania do tchawicy.
Główne właściwości fizykochemiczne: nieprzezroczysta ciecz o barwie od prawie białej do jasnobrunatnej.
Grupa farmakoterapeutyczna. Surfaktanty płucne. Kombinacje. Kod ATC R07AA30.
Właściwości farmakologiczne.
Beraktant – naturalny surfaktant płuca pochodzący z płuc bydła.
Farmakologia kliniczna. Endogenny surfaktant płuca obniża napięcie powierzchniowe powierzchni alweli podczas wdechu i stabilizuje alweli przed zapadaniem się przy obniżeniu ciśnienia przepłucnego. Niedobór surfaktantu płuca prowadzi do rozwoju zespołu ostrej niewydolności oddechowej (RDS) u niemowląt przedwcześnie urodzonych. Beraktant uzupełnia zasoby surfaktantu i przywraca powierzchniową aktywność płuc u dzieci. W badaniach in vitro wykazano, że beraktant istotnie obniża minimalne napięcie powierzchniowe do mniej niż 8 dyn/cm, co stwierdzono przy użyciu surfaktometru i metody Wilhelmy. In situ beraktant przywraca elastyczność płuc u szczurów z eksperymentalnie wywołanym niedoborem surfaktantu. In vivo pojedyncze dawki beraktantu poprawiają parametry ciśnienia i objętości płuc, elastyczność płuc oraz oxygenację, co potwierdzono w badaniach na przedwcześnie urodzonych królikach i owcach.
Metabolizm u zwierząt. Efekty biofizyczne beraktantu manifestują się na powierzchni alweli, ponieważ lek jest podawany bezpośrednio do narządu docelowego – płuc. U przedwcześnie urodzonych królików i jagniąt z niedoborem surfaktantu obserwuje się szybki klirens alwolarny znakowanych izotopem lipidów beraktantu. Większość leku wiąże się z tkanką płuc w ciągu kilku godzin po podaniu, a lipidy wchodzą w skład endogennych szlaków recyrkulacji surfaktantu. U dorosłych zwierząt z wystarczającą ilością surfaktantu klirens beraktantu jest szybszy niż u zwierząt przedwcześnie urodzonych i młodych. U dorosłych zwierząt obserwuje się niższy poziom recyrkulacji surfaktantu. Ograniczone badania na zwierzętach nie wykazały wpływu beraktantu na endogenny metabolizm surfaktantu.
Brak informacji na temat metabolizmu białek związanych z surfaktantem w beraktancie. Badania metaboliczne z udziałem ludzi nie były prowadzone.
Dane kliniczne.
Wskazania.
- Leczenie zespołu zaburzeń oddechowych (RDS – choroby błony szklistej) u noworodków przedwczesnych;
- Profilaktyka RDS u noworodków przedwczesnych z masą ciała mniejszą niż 1250 g, u których istnieje ryzyko rozwoju RDS i którzy wymagają intubacji do ustabilizowania stanu lub wykazują objawy niedoboru surfaktantu.
Przeciwwskazania. Nieznane.
Interakcje z innymi lekami i inne rodzaje interakcji. Nie ustalone.
Szczególne wskazania dotyczące stosowania.
Zastosowanie leku Suranta® należy przeprowadzać wyłącznie w warunkach klinicznych przez wykwalifikowanych lekarzy, którzy przeszli specjalistyczne szkolenie i mają doświadczenie w intubacji, sztucznej wentylacji płuc (WUP) oraz opiece medycznej nad przedwczesnie urodzonymi noworodkami. Procedurę podania można ułatwić, jeśli jeden specjalista podaje dawkę, podczas gdy inni zapewniają odpowiednią pozycję dziecka i monitorowanie.
Lek może szybko wpływać na utlenowanie i elastyczność płuc. Widoczna poprawa utlenienia może wystąpić w ciągu kilku minut po podaniu leku. Aby uniknąć hiperoksji, konieczne jest ciągłe i staranne obserwowanie kliniczne oraz kontrola utlenienia ogólnoustrojowego.
Opisywano przypadki przejściowej bradycardii oraz spadku nasycenia krwi tlenem podczas podawania leku. Jeśli wystąpią te objawy, należy przerwać procedurę podania i podjąć odpowiednie działania mające na celu złagodzenie stanu dziecka. Po ustabilizowaniu stanu pacjenta procedurę można wznowić.
