Spiriva® Respimat®
Ukraina
Spis treści
INSTRUKCJA dotyczÄ cza stosowania leku Spiriva® Respimat® (Spiriva® Respimat®)
SkÅ ad:
substancja czynna: tiotropium bromku monohydrat odpowiadajÄ cym tiotropium;
1 inhalacja zawiera 3,124 μg tiotropium bromku monohydratu odpowiadajÄ cego 2,5 μg tiotropium;
substancje pomocnicze: benzalkonium chlorid, dinatrium edetas, woda oczyszczona, kwas chlorowodorowy rozcieÅ niczony.
PostaÄ leku. RozwiÄ zanie do inhalacji.
GÅ wne wÅ asnoÅ ci fizykochemiczne: kartusze o pojemnoÅ ci do 4,5 ml wypeÅ nione pÅ ynnoÅ ciÄ , zamkniÄ te w cylindrach aluminiowych do inhalatora Respimat®, z wciÅ niÄ tÄ ochronnÄ wkÅ adkÄ .
Grupa farmakoterapeutyczna. Inne leki do leczenia chorób obturacyjnych dróg oddechowych, leki do inhalacji. Šrodki antycholinergiczne. Tiotropium bromku.
Kod ATC R03B B04.
Właściwości farmakologiczne.
Farmakodynamika.
Mechanizm działania
Bromek tiotropium jest specyficznym antagonistą receptora muskarynowego o długotrwałym działaniu. Tiotropium wykazuje podobne powinowactwo do podtypów receptorów od M1 do M5. W drogach oddechowych bromek tiotropium wiąże się konkurencyjnie i odwracalnie z receptorami M3 mięśni gładkich oskrzeli, przeciwdziałając cholinergicznemu (bronchokonstrykcyjnemu) działaniu acetylocholiny, co prowadzi do rozluźnienia mięśni gładkich oskrzeli. Efekt był zależny od dawki i trwał ponad 24 godziny. Ponieważ tiotropium jest miejscowym, bronchoselektywnym antycholinergikiem o czwórkowym charakterze azotu, po zastosowaniu wziewnym wykazuje odpowiedni zakres terapeutyczny przed wystąpieniem ogólnoustrojowych efektów antycholinergicznych.
Właściwości farmakodynamiczne
Dysocjacja bromku tiotropium, szczególnie od receptorów M3, jest bardzo powolna. W związku z tym okres półtrwania jest znacznie dłuższy niż w przypadku ipratropium. Dysocjacja od receptorów M2 jest szybsza niż od receptorów M3, co w badaniach funkcjonalnych in vitro wykazało większą selektywność (kontrolowaną kinetycznie) wobec podtypu receptora M3 niż M2. Stwierdzono, że wysoka aktywność, bardzo powolna dysocjacja z receptorów oraz miejscowe działanie selektywne po podaniu wziewnym korelują klinicznie z istotnym i długotrwałym działaniem rozszerzającym oskrzela u pacjentów z POCh i astmą.
Skuteczność kliniczna i bezpieczeństwo u pacjentów z POCh
Program klinicznej opracowywalności w fazie III obejmował dwa jednoroczne, dwa 12-tygodniowe oraz dwa 4-tygodniowe randomizowane, podwójnie ślepe badania z udziałem 2901 pacjentów z POCh (1038 pacjentów otrzymywało po 5 μg tiotropium). Program jednoroczny obejmował dwa badania kontrolowane placebo. Dwa 12-tygodniowe badania obejmowały kontrolę zarówno aktywnym lekiem (ipratropium), jak i placebo. Wszystkie sześć badań obejmowało pomiar funkcji płuc. Ponadto dwa jednoroczne badania obejmowały ocenę częstości duszności, jakości życia związaną ze zdrowiem oraz wpływ na częstość zaostrzeń.
Badania kontrolowane placebo. Funkcja płuc
Roztwór do inhalacji tiotropium podawany raz dziennie zapewnił istotne poprawienie funkcji płuc (objętość wymuszona w pierwszej sekundzie i całkowita pojemność życiowa płuc) w ciągu 30 minut po pierwszej dawce w porównaniu z placebo (średnie poprawienie OBJ1 po 30 minutach: 0,113 litra; 95% przedział ufności (PU): od 0,102 do 0,125 litra, p < 0,0001). Poprawa funkcji płuc utrzymywała się przez 24 godziny w stanie ustalonym w porównaniu z placebo (średnie poprawienie OBJ1: 0,122 litra; 95% PU: od 0,106 do 0,138 litra, p < 0,0001).
Stan farmakodynamiczny ustalony osiągnięto po jednym tygodniu.
Lek SPIRIVA RESPIMAT® istotnie poprawił wskaźnik porannej i wieczornej maksymalnej objętościowej prędkości wydechu według codziennych pomiarów wykonywanych przez pacjentów w porównaniu z placebo (średnie poprawienie maksymalnej objętościowej prędkości wydechu: rano o 22 l/min; 95% PU: od 18 do 55 l/min, p < 0,0001; wieczorem o 26 l/min; 95% PU: od 23 do 30 l/min, p < 0,0001). Stosowanie leku SPIRIVA RESPIMAT® prowadziło do zmniejszenia częstości stosowania leków rozszerzających oskrzela w nagłych przypadkach w porównaniu z placebo (średnie zmniejszenie częstości stosowania leków ratunkowych o 0,66 przypadku na dobę; 95% PU: od 0,51 do 0,81 przypadku na dobę, p < 0,0001).
Efekty bronchodylatacyjne leku SPIRIVA RESPIMAT® utrzymywały się przez cały jednoroczny okres stosowania bez oznak tolerancji.
Duszność, jakość życia związana ze zdrowiem, zaostrzenia POCh w długoterminowych jednorocznych badaniach.
Duszność. Lek SPIRIVA RESPIMAT® istotnie zmniejszył częstość występowania duszności (na podstawie oceny według indeksu przejściowej duszności) w porównaniu z placebo (średnie poprawienie o 1,05 punktu; 95% PU: od 0,73 do 1,38 punktu, p < 0,0001). Poprawa utrzymywała się przez cały okres leczenia.
Jakość życia związana ze zdrowiem. Poprawa średniej ogólnej oceny jakości życia przez pacjentów (mierzonej za pomocą kwestionariusza Hospital Saint George’s Respiratory Questionnaire) przy stosowaniu leku SPIRIVA RESPIMAT® w porównaniu z placebo w dwóch jednorocznych badaniach wyniosła 3,5 punktu (95% PU: od 2,1 do 4,9, p < 0,0001). Uznaje się, że zmniejszenie o 4 punkty jest klinicznie istotne.
Zaostrzenia POCh
Wyniki trzech jednorocznych randomizowanych, podwójnie ślepych, kontrolowanych placebo badań klinicznych leczenia lekiem SPIRIVA RESPIMAT® wykazały istotne zmniejszenie ryzyka zaostrzeń POCh w porównaniu z placebo. Zaostrzenia POCh definiowano jako wystąpienie co najmniej dwóch objawów/znaków w układzie oddechowym trwających trzy dni lub dłużej, wymagających zmiany terapii (przepisanie antybiotyków i/lub kortykosteroidów ogólnoustrojowych i/lub istotnej zmiany leków przepisanych na choroby układu oddechowego). Leczenie lekiem SPIRIVA RESPIMAT® prowadziło do zmniejszenia ryzyka hospitalizacji z powodu zaostrzenia POCh (istotne zmniejszenie ryzyka w dużym badaniu z udziałem pacjentów z zaostrzeniami).
