Spiriva Respimat
Ucrania
Contenido
INSTRUCCIONES para uso médico del medicamento SPIRIVA® RESPIMAT® (Spiriva® Respimat®)
Composición:
Principio activo: bromuro de tiotropio monohidrato, equivalente a tiotropio;
1 inhalación contiene 3,124 mcg de bromuro de tiotropio monohidrato, equivalente a 2,5 mcg de tiotropio;
Sustancias auxiliares: cloruro de benzalconio, edetato disódico, agua para preparaciones inyectables, ácido clorhídrico diluido.
Forma farmacéutica. Solución para inhalación.
Propiedades físico-químicas principales: cartuchos con un volumen de hasta 4,5 ml, llenos con líquido, sellados en cilindros de aluminio para el inhalador RESPIMAT®, con una junta protectora estampada.
Grupo farmacoterapéutico. Otros medicamentos para el tratamiento de enfermedades obstructivas de las vías respiratorias, agentes inhalatorios. Agentes anticolinérgicos. Bromuro de tiotropio.
Código ATC R03B B04.
Propiedades farmacológicas.
Farmacodinámica
Mecanismo de acción
El bromuro de tiotropio es un antagonista antimuscarínico de acción prolongada y específico. El tiotropio tiene una afinidad similar por los subtipos de receptores desde M1 hasta M5. En las vías respiratorias, el bromuro de tiotropio se une de forma competitiva y reversible a los receptores M3 del músculo liso bronquial, contrarrestando el efecto colinérgico (broncoconstrictor) de la acetilcolina, lo que conduce a la relajación de los músculos lisos bronquiales. Este efecto fue dependiente de la dosis y duró más de 24 horas. Dado que el tiotropio es un anticolinérgico N-cuaternario bronquioselectivo local, cuando se administra por inhalación presenta un rango terapéutico aceptable antes de la aparición de efectos anticolinérgicos sistémicos.
Efectos farmacodinámicos
La disociación del bromuro de tiotropio, especialmente de los receptores M3, es muy lenta. En consecuencia, el período de semivida de eliminación es significativamente más largo que el del ipratropio. La disociación de los receptores M2 es más rápida que de los M3, lo que en estudios funcionales in vitro mostró una mayor selectividad (controlada cinéticamente) por el subtipo de receptor M3 frente al M2. Se ha demostrado que la alta actividad, la disociación muy lenta de los receptores y la acción selectiva local tras la administración por inhalación se correlacionan clínicamente con una broncodilatación significativa y prolongada en pacientes con EPOC y asma.
Eficacia clínica y seguridad en pacientes con EPOC
El programa de desarrollo clínico de fase III incluyó dos estudios aleatorizados doble ciego de un año de duración, dos estudios de 12 semanas y dos estudios de 4 semanas, con la participación de 2901 pacientes con EPOC (1038 pacientes recibieron 5 mcg de tiotropio). El programa de un año incluyó dos ensayos controlados con placebo. Dos estudios de 12 semanas incluyeron comparación activa (ipratropio) y placebo. Todos los seis estudios incluyeron mediciones de la función pulmonar. Además, los dos estudios de un año incluyeron evaluaciones de la frecuencia de disnea, la calidad de vida relacionada con la salud y el impacto sobre la frecuencia de exacerbaciones.
Estudios controlados con placebo. Función pulmonar
La solución de tiotropio para inhalación, administrada una vez al día, proporcionó una mejora significativa de la función pulmonar (volumen espiratorio forzado en un segundo y capacidad vital forzada) a partir de los 30 minutos tras la primera dosis, en comparación con placebo (mejora media del VEF1 a los 30 minutos: 0,113 litros; intervalo de confianza (IC) del 95 %: de 0,102 a 0,125 litros, p < 0,0001). La mejora de la función pulmonar se mantuvo durante 24 horas en estado estable en comparación con placebo (mejora media del VEF1: 0,122 litros; IC del 95 %: de 0,106 a 0,138 litros, p < 0,0001).
Se alcanzó un estado farmacodinámico estable a la semana.