Podanie leku Suranta® za pomocą dwukanałowej rurki intubacyjnej jest funkcjonalnie równoważne stosowaniu zaworu do aspiracji, tj. uwalnianie beraktantu na końcu dystalnym rurki intubacyjnej odbywa się bez przerywania WUP. Ta metoda podania powinna zmniejszyć liczbę przypadków hipoksji i bradycardii, które mogą wystąpić bezpośrednio po podaniu dawki. Jednakże, na podstawie danych z badań klinicznych, nie stwierdzono różnic w wynikach krótko- i długoterminowych w porównaniu z innymi metodami podania.
Ogólne ostrzeżenia
Natychmiast po podaniu leku mogą tymczasowo występować świsty i chrypki, co nie jest objawem przedawkowania. Jeśli nie ma wyraźnych objawów obturacji dróg oddechowych, aspiracja endotchialna ani inne działania nagłego charakteru nie są konieczne.
W kontrolowanych badaniach klinicznych zaobserwowano zwiększone ryzyko rozwoju sepsy szpitalnej u niemowląt leczonych lekiem Suranta®. Zwiększony ryzyko sepsy u osób otrzymujących Surantę® nie było związane ze wzrostem śmiertelności. Patogeny były podobne u niemowląt leczonych i w grupie kontrolnej. Nie stwierdzono istotnych różnic między grupami pod względem częstości występowania innych infekcji po podanym leczeniu.
Nie prowadzono badań klinicznych dotyczących stosowania beraktantu u niemowląt o masie ciała przy urodzeniu mniejszej niż 600 g lub większej niż 1750 g.
Brak doświadczenia w stosowaniu beraktantu w połączeniu z eksperymentalnymi metodami leczenia zespołu zaburzeń oddechowych (wentylacja wysokiej częstotliwości lub ekstrakorpowalna oxygenacja membranowa).
Brak informacji dotyczącej stosowania leku w dawkach innych niż 100 mg/kg, stosowania więcej niż czterech dawek lub częstszego podawania niż co 6 godzin, a także podawania niemowlętom po 48 godzinach życia.
Nieotwarte, nieużywane fiolki leku Suranta®, ogrzane do temperatury pokojowej, należy jak najszybciej umieścić z powrotem w lodówce w ciągu 24 godzin od ogrzania i przechowywać do późniejszego użycia. Leku Suranta® nie należy ogrzewać i ponownie umieszczać w lodówce więcej niż jeden raz. Każdą jednorazową fiolkę można użyć tylko raz. Używane fiolki z resztką leku należy zutylizować.
Jeśli lek przypadkowo został zamrożony, należy zrezygnować z jego stosowania, a nieużywaną fiolkę zutylizować.
SURANTA® NIE WYMAGA ROZPUSZCZANIA ANI OBRÓBKI ULTRADŹWIĘKOWEJ PRZED ZASTOSOWANIEM.
Stosowanie w czasie ciąży lub karmienia piersią.
Nie wskazany do stosowania u dorosłych (patrz sekcja „Wskazania”).
Możliwość wpływania na szybkość reakcji przy prowadzeniu pojazdów lub obsługiwania innych mechanizmów.
Nie wskazany do stosowania u dorosłych (patrz sekcja „Wskazania”).
Sposób stosowania i dawki.
Stosować wyłącznie do podania do oskrzeli.
Profilaktyka zespołu zaburzeń oddechowych (RDS). Lek należy podać jak najszybciej, najlepiej w ciągu pierwszych 15 minut życia. Leczenie (pilna pomoc) zespołu zaburzeń oddechowych (RDS). Lek stosuje się jak najszybciej po rozpoczęciu wentylacji mechanicznej, najlepiej w ciągu pierwszych 8 godzin życia.
W ciągu 48 godzin można podać cztery dawki leku Suranta® w odstępach nie krótszych niż 6 godzin.
Dawka pojedyncza leku Suranta® wynosi 100 mg fosfolipidów (4 ml zawiesiny) na 1 kg masy ciała noworodka.