Wyniki analizy łącznej dwóch badań fazy III oraz oddzielnej analizy dodatkowego badania z udziałem pacjentów z zaostrzeniami przedstawiono w tabeli 1. Wszystkie leki na choroby układu oddechowego, z wyjątkiem leków antycholinergicznych i agonistów β o długotrwałym działaniu, były dozwolone jako terapia towarzysząca, tj. szybko działające agonisty β, glikokortykosteroidy wziewne i ksantyny. W badaniu oceniającym wpływ na zaostrzenia u pacjentów z zaostrzeniami dozwolone było również stosowanie agonistów β o długotrwałym działaniu.
Tabela 1
Analiza statystyczna przypadków zaostrzeń POCh i przypadków hospitalizacji z powodu zaostrzeń POCh u pacjentów z POCh o umiarkowanym do bardzo ciężkiego stopnia
| Badanie |
Punkt końcowy |
Spiriva Respimat |
Placebo |
% redukcja ryzyka (95 % CI)a |
Wartość p |
| Badania faz III roczne, |
Liczba dni do pierwszego zaostrzenia POChP |
160a |
86a |
29 (od 16 do 40)b |
< 0,0001b |
| Średnia częstość przypadków zaostrzeń na pacjenta-rok |
0,78c |
1,00c |
22 (od 8 do 33)c |
0,002c |
|
| Czas do pierwszej hospitalizacji z powodu zaostrzenia POChP |
25 (od -16 do 51)b |
0,20b |
|||
| Średnia częstość przypadków hospitalizacji z powodu zaostrzenia na pacjenta-rok |
0,09 c |
0,11 c |
20 (od -4 do 38) c |
0,096 c |
|
| Badanie jednoroczne fazy IIIb z udziałem pacjentów z zaostrzeniem |
Liczba dni do pierwszego zaostrzenia POChP |
169a |
119a |
31 (od 23 do 37)b |
< 0,0001b |
| Średnia częstość przypadków zaostrzeń na pacjenta-rok |
0,69c |
0,87c |
21 (od 13 do 28)c |
< 0,0001c |
|
| Czas do pierwszej hospitalizacji z powodu zaostrzenia POChP |
27 (od 10 do 41)b |
0,003b |
|||
| Średnia częstość przypadków hospitalizacji z powodu zaostrzenia na pacjenta-rok |
0,12c |
0,15c |
19 (od 7 do 30)c |
0,004c |
a Czas do pierwszego zdarzenia: liczba dni leczenia do momentu, w którym u 25 % pacjentów wystąpi przynajmniej jedno zaostrzenie POChP / hospitalizacja z powodu zaostrzenia POChP. W badaniu A u 25 % pacjentów otrzymujących placebo zaobserwowano zaostrzenie do dnia 112, podczas gdy u 25 % pacjentów leczonych lekiem Spiriva® Respimat® zaostrzenie wystąpiło do dnia 173 (p=0,09); w badaniu B u 25 % pacjentów otrzymujących placebo zaobserwowano zaostrzenie do dnia 74, podczas gdy u 25 % pacjentów leczonych lekiem Spiriva® Respimat® zaostrzenie wystąpiło do dnia 149 (p<0,0001).
b Stosunek ryzyka oszacowano na podstawie modelu proporcjonalnych ryzyk Coxa. Procentowy wskaźnik zmniejszenia ryzyka wynosi 100 (1 ─ stosunek ryzyka).
c Regresja Poissona. Zmniejszenie ryzyka wynosi 100 (1 ─ względne ryzyko).
d Połączenie wyników zostało określone podczas projektowania badań. Punkty końcowe oceny zaostrzeń znacząco poprawiły się w indywidualnych analizach wyników dwóch jednorocznych badań.
Długoterminowe badanie kontrolowane aktywnie tiotropium
Przeprowadzono długotrwałe, duże, randomizowane, podwójnie ślepe, kontrolowane aktywnie badanie z okresem obserwacji do 3 lat w celu porównania skuteczności i bezpieczeństwa stosowania leku Spiriva® Respimat® i Spiriva® z inhalatorem HandiHaler (Spiriva). W badaniu uczestniczyło 5711 pacjentów otrzymujących lek Spiriva® Respimat® i 5694 pacjentów otrzymujących lek Spiriva. Głównymi punktami końcowymi były czas do pierwszego zaostrzenia POChP, czas do śmierci z dowolnej przyczyny oraz – w podgrupie (906 pacjentów) – minimalna wartość FEV1 (przed podaniem dawki).
Czas do pierwszego zaostrzenia POChP podczas badania był liczbowo podobny przy stosowaniu leków Spiriva® Respimat® i Spiriva (współczynnik ryzyka (Spiriva® Respimat®/Spiriva) 0,98, 95 % przedział ufności (PU) od 0,93 do 1,03). Średni czas do pierwszego zaostrzenia POChP wyniósł 756 dni przy stosowaniu leku Spiriva® Respimat® i 719 dni przy stosowaniu leku Spiriva.
Efekt broncholityczny leku Spiriva® Respimat® utrzymywał się przez 120 tygodni i był podobny do efektu leku Spiriva. Średnia różnica w minimalnym FEV1 pomiędzy grupą leku Spiriva® Respimat® a grupą leku Spiriva wyniosła -0,010 l (95 % PU od -0,038 do 0,018 l).
W badaniu porównawczym po wprowadzeniu na rynek TIOSPIR leków Spiriva® Respimat® i Spiriva z inhalatorem HandiHaler stwierdzono podobne wskaźniki śmiertelności z przyczyn wszystkich, w tym kontrolę stanów życiowo ważnych, w badanych grupach leków (stosunek ryzyka (Spiriva z inhalatorem HandiHaler/Spiriva® Respimat®): 0,96, 95 % PU 0,84–1,09). Ekspozycja na leczenie wyniosła odpowiednio 13 135 i 13 050 pacjentów-let.
W placebo-kontrolowanych badaniach z monitorowaniem stanów życiowych do końca zaplanowanego okresu leczenia w grupie leku Spiriva® Respimat® zaobserwowano zwiększenie wskaźnika śmiertelności z dowolnej przyczyny w porównaniu z grupą placebo (stosunek wskaźników (95 % przedział ufności) 1,33 (0,93, 1,92) przy długości ekspozycji na lek Spiriva® Respimat® wynoszącej 2574 pacjentów-let; zwiększenie śmiertelności obserwowano u pacjentów z znanymi zaburzeniami rytmu serca. W grupie leku Spiriva zaobserwowano zmniejszenie ryzyka śmierci o 13 % (stosunek ryzyka, w tym kontrolę stanów życiowych (tiotropium/placebo) = 0,87; 95 % PU od 0,76 do 0,99). Ekspozycja na leczenie lekiem Spiriva wyniosła 10 927 pacjentów-let. Zwiększenia ryzyka śmierci nie zaobserwowano w podgrupie pacjentów z znanymi zaburzeniami rytmu serca w placebo-kontrolowanym badaniu leku Spiriva, jak również w badaniu TIOSPIR porównującym leki Spiriva® Respimat® i Spiriva.
Skuteczność kliniczna i bezpieczeństwo u pacjentów z astmą
Program kliniczny fazy III dla dorosłych pacjentów z astmą przewlekłą obejmował dwa jednoroczne, randomizowane, podwójnie ślepe, placebo-kontrolowane badania z udziałem 907 pacjentów z astmą (453 pacjentów otrzymujących lek Spiriva® Respimat®), którym podawano leczenie skojarzone z kortykosteroidem inhalacyjnym (ICS) (≥ 800 µg budezonidu na dobę lub równowartość) i agonistami β trwalego działania. Badania obejmowały pomiary funkcji płuc oraz ocenę ciężkich zaostrzeń jako główne punkty końcowe.