El medicamento SPIRIVA RESPIMAT mejoró significativamente el flujo espiratorio máximo matutino y vespertino, según mediciones diarias realizadas por los pacientes, en comparación con placebo (mejora media del flujo espiratorio máximo: por la mañana 22 l/min; IC del 95 %: de 18 a 55 l/min, p < 0,0001; por la tarde 26 l/min; IC del 95 %: de 23 a 30 l/min, p < 0,0001). El uso de SPIRIVA RESPIMAT redujo la frecuencia de uso de broncodilatadores de rescate en comparación con placebo (reducción media de uso de medicamentos de rescate de 0,66 episodios por día; IC del 95 %: de 0,51 a 0,81 episodios por día, p < 0,0001).
Los efectos broncodilatadores de SPIRIVA RESPIMAT se mantuvieron durante todo el período de un año sin signos de tolerancia.
Disnea, calidad de vida relacionada con la salud, exacerbaciones de EPOC en estudios a largo plazo de un año.
Disnea. El medicamento SPIRIVA RESPIMAT redujo significativamente la frecuencia de disnea (evaluada mediante el índice de disnea transitoria) en comparación con placebo (mejora media de 1,05 puntos; IC del 95 %: de 0,73 a 1,38 puntos, p < 0,0001). La mejora se mantuvo durante todo el período de tratamiento.
Calidad de vida relacionada con la salud. La mejora media global en la calidad de vida autoevaluada por los pacientes (medida mediante el cuestionario del Hospital de San Jorge para la evaluación de la función respiratoria) con SPIRIVA RESPIMAT en comparación con placebo, en dos estudios de un año, fue de 3,5 puntos (IC del 95 %: de 2,1 a 4,9, p < 0,0001). Una reducción de 4 puntos se considera clínicamente significativa.
Exacerbaciones de EPOC
Los resultados de tres ensayos clínicos aleatorizados doble ciego controlados con placebo de un año de duración con SPIRIVA RESPIMAT mostraron una reducción significativa del riesgo de exacerbaciones de EPOC en comparación con placebo. Las exacerbaciones de EPOC se definieron como la aparición de al menos dos síntomas respiratorios durante tres días o más, que requirieron un cambio en el tratamiento (prescripción de antibióticos y/o corticosteroides sistémicos y/o un cambio significativo en los medicamentos respiratorios prescritos). El tratamiento con SPIRIVA RESPIMAT redujo el riesgo de hospitalización por exacerbaciones de EPOC (reducción significativa del riesgo en un estudio amplio con pacientes con exacerbaciones).
Los resultados del análisis conjunto de dos ensayos de fase III y del análisis independiente de un estudio adicional con pacientes con exacerbaciones se presentan en la tabla 1. Todos los medicamentos respiratorios, excepto los anticolinérgicos y los agonistas β de acción prolongada, estaban permitidos como terapia concomitante, es decir, agonistas β de acción rápida, corticosteroides inhalados y xantinas. En el estudio sobre el impacto en exacerbaciones en pacientes con exacerbaciones, también se permitió el uso de agonistas β de acción prolongada.
Tabla 1
Análisis estadístico de episodios de exacerbación de EPOC y episodios de hospitalización por exacerbación de EPOC en pacientes con EPOC de moderada a muy grave.