Przygotowanie do podania
Sprawdzić kolor leku, który powinien być od niemal białego do jasnobrązowego. Jeżeli podczas przechowywania utworzył się osad, należy ostrożnie obrócić fiolkę, aby przywrócić zawiesinę. NIE WSTRZĄSAĆ. Może pojawić się pianka na powierzchni – jest to typowe dla natury leku. Przed zastosowaniem lek Suranta® należy ogrzać do temperatury pokojowej przez co najmniej 20 minut lub w dłoni przez co najmniej 8 minut. NIE WOLNO STOSOWAĆ SZTUCZNYCH METOD OGRZEWANIA. Jeżeli planowane jest zastosowanie leku w celu profilaktyki, przygotowanie środka leczniczego do podania należy rozpocząć wcześnie, przed porodem dziecka.
Procedura podania
Lek Suranta® podaje się do oskrzeli za pomocą kaniuli o kalibrze 5 French z otworem na końcu dystalnym. Kaniulę wprowadza się do rurki intubacyjnej po szybkim odłączeniu jej od aparatu do wentylacji mechanicznej lub przez zawór do ssania, bez odłączania rurki intubacyjnej od aparatu do wentylacji mechanicznej; alternatywnie instylację można przeprowadzić przez dodatkowy kanał dwukanałowej rurki intubacyjnej.
Obliczyć dawkę leku zgodnie z masą ciała noworodka. Powoli za pomocą dużej igły (co najmniej 20 G) nabrać całą zawartość fiolki(i) do plastikowej strzykawki. NIE FILTROWAĆ ŚRODKU LECZNICZEGO I UNIKAĆ INTENSYWNEGO WSTRZĄSANIA.
Aby zapewnić jednolite rozmieszczenie beraktantu w płucach, każdą dawkę dzieli się na dawki frakcjonowane (częściowe). Każdą dawkę można podzielić na dwie lub cztery dawki frakcjonowane. Każdą dawkę frakcjonowaną podaje się dziecku w różnych pozycjach ciała.
Do podania leku w dwóch dawkach frakcjonowanych zalecane są następujące pozycje:
- Głowa i tułów obrócone o około 45° w prawo.
- Głowa i tułów obrócone o około 45° w lewo.
Pozycje do podania czterech dawek frakcjonowanych zilustrowane są poniżej:
|
|
|
|
|
|
|
|
|
|

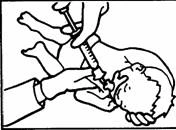
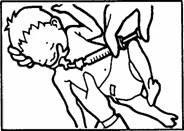
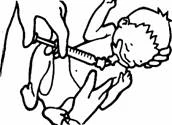
Podanie pierwszej dawki.
Podanie przez kaniulę z otworem końcowym
Skrócić kaniulę tak, aby jej koniec ledwo wystawał poza rurkę intubacyjną, nieco powyżej rozgałęzienia tchawicy. Nie wolno podawać leku do oskrzela głównego.
Dołączyć kaniulę do strzykawki. Napełnić kaniulę lekiem. Usunąć nadmiar leku przez kaniulę, tak aby w strzykawce pozostał tylko pełny dawkę do podania. W przypadku nagłej pomocy w ZRL przed podaniem pierwszej dawki frakcjonowanej ustawić następujące parametry respiratora: częstość – 60/min, czas wdechu – 0,5 sek, zawartość tlenu (FiO₂) – 1,0. Ułożyć dziecko w odpowiedniej pozycji i powoli podać pierwszą dawkę frakcjonowaną przez kaniulę w ciągu 2–3 sekund. Po podaniu pierwszej dawki frakcjonowanej usunąć kaniulę z rurki intubacyjnej. W przypadku podania profilaktycznego prowadzić wentylację ręczną za pomocą worka z odpowiednią podażą tlenu, aby zapobiec wystąpieniu cyjanозy, z częstością około 60/min i wystarczającym ciśnieniem dodatnim, aby zapewnić adekwatny wymianę powietrza i ekspansję klatki piersiowej; w przypadku nagłej pomocy w ZRL wznowić wentylację mechaniczną za pomocą respiratora. Między podawaniem dawek frakcjonowanych należy wentylować dziecko przez co najmniej 30 sekund lub do ustabilizowania stanu. Zmienić pozycję dziecka przed instylacją kolejnej dawki frakcjonowanej. Pozostałe dawki frakcjonowane podaje się, stosując opisany powyżej ciąg czynności. Po instylacji końcowej dawki frakcjonowanej usunąć kaniulę bez płukania. Nie przeprowadzać aspiracji zawartości oskrzeli przez 1 godzinę po procedurze podania leku, z wyjątkiem przypadków wystąpienia objawów znacznej obturacji dróg oddechowych.