Badania PrimoTinA z udziałem pacjentów z astmą
W dwóch jednorocznych badaniach u pacjentów, u których nadal występowały objawy astmy mimo leczenia podtrzymującego obejmującego co najmniej ICS (≥ 800 µg budezonidu na dobę lub równowartość) w połączeniu z agonistami β trwalego działania, lek Spiriva® Respimat® wykazał klinicznie istotne poprawienie funkcji płuc w porównaniu z placebo, gdy stosowano go w połączeniu z podstawowym leczeniem.
Po 24 tygodniach średnie poprawienie wartości FEV1 szczytowego i minimalnego wyniosło odpowiednio 0,110 litra (95 % PU: od 0,063 do 0,158 litra, p<0,0001) i 0,093 litra (95 % PU: od 0,050 do 0,137 litra, p<0,0001). Poprawa funkcji płuc w porównaniu z placebo utrzymywała się przez 24 godziny.
W badaniach PrimoTinA u pacjentów z astmą leczenie pacjentów (N=453) kombinacją ICS, agonistów β trwalego działania i tiotropium zmniejszyło ryzyko wystąpienia ciężkich zaostrzeń astmy o 21 % w porównaniu z leczeniem pacjentów (N=454) kombinacją ICS, agonistów β trwalego działania i placebo. Zmniejszenie ryzyka pod względem średniej częstości ciężkich zaostrzeń astmy na jednego pacjenta-rok wyniosło 20 %.
Te wyniki potwierdzone zostały przez zmniejszenie ryzyka nasilenia objawów astmy o 31 % oraz zmniejszenie ryzyka o 24 % pod względem średniej częstości nasilenia objawów astmy na jednego pacjenta-rok (patrz tabela 2).
Tabela 2
Zaostrzenia u pacjentów, u których nadal występowały objawy astmy mimo stosowania ICS (≥800 µg budezonidu na dobę lub równowartość) w połączeniu z agonistami β trwalego działania (badania PrimoTinA z udziałem pacjentów z astmą)
| Badanie |
Punkt końcowy |
Spiriva® Respimat® w kombinacji z co najmniej jednym IKSa/agonistą β długodziałającym |
Placebo w kombinacji z co najmniej jednym IKSa/agonistą β długodziałającym |
% redukcji ryzyka (95 % CI) |
Wartość p |
| Dwa jednoroczne badania fazy III, |
Liczba dni do pierwszego poważnego zaostrzenia astmy |
282c |
226c |
21b |
0,0343 |
| Średnia liczba przypadków ciężkich zaostrzeń astmy na jednego pacjenta-rok |
0,530 |
0,663 |
20d |
0,0458 |
|
| Liczba dni do pierwszego nasilenia objawów astmy |
315c |
181c |
31b |
<0,0001 |
|
| Średnia liczba przypadków nasilenia objawów astmy na jednego pacjenta-rok |
2,145 |
2,835 |
24d |
0,0031 |
a ≥ 800 µg budesonidu na dobę lub równowartość.
b Stosunek ryzyka, przedział ufności i wartość p uzyskane na podstawie modelu proporcjonalnych ryzyk Coxa, z uwzględnieniem jedynie efektywnej terapii. Procentowy stosunek zmniejszenia ryzyka wynosi 100 (1 – stosunek ryzyka).
c Czas do pierwszego zdarzenia: liczba dni do leczenia w momencie, gdy u 25 %/50 % pacjentów wystąpiło co najmniej jedno poważne nasilenie astmy/zaostrzenie astmy.
d Ryzyko względne ustalono na podstawie regresji Poissona z wykorzystaniem logarytmu ekspozycji (w latach) jako korekty. Procentowe zmniejszenie ryzyka wynosi 100 (1 – ryzyko względne).
Pacjenci w wieku pediatrycznym
POChP
Europejska Agencja Leków wyraziła odmowę obowiązku przedstawiania wyników badań leku Spiriva® Respimat® we wszystkich podgrupach pacjentów w wieku pediatrycznym z POChP (patrz sekcja „Sposób stosowania i dawki” w celu uzyskania informacji na temat stosowania u pacjentów w wieku pediatrycznym).
Asthma
Wszystkie badania fazy III u pacjentów w wieku pediatrycznym z astmą przewlekłą (1–17 lat) były randomizowanymi, podwójnie ślepymi, placebo-kontrolowanymi badaniami. Wszyscy pacjenci otrzymywali leczenie podstawowe, które obejmowało ICS (glikokortykosteroidy wziewne).
Ciężka astma
Dorośli w wieku 12–17 lat
W 12-tygodniowym badaniu PensieTinA-asthma włączono 392 pacjentów (130 leczonych lekiem Spiriva® Respimat®), u których objawy astmy utrzymywały się pomimo stosowania wysokiej dawki ICS (glikokortykosteroidy wziewne) z jednym lekiem wspomagającym lub średniej dawki ICS z dwoma lekami wspomagającymi.
U pacjentów w wieku 12–17 lat wysoka dawka ICS wynosiła > 800–1600 µg budesonidu na dobę lub równowartość, a średnia dawka ICS (glikokortykosteroidy wziewne) – 400–800 µg budesonidu na dobę lub równowartość. Ponadto pacjenci w wieku 12–14 lat mogli otrzymywać dawkę ICS > 400 µg budesonidu na dobę lub równowartość i co najmniej jeden lek wspomagający lub ≥ 200 µg budesonidu na dobę lub równowartość i co najmniej dwa leki wspomagające.
W tym badaniu stosowanie leku Spiriva® Respimat® jako terapii wspomagającej do leczenia podstawowego wiązało się z istotnym poprawieniem funkcji płuc w porównaniu z placebo, jednak różnice w wartościach szczytowych i resztkowych OBJ (objętości oddechowej wydechu siłowego w 1 sekundzie) nie były statystycznie istotne.
- W 12. tygodniu średnia poprawa wartości szczytowych i resztkowych OBJ wynosiła odpowiednio 0,090 l (95 % CI: od –0,019 do 0,198 l, p=0,1039) oraz 0,054 l (95 % CI: od –0,061 do 0,168 l, p=0,3605).
- W 12. tygodniu lek Spiriva® Respimat® istotnie poprawił poranną i wieczorną maksymalną prędkość wydechu (MPW) (poranna 17,4 l/min; 95 % CI: od 5,1 do 29,6 l/min; wieczorna 17,6 l/min; 95 % CI: od 5,9 do 29,6 l/min).
Dzieci (6–11 lat)
W 12-tygodniowym badaniu VivaTinA-asthma włączono 400 pacjentów (130 leczonych lekiem Spiriva® Respimat®), u których objawy astmy utrzymywały się pomimo stosowania wysokiej dawki ICS z jednym lekiem wspomagającym lub średniej dawki ICS z dwoma lekami wspomagającymi. Wysoka dawka ICS wynosiła > 400 µg budesonidu na dobę lub równowartość, a średnia dawka ICS – 200–400 µg budesonidu na dobę lub równowartość.
W tym badaniu stosowanie leku Spiriva® Respimat® jako terapii wspomagającej do leczenia podstawowego wiązało się z istotnym poprawieniem funkcji płuc w porównaniu z placebo.
- W 12. tygodniu średnia poprawa wartości szczytowych i resztkowych OBJ wynosiła odpowiednio 0,139 l (95 % CI: od 0,075 do 0,203 l, p< 0,0001) oraz 0,087 l (95 % CI: od 0,019 do 0,154 l, p=0,0117).