| Estudio |
Punto final |
Spiriva Respimat |
Placebo |
% de reducción del riesgo (IC 95 %)a |
valor p |
| Estudios de fase III de 1 año, |
Número de días hasta la primera exacerbación de EPOC |
160a |
86a |
29 (del 16 al 40)b |
< 0,0001b |
| Frecuencia media de episodios de exacerbación por paciente-año |
0,78c |
1,00c |
22 (del 8 al 33)c |
0,002c |
|
| Tiempo hasta la primera hospitalización por exacerbación de EPOC |
25 (del -16 al 51)b |
0,20b |
|||
| Frecuencia media de episodios de hospitalización por exacerbación por paciente-año |
0,09 c |
0,11 c |
20 (del -4 al 38) c |
0,096 c |
|
| Estudio de fase III de 1 año con participación de pacientes con exacerbación |
Número de días hasta la primera exacerbación de EPOC |
169a |
119a |
31 (del 23 al 37)b |
< 0,0001b |
| Frecuencia media de episodios de exacerbación por paciente-año |
0,69c |
0,87c |
21 (del 13 al 28)c |
< 0,0001c |
|
| Tiempo hasta la primera hospitalización por exacerbación de EPOC |
27 (del 10 al 41)b |
0,003b |
|||
| Frecuencia media de episodios de hospitalización por exacerbación por paciente-año |
0,12c |
0,15c |
19 (del 7 al 30)c |
0,004c |
a Tiempo hasta el primer evento: número de días de tratamiento hasta que al menos el 25 % de los pacientes experimente una exacerbación de EPOC o un caso de hospitalización por exacerbación de EPOC. En el estudio A, el 25 % de los pacientes que recibieron placebo experimentaron una exacerbación hasta el día 112, mientras que en el 25 % de los pacientes que recibieron SPIRIVA RESPIMAT se observó una exacerbación hasta el día 173 (p=0,09); en el estudio B, el 25 % de los pacientes que recibieron placebo experimentaron una exacerbación hasta el día 74, mientras que en el 25 % de los pacientes que recibieron SPIRIVA RESPIMAT se observó una exacerbación hasta el día 149 (p<0,0001).
b El cociente de riesgos se evaluó basándose en el modelo de riesgos proporcionales de Cox. El porcentaje de reducción del riesgo se calcula como 100 (1 ─ cociente de riesgos).
c Regresión de Poisson. La reducción del riesgo se calcula como 100 (1 ─ riesgo relativo).
d La combinación de resultados fue preespecificada durante el diseño de los estudios. Los puntos finales de evaluación de exacerbaciones mejoraron significativamente en los análisis individuales de los dos estudios de un año de duración.
Estudio controlado activo a largo plazo con tiotropio
Se realizó un estudio a largo plazo, amplio, aleatorizado, doble ciego y controlado activamente, con un periodo de observación de hasta 3 años, para comparar la eficacia y seguridad del uso de SPIRIVA RESPIMAT frente a SPIRIVA con el dispositivo inhalador HANDIHALER (SPIRIVA) (5 711 pacientes recibieron SPIRIVA RESPIMAT; 5 694 pacientes recibieron SPIRIVA). Los puntos finales principales fueron el tiempo hasta la primera exacerbación de EPOC, el tiempo hasta la muerte por cualquier causa y, en un subestudio (906 pacientes), el VEF1 mínimo (antes de la dosis).
El tiempo hasta la primera exacerbación de EPOC durante el estudio fue numéricamente similar con SPIRIVA RESPIMAT y SPIRIVA (cociente de riesgos [SPIRIVA RESPIMAT/SPIRIVA] 0,98 con IC del 95 % de 0,93 a 1,03). El tiempo medio hasta la primera exacerbación de EPOC fue de 756 días con SPIRIVA RESPIMAT y de 719 días con SPIRIVA.
El efecto broncodilatador de SPIRIVA RESPIMAT se mantuvo durante 120 semanas y fue similar al de SPIRIVA. La diferencia media en el VEF1 mínimo entre el grupo que recibió SPIRIVA RESPIMAT y el grupo que recibió SPIRIVA fue de -0,010 l (IC del 95 %: -0,038 a 0,018 l).
En el estudio comparativo poscomercialización TIOSPIR de SPIRIVA RESPIMAT frente a SPIRIVA con el dispositivo HANDIHALER, se observaron tasas similares de mortalidad por todas las causas, incluyendo el control de parámetros vitales, en los grupos estudiados (cociente de riesgos [SPIRIVA con dispositivo HANDIHALER/SPIRIVA RESPIMAT]: 0,96; IC del 95 %: 0,84-1,09). La exposición al tratamiento fue de 13 135 y 13 050 paciente-años, respectivamente.