Instylacja przez drugi światłowiec dwuświatłowej rurki intubacyjnej
Wykonuje się według tej samej procedury frakcjonowanej przez drugi otwór rurki bez przerywania wentylacji mechanicznej. Po instylacji końcowej dawki frakcjonowanej usunąć strzykawkę z drugiego światłowodu, Wprowadzić 0,5 ml POWIETRZA, ABY PRZEDMUCHAĆ DRUGI ŚWIATŁOWÓD RURKI, I ZAMKNIĘĆ GO.
Podawanie dawek powtórzonych
Potrzeba dodatkowych dawek beraktantu ustala się w przypadku utrzymywania się objawów ZRL. Parametry respiratora przy podawaniu dawek powtórzonych różnią się od tych stosowanych przy pierwszej dawce: FiO₂ zwiększyć o 0,20 (lub o wartość wystarczającą do zapobiegania cyanozie), czas wdechu < 1,0 sek, częstość oddechów – 30/min, która nie zmienia się w czasie podania leku, jeśli przed podaniem częstość oddechów była większa niż 30/min.
Do podania dawek powtórzonych nie wolno stosować wentylacji ręcznej za pomocą worka.
W TRAKCIE PROCEDURY PODANIA PARAMETRY RESPIRATORA MOGĄ BYĆ SKORYGOWANE PRZEZ LEKARZA W CELU ZAPEWNIENIA ADEKWATNEJ OXYGENACJI I WENTYLACJI.
Podawanie noworodkom z oddychaniem spontanicznym
Intubacja – surfaktant – ekstubacja (INSURE)
Po intubacji i wprowadzeniu kaniuli, jak opisano powyżej, ułożyć noworodka w pozycji neutralnej i ostrożnie podać dawkę leku jednym strzykawkowym wstrząśnieniem w ciągu 1–3 minut w sali porodowej lub po przeniesieniu do oddziału neonatologicznego. Po podaniu rozpocząć wentylację workiem oraz ekstubację i CPAP zgodnie z wskazaniami klinicznymi.
Dzieci.
Lek wskazany jest do stosowania u noworodków przedwczesnych (patrz sekcja „Wskazania”).
Przedawkowanie.
Nie odnotowano przypadków przedawkowania beraktantu. Przedawkowanie może objawiać się ostrą obturacją dróg oddechowych. Leczenie powinno być objawowe i wspierające.
Niepożądane działania.
Najczęściej zgłaszane niepożądane reakcje były związane z procedurą podania leku. W kontrolowanych badaniach klinicznych tymczasowa bradykardia występowała w 11,9% przypadków, a obniżenie stężenia tlenu – w 9,8% przypadków. Inne reakcje obserwowane podczas procedury podania wystąpiły poniżej 1% wszystkich podań i obejmowały refluks z rurki intubacyjnej, bladość, wazokonstrykcję, hipotensję tętniczą, zablokowanie rurki intubacyjnej, nadciśnienie tętnicze, hipokapnię, hiperkapnię oraz apneę. Nie odnotowano przypadków śmiertelnych podczas procedury podania; wszystkie reakcje ustępowały po leczeniu objawowym.
W kontrolowanych badaniach klinicznych oceniano częstość chorób typowych dla noworodków przedwczesnych. Dane ze wszystkich kontrolowanych badań przedstawiono w poniższej tabeli.