Umiarkowana astma
Dorośli (12–17 lat)
W jednorocznym badaniu RubaTinA-asthma u 397 pacjentów (134 leczonych lekiem Spiriva® Respimat®), u których objawy astmy utrzymywały się pomimo stosowania średniej dawki ICS (200–800 µg budesonidu na dobę lub równowartość u pacjentów w wieku 12–14 lat lub 400–800 µg budesonidu na dobę lub równowartość u pacjentów w wieku 15–17 lat), stosowanie leku Spiriva® Respimat® jako terapii wspomagającej do leczenia podstawowego wiązało się z istotnym poprawieniem funkcji płuc w porównaniu z placebo.
Dzieci (6–11 lat)
W jednorocznym badaniu CanoTinA-asthma u 401 pacjentów (135 leczonych lekiem Spiriva® Respimat®), u których objawy utrzymywały się pomimo stosowania średniej dawki ICS (200–400 µg budesonidu na dobę lub równowartość), stosowanie leku Spiriva® Respimat® jako terapii wspomagającej do leczenia podstawowego wiązało się z istotnym poprawieniem funkcji płuc w porównaniu z placebo.
Dzieci (1–5 lat)
Przeprowadzono jedno 12-tygodniowe, randomizowane, podwójnie ślepe, placebo-kontrolowane badanie kliniczne fazy II/III (NinoTinA-asthma) z udziałem 101 dzieci (31 leczonych lekiem Spiriva® Respimat®) z astmą, które otrzymywały leczenie podstawowe, w tym ICS. U 98 pacjentów do podania badanego leku użyto przenośnego komory zaworowej Aerochamber Plus Flow-Vu® z maską twarzową.
Pierwotnym celem badania była ocena bezpieczeństwa, a ocena skuteczności była celem eksploracyjnym.
Liczba i odsetek pacjentów, u których zaobserwowano działania niepożądane, niezależnie od związku z lekiem, przedstawiono w tabeli 3. W grupie leku Spiriva® Respimat® liczba działań niepożądanych związanych z astmą była niższa niż przy placebo. Eksploracyjna ocena skuteczności nie wykazała różnic między lekiem Spiriva® Respimat® a placebo.
Tabela 3
Liczba pacjentów z działaniami niepożądanymi zarejestrowanymi u ≥ 5 pacjentów w badaniu NinoTinA-asthma (dzieci w wieku od 1 do 5 lat)
| Wskaźnik |
Placebo, N (%) |
Spiriva® Respimat®, N (%) |
| Liczba pacjentów |
34 (100,0) |
31 (100,0) |
| Pacjenci z dowolnymi NZ |
25 (73,5) |
18 (58,1) |
| Nowotwór nosowo-gardłowy |
5 (14,7) |
2 (6,5) |
| Infekcja dróg oddechowych górnych |
1 (2,9) |
5 (16,1) |
| Astma oskrzelowa* |
10 (29,4) |
2 (6,5) |
| Hipertermia |
6 (17,6) |
3 (9,7) |
*Terminy niskiego poziomu według MedDRA w kontekście głównego terminu „astma oskrzelowa”: „nasilenie astmy oskrzelowej” lub „zaostrzenie astmy oskrzelowej”
Europejska Agencja Leków odmówiła obowiązku przedstawiania wyników badań leku Spiriva® Respimat® w podpopulacji pediatrycznej pacjentów w wieku do 1 roku życia (patrz sekcja „Sposób stosowania i dawki” w celu uzyskania informacji dotyczącej stosowania u dzieci).
Farmakokinetyka.
Tiotropiumu bromek jest niechiralnym związkiem amonowym czwartorzędowym o umiarkowanej rozpuszczalności w wodzie. Tiotropiumu bromek dostępny jest w postaci roztworu do inhalacji, stosowanego za pomocą inhalatora Respimat®. Oszacowano, że około 40 % dawki wdychanej osiąga płuca, a pozostała część dawki osiąda w przewodzie pokarmowym. Niektóre dane farmakokinetyczne opisane poniżej uzyskano po podaniu dawek wyższych niż zalecane w terapii.
Wchłanianie. Dane dotyczące wydalania z moczem po inhalacji roztworu u zdrowych ochotników pozwalają przypuszczać, że około 33 % dawki inhalowanej dociera do krążenia ogólnego. Nie oczekuje się wpływu pokarmu na wchłanianie tej czwartorzędowej substancji amonowej. Biodostępność doustna roztworu tiotropiumu wynosi 2–3 %. Maksymalne stężenie tiotropiumu w osoczu obserwowano 5–7 minut po inhalacji. W stanie stacjonarnym maksymalny poziom tiotropiumu w osoczu u pacjentów z POChP wynosił 10,5 pg/ml i szybko obniżał się w sposób wieloetapowy. Stężenie stacjonarne w osoczu wynosiło 1,60 pg/ml. Stan stacjonarny maksymalnego stężenia tiotropiumu w osoczu wynoszący 5,15 pg/ml osiągany był po 5 minutach od inhalacyjnego podania tej samej dawki pacjentom z astmą.
Działanie systemowe tiotropiumu po inhalacyjnym podaniu za pomocą inhalatora Respimat® było podobne do działania systemowego tiotropiumu po inhalacyjnym podaniu za pomocą urządzenia HandiHaler.
Rozkład. Lek wiąże się z białkami osocza w 72 %, objętość rozkładu wynosi 32 l/kg. Lokalne stężenie w płucach jest nieznane, ale ze względu na sposób stosowania przypuszcza się wysokie stężenie w płucach. Badania na szczurach wykazały, że tiotropium nie przenika przez barierę krew–mózg w żaden sposób.
Biotransformacja. Stopień biotransformacji jest niewielki. Świadczy o tym wydalanie z moczem 74 % niezmienionej substancji po dożylnej dawce podanej zdrowym młodym ochotnikom. Eter tiotropiumu bromku ulega niel enzymatycznemu rozkładowi do alkoholu (N-metyloskopinu) i kwasu (kwasu ditieniloglikolowego), które nie wiążą się z receptorami muskarynowymi. Wyniki badań in vitro z mikrosomami wątroby człowieka i hepatocytami człowieka wskazują, że pewna część leku (< 20 % dawki po podaniu dożylnym) metabolizowana jest drogą zależną od cytochromu P450 (CYP) przez utlenienie oraz dalszą koniugację z glutationem i powstawanie różnych metabolitów w fazie II biotransformacji.
Badania in vitro z mikrosomami wątroby wykazały, że droga enzymatyczna może być hamowana przez inhibitory CYP 2D6 (i 3A4) takie jak chinidyna, ketokonazol i gestoden. W związku z tym CYP 2D6 i 3A4 uczestniczą w szlaku metabolicznym odpowiedzialnym za wyeliminowanie mniejszej części dawki.
Tiotropiumu bromek, nawet w stężeniach przekraczających terapeutyczne, nie hamuje CYP 1A1, 1A2, 2B6, 2C9, 2C19, 2D6, 2E1 ani 3A4 w mikrosomach wątroby człowieka.
Eliminacja. Wartość efektywnego okresu półtrwania tiotropiumu mieści się w granicach 27–45 godzin po inhalacji u zdrowych ochotników i pacjentów z POChP. Efektywny okres półtrwania tiotropiumu wynosił 34 godziny po inhalacji u pacjentów z astmą. Po podaniu dożylnym zdrowym młodym ochotnikom całkowity klirens wynosił 880 ml/min. Tiotropium podany dożylnie wydalał się głównie z moczem (74 %). Po inhalacji roztworu u pacjentów z POChP w stanie równowagi wydalanie z moczem wynosiło 18,6 % dawki (0,93 μg), reszta nie była wchłaniana przez jelita i wydalała się z kałem.