En estudios controlados con placebo con seguimiento de parámetros vitales hasta el final del periodo de tratamiento planificado, en el grupo de SPIRIVA RESPIMAT se registró un aumento de la tasa de mortalidad por cualquier causa en comparación con el grupo de placebo (cociente de tasas [IC del 95 %] 1,33 [0,93, 1,92]) con una duración del tratamiento con SPIRIVA RESPIMAT de 2574 paciente-años; el aumento de la mortalidad se observó en pacientes con alteraciones conocidas del ritmo cardíaco. En el grupo de SPIRIVA se observó una reducción del 13 % en el riesgo de muerte (cociente de riesgos, incluyendo el control de parámetros vitales [tiotropio/placebo] = 0,87; IC del 95 %: 0,76 a 0,99). La exposición al tratamiento con SPIRIVA fue de 10 927 paciente-años. No se observó aumento del riesgo de muerte en el subgrupo de pacientes con alteraciones conocidas del ritmo cardíaco en el estudio controlado con placebo de SPIRIVA, ni tampoco en el estudio TIOSPIR de comparación entre SPIRIVA RESPIMAT y SPIRIVA.
Eficacia clínica y seguridad en pacientes con asma
El programa clínico de fase III en adultos con asma persistente incluyó dos estudios aleatorizados, doble ciego, controlados con placebo, de un año de duración, con 907 pacientes asmáticos (453 pacientes que recibieron SPIRIVA RESPIMAT), que recibieron tratamiento combinado con corticosteroides inhalados (CSI) (≥ 800 µg de budesonida/día o equivalente) y agonistas β2 de acción prolongada. Los estudios incluyeron mediciones de la función pulmonar y evaluación de exacerbaciones graves como puntos finales primarios.
Estudios PrimoTinA con pacientes asmáticos
En dos estudios de un año de duración en pacientes con síntomas persistentes de asma a pesar del tratamiento de mantenimiento, que incluía al menos CSI (≥ 800 µg de budesonida/día o equivalente) en combinación con agonistas β2 de acción prolongada, SPIRIVA RESPIMAT demostró una mejora clínicamente significativa de la función pulmonar en comparación con placebo cuando se administró en combinación con el tratamiento de fondo.
A las 24 semanas, la mejora media en el pico de VEF1 y en el VEF1 mínimo fue de 0,110 litros (IC del 95 %: 0,063 a 0,158 litros, p<0,0001) y 0,093 litros (IC del 95 %: 0,050 a 0,137 litros, p<0,0001), respectivamente. La mejora en la función pulmonar en comparación con placebo se mantuvo durante 24 horas.
En los estudios PrimoTinA en pacientes con asma, el tratamiento de los pacientes (N=453) con la combinación de CSI, agonistas β2 de acción prolongada y tiotropio redujo el riesgo de exacerbaciones graves de asma en un 21 % en comparación con el tratamiento de los pacientes (N=454) con la combinación de CSI, agonistas β2 de acción prolongada y placebo. La reducción del riesgo en términos de frecuencia media de exacerbaciones graves de asma por paciente-año fue del 20 %.
Estos resultados se confirmaron con una reducción del riesgo de empeoramiento del asma del 31 % y una reducción del 24 % en términos de frecuencia media de empeoramiento del asma por paciente-año (véase la tabla 2).
Tabla 2
Exacerbaciones en pacientes con síntomas persistentes de asma a pesar del uso de CSI (≥800 µg de budesonida/día o equivalente) en combinación con agonistas β2 de acción prolongada (estudios PrimoTinA en pacientes con asma)
| Estudio |
Punto final |
SPRIVATO RESPIRATOR |
Placebo |
% de reducción del riesgo |
Valor de p |
| Dos estudios de fase III de un año, |
Número de días hasta la primera exacerbación grave de asma |
282c |
226c |
21b |
0,0343 |
| Número medio de episodios de exacerbaciones graves de asma por paciente-año |
0,530 |
0,663 |
20d |
0,0458 |
|
| Número de días hasta la primera descompensación de asma |
315c |
181c |
31b |
<0,0001 |
|
| Número medio de episodios de descompensación de asma por paciente-año |
2,145 |
2,835 |
24d |
0,0031 |
a ≥ 800 mcg de budesonida/día o equivalente.
b Relación de riesgos, intervalo de confianza y valor p obtenidos a partir del modelo de riesgos proporcionales de Cox, considerando únicamente el tratamiento efectivo. El porcentaje de reducción del riesgo se calcula como 100 (1 ─ relación de riesgos).
c Tiempo hasta el primer evento: número de días de tratamiento hasta el momento en que al menos un 25 %/50 % de los pacientes experimentaron al menos una exacerbación grave de asma/empeoramiento del asma.
d El riesgo relativo se determinó mediante regresión de Poisson utilizando la exposición logarítmica (en años) como ajuste. El porcentaje de reducción del riesgo es 100 (1 ─ riesgo relativo).