Choroby towarzyszące typowe dla noworodków przedwczesnych, według danych ze wszystkich kontrolowanych badań.
| Powiązane choroby |
Dzieci przedwczesne otrzymujące beraktant (%) |
Grupa kontrolna (%) |
Wartość P |
| Przetrwały przewód tętniczy |
46,9 |
47,1 |
0,814 |
| Krwawienie wewnątrzczaszkowe |
48,1 |
45,2 |
0,241 |
| Ciężkie krwawienie wewnątrzczaszkowe |
24,1 |
23,3 |
0,693 |
| Zespół wycieku powietrza |
10,9 |
24,7 |
<0,001 |
| Interstycjalna emfizema płuc |
20,2 |
38,4 |
<0,001 |
| Nekrotyzujące zapalenie jelit |
6,1 |
5,3 |
0,427 |
| Apnea |
65,4 |
59,6 |
0,283 |
| Ciężka apnea |
46,1 |
42,5 |
0,114 |
| Sepsa (po leczeniu) |
20,7 |
16,1 |
0,019 |
| Zakażenie (po leczeniu) |
10,2 |
9,1 |
0,345 |
| Krwawienie do płuc |
7,2 |
5,3 |
0,166 |
Częstotliwość występowania krwawienia śródczaszkowego u dzieci otrzymujących beraktant nie różniła się od częstości w ogólnej populacji takich pacjentów. Zgłaszano również krwawienie do płuc. Nie odnotowano innych poważnych działań niepożądanych.
W kontrolowanych badaniach klinicznych wykazano brak wpływu leku Suranta® na wyniki ogólnych badań laboratoryjnych: liczbę leukocytów, stężenie sodu, potasu, bilirubiny i kreatyniny w osoczu krwi.
Nie wykryto przeciwciał wobec białek leku Suranta®.
W kontrolowanych badaniach klinicznych zgłaszano poniżej opisane stany, których częstość nie różniła się u dzieci otrzymujących leczenie w porównaniu z grupą kontrolną, a żadne z powikłań nie było związane z beraktantem.
Ze strony układu krwiotwórczego i chłonnego: koagulopatia, trombocytopenia, rozsiane wewnątrznaczyniowe krzepnięcie krwi.
Ze strony układu endokrynnego: krwawienie do nadnerczy, niedostateczna sekrecja ADH.
Zaburzenia metabolizmu i trawienia: hiperfosfatemia, nietolerancja pokarmu.
Zaburzenia neurologiczne: drgawki.
Ze strony serca: tachykardia, tachykardia komorowa, niewydolność serca, zatrzymanie akcji serca, nasilony puls wierzchołkowy, utrzymujący się krążenie płodowe, całkowity nietypowy odpływ żył płucnych.
Ze strony naczyń: nadciśnienie tętnicze, nadciśnienie tętnicze, zakrzepica aorty, zakrzepica powietrzna.
Ze strony układu oddechowego, organów klatki piersiowej i jamy międzybłoniowej: konsolidacja płuc, wyciek krwi z rurki intubacyjnej, pogorszenie stanu po odstawieniu od respiratora, dekompensacja oddechowa, zwężenie podgłośniowe, porażenie przepony, niewydolność oddechowa.
Ze strony przewodu pokarmowego: rozdęcie brzucha, krwawienie z przewodu pokarmowego, perforacje jelita, zwoj przewodu pokarmowego, zawał jelita, wrzód stresowy, przepuklina pachwinowa.
Ze strony układu wątrobowo-żółciowego: niewydolność wątroby.
Ze strony układu moczowego: niewydolność nerek, hematuria.
Ogólne zaburzenia i reakcje w miejscu podania: gorączka, załamanie mechanizmów kompensacyjnych.
Długoterminowe badania. Do chwili obecnej nie stwierdzono żadnych późnych powikłań ani skutków terapii beraktantem.
Technika INSURE
Wyniki badania bezpieczeństwa stosowania techniki INSURE były porównywalne z wynikami w grupach kontrolnych.
Okres ważności. 18 miesięcy.
Warunki przechowywania.
Przechowywać w miejscu niedostępnym dla dzieci w temperaturze od 2 do 8 °C (w lodówce) we wkładzie oryginalnym z tekturowej puszki. Nie zamrażać!
Niezgodność. Nie określono.
Opakowanie. 4 ml lub 8 ml w fiolkach szklanych, zamkniętych butelkowymi korkami gumowymi i uszczelnionych aluminiowymi kapturkami. Po 1 fiolce w pudełku tekturowym.
Kategoria wydawania. Na receptę.
Producent. Ebbvі Inc., USA/AbbVie Inc., USA.
Miejsce położenia producenta oraz adres miejsca prowadzenia jego działalności. 1401 Sheridan Road, North Chicago, Illinois (IL) 60064, USA.