Po inhalacji roztworu u zdrowych ochotników wydalanie z moczem wynosiło 20,1–29,4 % dawki, reszta nie była wchłaniana przez jelita i wydalała się z kałem.
U pacjentów z astmą 11,9 % (0,595 μg) dawki wydalało się w niezmienionej postaci z moczem ponad 24 godziny po podaniu w stanie równowagi.
Nerkowy klirens tiotropiumu przekracza klirens kreatyniny, co wskazuje na wydalanie z moczem.
Po długotrwałym stosowaniu inhalacji raz dziennie u pacjentów z POChP stan równowagi farmakokinetycznej osiągany był w 7. dniu bez dalszej kumulacji tiotropiumu.
Liniowość/nieliniowość. Tiotropium wykazuje liniowe właściwości farmakokinetyczne w zakresie terapeutycznym niezależnie od postaci leku.
Pacjenci w podeszłym wieku. Jak w przypadku wszystkich innych leków wydalanych głównie przez nerki, stosowanie tiotropiumu u pacjentów w podeszłym wieku wiąże się ze zmniejszeniem klirensu nerkowego (347 ml/min u pacjentów z POChP < 65 lat w porównaniu z 275 ml/min u pacjentów z POChP > 65 lat). Nie prowadziło to do odpowiedniego zwiększenia wartości AUC0-6,ss ani Cmax,ss. U pacjentów z astmą nie stwierdzono różnic w działaniu tiotropiumu w zależności od wieku pacjenta.
Pacjenci z zaburzeniami funkcji nerek.
Po podaniu tiotropiumu (raz dziennie przez inhalację) pacjentom z POChP do osiągnięcia stanu równowagi, niewielkie niewydolności nerek (klirens kreatyniny 50–80 ml/min) powodowały pewne zwiększenie parametru AUC0-6,ss (o 1,8–30 %) i podobne wartości Cmax,ss w porównaniu z pacjentami z prawidłową funkcją nerek (klirens kreatyniny >80 ml/min).
U pacjentów z POChP z umiarkowanymi do ciężkich zaburzeniami funkcji nerek (klirens kreatyniny < 50 ml/min) dożylne podanie pojedynczej dawki tiotropiumu doprowadziło do podwojenia ogólnego wpływu (AUC0-4h wyższe o 82 % i Cmax wyższe o 52 %) w porównaniu z pacjentami z POChP i prawidłową funkcją nerek, co potwierdzono stężeniem tiotropiumu w osoczu po inhalacji postaci leku w postaci proszku do inhalacji. U pacjentów z astmą z łagodnym zaburzeniem funkcji nerek (klirens kreatyniny 50–80 ml/min) tiotropium w postaci inhalacji nie prowadził do odpowiedniego zwiększenia ekspozycji w porównaniu z pacjentami z prawidłową funkcją nerek.
Pacjenci z zaburzeniami funkcji wątroby. Niewydolność wątroby nie ma istotnego wpływu na farmakokinetykę tiotropiumu. Tiotropium wydala się głównie drogą nerkową (do 74 % u młodych zdrowych ochotników) oraz drogą prostego niel enzymatycznego rozszczepienia eteru do farmakologicznie nieaktywnych produktów.
Pacjenci z POChP narodowości japońskiej. W trakcie badania krzyżowego średnie maksymalne stężenia tiotropiumu w osoczu 10 minut po podaniu dawki w stanie równowagi były o 20–70 % wyższe u pacjentów japońskich niż u europejskich po inhalacji tiotropiumu, jednak nie stwierdzono oznak zwiększonej śmiertelności ani zwiększonego ryzyka powikłań sercowych u pacjentów japońskich w porównaniu z europejskimi. W odniesieniu do innych ras lub grup etnicznych dane farmakokinetyczne są niewystarczające.
Pacjenci w wieku dziecięcym
Astma oskrzelowa
Szczyciowa i ogólna ekspozycja (AUC i wydalanie z moczem) tiotropiumu u pacjentów z astmą w wieku 6–11 lat, 12–17 lat oraz ≥18 lat była porównywalna. Ze względu na wydalanie z moczem ogólna ekspozycyjność tiotropiumu u pacjentów w wieku 1–5 lat była o 52–60 % niższa niż w innych grupach wiekowych. Po skorygowaniu względem powierzchni ciała dane dotyczące ogólnej ekspozycji we wszystkich grupach wiekowych były porównywalne. Pacjentom w wieku 1–5 lat do stosowania leku Spiriva® Respimat® używano przenośnego zaworowego dychawka z medyczną maską twarzową.
POChP
W programie POChP nie uczestniczyli pacjenci w wieku dziecięcym (patrz sekcja „Sposób stosowania i dawki”).
Związek farmakokinetyka/farmakodynamika. Nie ma bezpośredniego związku między farmakokinetyką a farmakodynamiką.
Właściwości kliniczne.
Wskazania.
POChP
Preparat Spiriva Respimat stosuje się jako wspomagającą terapię rozszerzającą oskrzela w celu złagodzenia objawów u pacjentów z przewlekłą obturacyjną chorobą płuc (POChP).
Astma
Preparat Spiriva® Respimat® stosuje się jako dodatkową wspomagającą terapię rozszerzającą oskrzela u dorosłych i dzieci w wieku od 6 lat z ciężką astmą, u których wystąpiło jedno lub więcej ciężkich zaostrzeń astmy w ciągu poprzedniego roku (patrz punkty „Sposób stosowania i dawki”, „Farmakodynamika”).
Przeciwwskazania.
Nadwrażliwość na bromek tiotropium, atropinę lub jej pochodne, np. ipratropium lub oxytripium, lub na inne składniki preparatu.
Interakcje z innymi lekami i inne rodzaje interakcji.
Chociaż nie przeprowadzono specjalnych badań interakcji z innymi lekami, bromek tiotropium stosowano równolegle z innymi lekami stosowanymi w leczeniu POChP i astmy, w tym z sympatykomimetykami rozszerzającymi oskrzela, metylksantanami, kortykosteroidami doustnymi i do inhalacji, lekami przeciwhistaminowymi, lekami mukolitycznymi, modyfikatorami leukotrienów, krotonami, przeciwciałami anty-IgE, bez danych klinicznych o reakcjach niepożądanych.
Stosowanie długodziałających agonistów receptora β-adrenergicznego i kortykosteroidów do inhalacji nie wykazało wpływu na tiotropium.
Równoległe stosowanie bromku tiotropium z innymi lekami przeciwbłoniopodobnymi nie było badane i dlatego nie jest zalecane.
Szczególne ostrzeżenia i środki ostrożności podczas stosowania.
Tiotropium bromek jest lekiem rozszerzającym oskrzela, stosowanym raz dziennie w leczeniu wspomagającym. Lek nie jest przeznaczony do wstępnego leczenia napadów astmy oskrzelowej, tzn. do pomocy w nagłych przypadkach. W przypadku nagłego napadu należy stosować szybko działające agonisty β-2.
Nie można stosować leku Spiriva® Respimat® jako monoterapii w leczeniu astmy. Pacjenci z astmą powinni kontynuować przyjmowanie leków przeciwzapalnych, np. kortykosteroidów do inhalacji, bez zmiany dawki po wprowadzeniu leku Spiriva® Respimat®, nawet jeśli objawy mogą ulec złagodzeniu.