Pacientes pediátricos
EPOC
La Agencia Europea de Medicamentos ha eximido al solicitante de la obligación de presentar los resultados de los estudios con Spiriva Respimat en todas las subpoblaciones pediátricas con EPOC (véase la sección «Posología y forma de administración» para obtener información sobre el uso en pacientes pediátricos).
Asma
Todos los estudios de Fase III en pacientes pediátricos con asma persistente (1-17 años) fueron ensayos aleatorizados, doble ciego y controlados con placebo. Todos los pacientes recibieron tratamiento de fondo que incluía corticosteroides inhalados (CIS).
Asma grave
Adolescentes de 12 a 17 años
En un estudio de 12 semanas, PensieTinA-asthma, se incluyeron 392 pacientes (130 tratados con Spiriva Respimat) que presentaban síntomas persistentes de asma a pesar de recibir dosis altas de CIS (corticosteroides inhalados) más un fármaco de mantenimiento o dosis medias de CIS más dos fármacos de mantenimiento.
Para pacientes de 12 a 17 años, la dosis alta de CIS fue > 800-1600 mcg de budesonida/día o equivalente, y la dosis media de CIS (corticosteroides inhalados) fue de 400-800 mcg de budesonida/día o equivalente. Además, los pacientes de 12 a 14 años podían recibir una dosis de CIS > 400 mcg de budesonida/día o equivalente y al menos un fármaco de mantenimiento, o ≥ 200 mcg de budesonida/día o equivalente y al menos dos fármacos de mantenimiento.
En este estudio, el uso de Spiriva Respimat como tratamiento adicional al tratamiento de fondo mostró una mejora significativa en la función pulmonar en comparación con placebo, aunque las diferencias en los valores pico y de fondo del VEF1 (volumen espiratorio forzado en 1 segundo) no fueron estadísticamente significativas.
- A las 12 semanas, la mejora media en los valores pico y de fondo del VEF1 fue de 0,090 l (IC 95 %: de -0,019 a 0,198 l, p=0,1039) y 0,054 l (IC 95 %: de -0,061 a 0,168 l, p=0,3605), respectivamente.
- A las 12 semanas, Spiriva Respimat mejoró significativamente la velocidad máxima espiratoria matutina y vespertina (VMEm) (matutina 17,4 l/min; IC 95 %: de 5,1 a 29,6 l/min; vespertina 17,6 l/min; IC 95 %: de 5,9 a 29,6 l/min).
Niños (6-11 años)
En un estudio de 12 semanas, VivaTinA-asthma, se incluyeron 400 pacientes (130 tratados con Spiriva Respimat) que presentaban síntomas persistentes de asma a pesar de recibir dosis altas de CIS más un fármaco de mantenimiento o dosis medias de CIS más dos fármacos de mantenimiento. La dosis alta de CIS fue > 400 mcg de budesonida/día o equivalente, y la dosis media de CIS fue de 200-400 mcg de budesonida/día o equivalente.
En este estudio, el uso de Spiriva Respimat como tratamiento adicional al tratamiento de fondo mostró una mejora significativa en la función pulmonar en comparación con placebo.
- A las 12 semanas, la mejora media en los valores pico y de fondo del VEF1 fue de 0,139 l (IC 95 %: de 0,075 a 0,203 l, p< 0,0001) y 0,087 l (IC 95 %: de 0,019 a 0,154 l, p=0,0117), respectivamente.
Asma moderada
Adolescentes (12-17 años)
En un estudio de un año, RubaTinA-asthma, con 397 pacientes (134 tratados con Spiriva Respimat) que presentaban síntomas persistentes de asma a pesar de recibir dosis medias de CIS (200-800 mcg de budesonida/día o equivalente en pacientes de 12-14 años o 400-800 mcg de budesonida/día o equivalente en pacientes de 15-17 años), el uso de Spiriva Respimat como tratamiento adicional al tratamiento de fondo mostró una mejora significativa en la función pulmonar en comparación con placebo.