Po zastosowaniu roztworu do inhalacji tiotropium bromku mogą wystąpić reakcje nadwrażliwości typu natychmiastowego.
Z uwagi na działanie przeciwpodobne do działania antycholinergicznego tiotropium bromek należy stosować z ostrożnością u pacjentów z jaskrą zamkniętego kąta, przerośnięciem gruczołu krokowego lub obturacją szyjki pęcherza moczowego.
Stosowanie leków do inhalacji może powodować skurcz oskrzeli wywołany samą inhalacją.
Tiotropium należy stosować z ostrożnością u pacjentów po niedawnym zawałcie mięśnia sercowego (< 6 miesięcy), z niestabilną lub zagrożeniem życia arytmią serca lub z arytmią serca, która wymagała interwencji lub zmiany terapii w ciągu ostatniego roku; hospitalizacją z powodu niewydolności serca (klasa NYHA III lub IV) w ciągu ostatniego roku. Pacjenci ci byli wykluczeni z badań klinicznych, ponieważ mogą doświadczać działania antycholinergicznego leku.
Ponieważ stężenie leku w osoczu rośnie wraz ze spadkiem funkcji nerek u pacjentów z umiarkowaną do ciężkiej niewydolnością nerek (klirens kreatyniny ≤ 50 ml/min), tiotropium bromek można stosować tylko wtedy, gdy oczekiwana korzyść przeważa nad potencjalnym ryzykiem. Brak długotrwałego doświadczenia w stosowaniu leku u pacjentów z ciężką niewydolnością nerek (patrz sekcja „Farmakokinetyka”).
Pacjentów należy poinstruować, aby nie dopuścić do dostania się aerozolu do oczu, ponieważ może to prowadzić do przyspieszenia rozwoju lub nasilenia jaskry zamkniętego kąta, bólu lub dyskomfortu w oku, tymczasowej nieostrości widzenia, uczucia pojawienia się aureoli lub plam barwnych przed oczami w połączeniu z zaczerwienieniem oka w postaci hiperemii spojówek oraz obrzęku rogówki. W przypadku wystąpienia wymienionych objawów w dowolnej kombinacji należy przerwać stosowanie tiotropium bromku i skonsultować się z lekarzem.
Suchość błony śluzowej jamy ustnej, obserwowana podczas terapii antycholinergicznym, może być związana z próchnicą zębów w przypadku długotrwałej terapii.
Tiotropium bromek nie należy stosować częściej niż raz dziennie (patrz sekcja „Przedawkowanie”).
Leku Spiriva® Respimat® nie zaleca się stosować w przypadku mukowiscydozy. W przypadku stosowania u pacjentów z mukowiscydozą lek Spiriva® Respimat® może nasilić objawy i dolegliwości związane z mukowiscydozą (np. poważne działania niepożądane, nasilenie chorób płuc, infekcje dróg oddechowych).
Substancje pomocnicze
Chlorek benzalkonium może powodować obturację oskrzeli i trudności w oddychaniu. Pacjenci z astmą są bardziej narażeni na te działania niepożądane. Lek zawiera 0,0011 mg chlorku benzalkonium na każdą inhalację. Inhalacja to dawka dostępna dla pacjenta po przejściu przez ustnik. 2 inhalacje odpowiadają jednej dawce terapeutycznej.
Stosowanie w czasie ciąży lub karmienia piersią.
Ciąża
Istnieje bardzo mało danych dotyczących stosowania tiotropium u kobiet w ciąży. Badania na zwierzętach nie wykazały braku bezpośredniego lub pośredniego negatywnego wpływu z punktu widzenia toksyczności rozrodczej przy stosowaniu leku w dawkach klinicznie istotnych.
Z ostrożności należy unikać stosowania leku Spiriva® Respimat® w czasie ciąży.
Karmienie piersią
Nie wiadomo, czy tiotropium bromek wydzielany jest z mlekiem matki. Choć badania na gryzoniach wykazały, że tiotropium bromek wydzielany jest z mlekiem matki jedynie w niewielkich ilościach, stosowanie leku Spiriva® Respimat® nie jest zalecane w czasie karmienia piersią. Tiotropium bromek jest związkiem o długim działaniu. Decyzję o kontynuowaniu/przerwaniu karmienia piersią lub o kontynuowaniu/przerwaniu terapii lekiem Spiriva® Respimat® należy podjąć, biorąc pod uwagę korzyści wynikające z karmienia piersią dla dziecka oraz korzyści z terapii lekiem dla matki.
Plodność
Brak danych klinicznych dotyczących wpływu tiotropium na płodność. Badania przedkliniczne tiotropium wykazały brak jakiegokolwiek negatywnego wpływu na płodność.
Wpływ na zdolność prowadzenia pojazdów i obsługi urządzeń.
Nie przeprowadzono badań dotyczących wpływu na zdolność prowadzenia pojazdów lub pracy z innymi urządzeniami.
Zawroty głowy lub zamazanie widzenia mogą wpływać na szybkość reakcji podczas prowadzenia pojazdów i pracy z innymi urządzeniami.
Sposób stosowania i dawki.
Sposób stosowania
Lek jest przeznaczony wyłącznie do inhalacji. Pojemnik może być używany wyłącznie z inhalatorem RESPIMAT®.
Dwa wdechy za pomocą inhalatora RESPIMAT® zawierają jedną dawkę.
Zalecana dawka dla dorosłych to 5 µg tiotropiumu w postaci dwóch inhalacji za pomocą inhalatora RESPIMAT® jeden raz dziennie o tej samej porze dnia.
Nie należy przekraczać zalecanej dawki.
W leczeniu astmy korzyść z leku ujawnia się w pełni po kilku dawkach. U dorosłych pacjentów z ciężką astmą tiotropium należy stosować jako lek uzupełniający do inhalacyjnych kortykosteroidów (ICS) (≥ 800 µg budesonidu na dobę lub równowartość) oraz co najmniej jednego leku wspomagającego terapię.
Osoby z grup szczególnych
Pacjenci w wieku podeszłym mogą stosować lek SPIRIVA® RESPIMAT® w zalecanych dawkach.
Pacjenci z niewydolnością nerek mogą stosować lek SPIRIVA® RESPIMAT® w zalecanych dawkach. W przypadku pacjentów z niewydolnością nerek od umiarkowanej do ciężkiej (klirens kreatyniny ≤ 50 ml/min, patrz sekcje „Szczególne środki ostrożności” i „Właściwości farmakologiczne”).
Pacjenci z niewydolnością wątroby mogą stosować lek w zalecanych dawkach (patrz sekcja „Właściwości farmakologiczne”).
Dzieci
Astma
Zalecana dawka dla pacjentów w wieku od 6 do 17 lat to 5 µg tiotropiumu w postaci dwóch inhalacji za pomocą inhalatora RESPIMAT® jeden raz dziennie o tej samej porze dnia.
U młodzieży (12–17 lat) z ciężką astmą tiotropium należy stosować jako lek uzupełniający do inhalacyjnych kortykosteroidów (> 800–1600 µg budesonidu na dobę lub równowartość) oraz jednego leku wspomagającego terapię lub jako lek uzupełniający do inhalacyjnych kortykosteroidów (400–800 µg budesonidu na dobę lub równowartość) z dwoma lekami wspomagającymi terapię.
U dzieci (6–11 lat) z ciężką astmą tiotropium należy stosować jako lek uzupełniający do inhalacyjnych kortykosteroidów (> 400 µg budesonidu na dobę lub równowartość) oraz jednego leku wspomagającego terapię lub jako lek uzupełniający do inhalacyjnych kortykosteroidów (200–400 µg budesonidu na dobę lub równowartość) z dwoma lekami wspomagającymi terapię.
Nie ustalono bezpieczeństwa i skuteczności leku SPIRIVA® RESPIMAT® u dzieci w wieku 6–17 lat z umiarkowaną astmą. Nie ustalono bezpieczeństwa i skuteczności leku SPIRIVA® RESPIMAT® u dzieci poniżej 6 roku życia. Dostępne informacje zawarte są w sekcjach „Farmakodynamika” i „Farmakokinetyka”, jednak nie można podać zaleceń dotyczących dawkowania.
POChP
Nie ma doświadczenia w stosowaniu leku SPIRIVA® RESPIMAT® u dzieci (do 18 roku życia).
Mukowiscydoza
Nie ustalono skuteczności i bezpieczeństwa stosowania leku SPIRIVA® RESPIMAT® (patrz sekcja „Szczególne środki ostrożności”).
Sposób stosowania
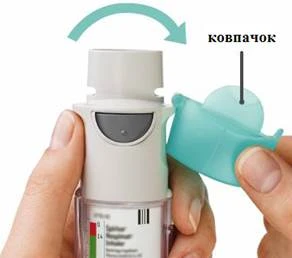
Ten lek jest przeznaczony wyłącznie do inhalacji. RESPIMAT® to inhalator generujący aerozol do inhalacji. Jest przeznaczony do użytku przez jednego pacjenta i wielokrotnego stosowania dawek wydzielanych z jednego pojemnika.
Pacjent powinien zapoznać się z instrukcją obsługi inhalatora RESPIMAT® przed rozpoczęciem stosowania leku SPIRIVA® RESPIMAT®.
Aby zapewnić właściwe stosowanie leku, lekarz lub inny pracownik ochrony zdrowia powinien zademonstrować pacjentowi sposób stosowania inhalatora.
Instrukcje dotyczące użytkowania i pielęgnacji inhalatora RESPIMAT®
Dzieci powinny stosować lek SPIRIVA® RESPIMAT® przy pomocy dorosłego.
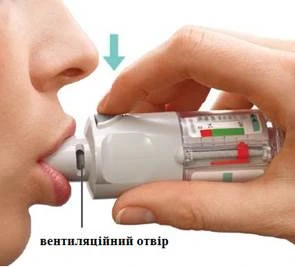
Pacjent powinien stosować inhalator RAZ NA DOBĘ.
Za każdym razem należy wykonać DWA WDECHY.
**

**
- Jeśli lek SPIRIVA® RESPIMAT® nie był stosowany dłużej niż 7 dni, wyzwól jedną inhalację, kierując inhalator w dół.
- Jeśli lek SPIRIVA® RESPIMAT® nie był stosowany dłużej niż 21 dni, powtórz kroki 4–6 „Przygotowanie do pierwszego użycia”, aż pojawi się chmura aerozolu. Następnie powtórz kroki 4–6 jeszcze trzy razy.
Jak pielęgnować inhalator RESPIMAT®
- Mundułek, w tym metalową część wewnątrz mundułka, należy czyścić wyłącznie wilgotną chusteczką z tkaniny lub tkaniną co najmniej raz w tygodniu.
- Jakiekolwiek niewielkie zmiany koloru mundułka nie wpływają na działanie inhalatora RESPIMAT®.
- W razie potrzeby przetrzyj zewnętrzne części inhalatora RESPIMAT® wilgotną chusteczką z tkaniny.
Kiedy należy zakupić nowy lek SPIRIVA® RESPIMAT®

-
Inhalator z lekiem SPIRIVA® RESPIMAT® zawiera 60 wdechów (30 dawek) przy stosowaniu zgodnie z zaleceniami (dwa wdechy raz dziennie).
-
Wskaźnik dawek pokazuje przybliżoną ilość pozostałego leku.
-
Gdy wskaźnik dawek osiągnie czerwoną strefę skali, oznacza to, że roztworu pozostało na około 7 dni (14 wdechów).
-
Gdy wskaźnik dawek osiągnie koniec czerwonej skali, inhalator z lekiem SPIRIVA® RESPIMAT® zostanie automatycznie zablokowany – nie będzie więcej dawek do wyzwolenia. Od tego momentu nie będzie możliwe obrócenie przezroczystej podstawy.
-
Nie później niż po trzech miesiącach od pierwszego użycia inhalator z lekiem SPIRIVA® RESPIMAT® należy wyrzucić, nawet jeśli nie cały roztwór został zużyty.
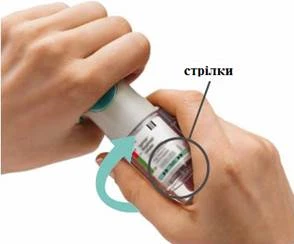
Przygotowanie do pierwszego użycia
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
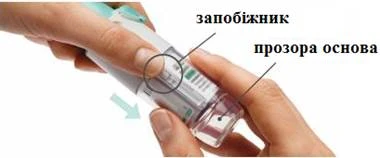
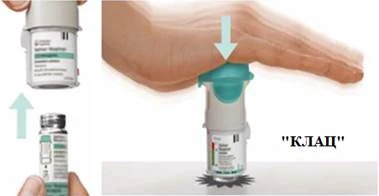
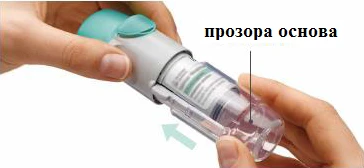
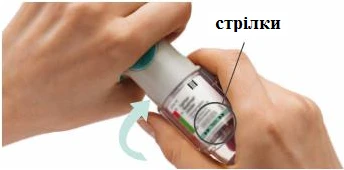


 KROKI 4-6
KROKI 4-6
Stosowanie codzienne
| POWRÓĆ
|
|
| OTWÓRZ
|
|
| NACIŚNIJ
|
2 WDYCHANIA RAZ NA DZIEŃ |
Dzieci.
Asthma
Bezpieczeństwo i skuteczność leku Spiriva Respimat u dzieci poniżej 6. roku życia nie zostały ustalone (patrz punkty „Właściwości farmakologiczne” oraz „Sposób stosowania i dawki”).
POChP
Brak doświadczenia w stosowaniu leku Spiriva Respimat u dzieci (poniżej 18. roku życia).
Przedawkowanie.
Wysokie dawki leku mogą powodować objawy i objawy działania antycholinergicznego.
Nie zaobserwowano jednak systemowych skutków ubocznych o charakterze antycholinergicznym u zdrowych ochotników po jednorazowym inhalowaniu dawki do 340 µg bromku tiotropium. Ponadto po 14 dniach stosowania w dawkach do 40 µg tiotropium, roztwór do inhalacji, u zdrowych ochotników nie zaobserwowano żadnych innych istotnych skutków ubocznych poza suchością błon śluzowych jamy ustnej/gardła i nosa, z wyjątkiem wyraźnego zmniejszenia wydzielania śliny po 7 dniach stosowania.
Działania niepożądane.
Skrócona informacja o bezpieczeństwa leku
Wiele z wymienionych efektów niepożądanych może być związane z działaniem antycholinergicznym bromku tiotropium.
Podsumowanie tabeli działań niepożądanych
Częstość występowania poniżej wymienionych działań niepożądanych obserwowanych u pacjentów przyjmujących bromek tiotropium została obliczona na podstawie zestawionych danych uzyskanych w wyniku przeprowadzenia 7 placebo-kontrolowanych badań klinicznych z udziałem chorych na POChP (3 282 pacjentów) oraz 12 placebo-kontrolowanych badań klinicznych z udziałem dorosłych i dzieci chorych na astmę (1930 pacjentów), z okresami leczenia od czterech tygodni do jednego roku.
Kryteria oceny częstości występowania działań niepożądanych:
bardzo często (≥1/10); często (≥1/100 – <1/10); nieczęsto (≥1/1 000 – <1/100); rzadko (≥1/10 000 – <1/1 000); bardzo rzadko (<1/10 000); nieznane (nie można oszacować na podstawie dostępnych danych).
Tabela 4
| Klasyfikacja systemów narządowych / termin preferencyjny według klasyfikacji MedDRA |
Częstość |
|
| POChP |
Astma |
|
| Zaburzenia metabolizmu i odżywiania |
||
| Odewodnienie |
Nieznane |
Nieznane |
| Zaburzenia układu nerwowego |
||
| Zawroty głowy |
Niekorzystne |
Niekorzystne |
| Ból głowy |
Niekorzystne |
Niekorzystne |
| Zaburzenia snu |
Pojedyncze |
Niekorzystne |
| Zaburzenia narządu wzroku |
||
| Glaukoma |
Pojedyncze |
Nieznane |
| Zwiększone ciśnienie wewnątrzgałkowe |
Pojedyncze |
Nieznane |
| Wzrok zamazany |
Pojedyncze |
Nieznane |
| Zaburzenia układu sercowo-naczyniowego |
||
| Migotanie przedsionków |
Pojedyncze |
Nieznane |
| Odczucie przyspieszonego rytmu serca |
Pojedyncze |
Niekorzystne |
| Tachykardia nadkomorowa |
Pojedyncze |
Nieznane |
| Tachykardia |
Pojedyncze |
Nieznane |
| Zaburzenia układu oddechowego, narządów klatki piersiowej i jamy śródpiersia |
||
| Kaszel |
Niekorzystne |
Niekorzystne |
| Przeziębienie |
Niekorzystne |
Niekorzystne |
| Dysfonia |
Niekorzystne |
Niekorzystne |
| Krwawienie z nosa |
Pojedyncze |
Pojedyncze |
| Bronchospazm |
Pojedyncze |
Niekorzystne |
| Zapalenie krtani |
Pojedyncze |
Nieznane |
| Zapalenie zatok |
Nieznane |
Nieznane |
| Zaburzenia przewodu pokarmowego |
||
| Sucha jamy ustnej |
Częste |
Niekorzystne |
| Wstyd |
Niekorzystne |
Pojedyncze |
| Kandydoza gardła i jamy ustnej |
Niekorzystne |
Niekorzystne |
| Dysfagia |
Pojedyncze |
Nieznane |
| Choroba refluksowa przełyku |
Pojedyncze |
Nieznane |
| Próchnica zębów |
Pojedyncze |
Nieznane |
| Zapalenie dziąseł |
Pojedyncze |
Pojedyncze |
| Zapalenie języka |
Pojedyncze |
Nieznane |
| Stomatyt |
Nieznane |
Pojedyncze |
| Przesytka jelita, w tym przesytka jelita o charakterze porażenia |
Nieznane |
Nieznane |
| Światr |
Nieznane |
Nieznane |
| Zaburzenia skóry, tkanek podskórnych i układu odpornościowego |
||
| Wysypka |
Niekorzystne |
Niekorzystne |
| Świąd |
Niekorzystne |
Pojedyncze |
| Obwód naczynioruchowy |
Pojedyncze |
Pojedyncze |
| Krzypa |
Pojedyncze |
Pojedyncze |
| Zakażenie skóry / owrzodzenie skóry |
Pojedyncze |
Nieznane |
| Susza skóry |
Pojedyncze |
Nieznane |
| Podatność (w tym reakcje alergiczne typu natychmiastowego) |
Nieznane |
Pojedyncze |
| Reakcja anafilaktyczna |
Nieznane |
Nieznane |
| Zaburzenia układu mięśniowo-szkieletowego i tkanki łącznej |
||
| Obwód stawów |
Nieznane |
Nieznane |
| Zaburzenia nerek i dróg moczowych |
||
| Zatrzymanie moczu |
Niekorzystne |
Nieznane |
| Zaburzenia oddawania moczu |
Niekorzystne |
Nieznane |
| Zakażenie dróg moczowych |
Pojedyncze |
Pojedyncze |
Opis wybranych działań niepożądanych
W przebiegu kontrolowanych badań klinicznych z udziałem pacjentów z POChP częstymi niepożądanymi zjawiskami związanymi z działaniem antycholinergicznym była suchość w ustach. Pojawiała się ona u około 2,9% pacjentów. U chorych na astmę częstość występowania suchości w ustach wyniosła 0,83%.
W trakcie 7 badań klinicznych z udziałem chorych na POChP suchość w ustach była powodem przedwczesnego wycofania się z badania u 3 spośród 3282 pacjentów otrzymujących tiotropium (0,1%). W trakcie 12 badań klinicznych z udziałem chorych na astmę nie odnotowano żadnego przypadku przedwczesnego wycofania się z badania z powodu suchości w ustach (1930 pacjentów).
Poważne działania niepożądane związane z działaniem antycholinergicznym obejmują jaskrę, zaparcia, niedrożność jelit, w tym niedrożność jelit paralityczną oraz zatrzymanie moczu.
Pacjenci w wieku dziecięcym
Dane dotyczące bezpieczeństwa obejmują 560 pacjentów w wieku dziecięcym (296 pacjentów w wieku 1\–11 lat oraz 264 pacjentów w wieku 12\–17 lat), którzy wzięli udział w 5 placebo-kontrolowanych badaniach klinicznych, w których okresy leczenia wahały się od 12 tygodni do jednego roku. Częstość, rodzaj i nasilenie reakcji niepożądanych u pacjentów w wieku dziecięcym były podobne jak u dorosłych.
Inne kategorie pacjentów.
Wzmożenie działania antycholinergicznego leku może się rozwijać wraz ze wzrostem wieku pacjenta.
Zgłaszanie podejrzewanych działań niepożądanych
Zgłaszanie działań niepożądanych po rejestracji leku ma istotne znaczenie. Umożliwia to monitorowanie stosunku korzyści do ryzyka w przypadku stosowania tego leku. Pracownicy medyczni i farmaceutyczni, a także pacjenci lub ich ustawieni reprezentanci powinni zgłaszać wszystkie przypadki podejrzewanych działań niepożądanych oraz braku skuteczności leku poprzez Zautomatyzowany System Informacyjny Nadzoru Farmakologicznego pod adresem: https://aisf.dec.gov.ua».
Okres ważności. 3 lata.
Okres ważności po pierwszym użyciu – 3 miesiące.
Warunki przechowywania.
Przechowywać w temperaturze nie wyższej niż 25°C. Nie zamrażać! Przechowywać w miejscu niedostępnym dla dzieci!
Opakowanie.
Po 4 ml w kartuszu (60 inhalacji); po 1 kartuszu w zestawie z 1 inhalatorem RESPIMAT w pudełku kartonowym.
Kategoria wydawania.
Na receptę.
Producent.
Boehringer Ingelheim Pharma GmbH & Co. KG
lub
Boehringer Ingelheim Espana, SA.
Miejsce produkcji oraz adres miejsca prowadzenia działalności.
Binger Strasse 173, 55216 Ingelheim am Rhein, Niemcy
lub
Prat de la Riba, 50, 08174 Sant Cugat del Valles (Barcelona), Hiszpania.