Niños (6-11 años)
En un estudio de un año, CanoTinA-asthma, con 401 pacientes (135 tratados con Spiriva Respimat) que presentaban síntomas persistentes a pesar de recibir dosis medias de CIS (200-400 mcg de budesonida/día o equivalente), el uso de Spiriva Respimat como tratamiento adicional al tratamiento de fondo mostró una mejora significativa en la función pulmonar en comparación con placebo.
Niños (1─5 años)
Se realizó un único estudio clínico aleatorizado, doble ciego y controlado con placebo de Fase II/III de 12 semanas (NinoTinA-asthma) con 101 niños (31 tratados con Spiriva Respimat) con asma que recibían tratamiento de fondo que incluía CIS. En 98 pacientes, el medicamento en estudio se administró mediante un espaciador portátil con válvula Aerochamber Plus Flow-Vu® con mascarilla médica.
El objetivo primario del estudio fue evaluar la seguridad, y la evaluación de la eficacia fue un objetivo exploratorio.
El número y porcentaje de pacientes que presentaron reacciones adversas, independientemente de su relación con el fármaco, se muestran en la Tabla 3. En el grupo de Spiriva Respimat, el número de eventos adversos relacionados con el asma fue menor que con placebo. La evaluación exploratoria de la eficacia no mostró diferencias entre Spiriva Respimat y placebo.
Tabla 3
Número de pacientes con reacciones adversas registradas en ≥ 5 pacientes en el estudio NinoTinA-asthma (niños de 1 a 5 años)
| Indicador |
Placebo, N (%) |
SPIRIVA RESPIMAT, N (%) |
| Número de pacientes |
34 (100,0) |
31 (100,0) |
| Pacientes con cualquier evento adverso |
25 (73,5) |
18 (58,1) |
| Nasofaringitis |
5 (14,7) |
2 (6,5) |
| Infección de las vías respiratorias superiores |
1 (2,9) |
5 (16,1) |
| Asma bronquial* |
10 (29,4) |
2 (6,5) |
| Hipertermia |
6 (17,6) |
3 (9,7) |
*Términos de bajo nivel según MedDRA en el contexto del término preferente «asma bronquial»: «empeoramiento del asma bronquial» o «exacerbación del asma bronquial»
La Agencia Europea de Medicamentos ha eximido del requisito de presentar resultados de estudios del medicamento SPIRIVA RESPIMAT en la subpoblación pediátrica de pacientes menores de 1 año (véase la sección «Posología y forma de administración» para obtener información sobre el uso en pacientes pediátricos).
Farmacocinética.
El bromuro de tiotropio es un compuesto amónico cuaternario no quiral, moderadamente soluble en agua. El bromuro de tiotropio está disponible en forma de solución para inhalación, que se administra mediante el inhalador RESPIMAT. Aproximadamente el 40 % de la dosis inhalada se deposita en los pulmones, mientras que el resto de la dosis se deposita en el tracto gastrointestinal. Algunos datos farmacocinéticos descritos a continuación se obtuvieron tras la administración de dosis superiores a las recomendadas para tratamiento.
Absorción. Los datos de excreción urinaria tras la inhalación de la solución en voluntarios sanos permiten suponer que aproximadamente el 33 % de la dosis inhalada alcanza la circulación sistémica. No se espera que la comida influya en la absorción de este compuesto amónico cuaternario. La biodisponibilidad absoluta de la solución de tiotropio por vía oral es del 2-3 %. La concentración máxima de tiotropio en plasma se observó entre 5 y 7 minutos tras la inhalación. En estado de equilibrio, la concentración máxima de tiotropio en plasma en pacientes con EPOC fue de 10,5 pg/ml y disminuyó rápidamente mediante un proceso multifásico. La concentración en estado de equilibrio en plasma fue de 1,60 pg/ml. El estado de equilibrio con una concentración máxima de tiotropio en plasma de 5,15 pg/ml se alcanzó a los 5 minutos tras la administración inhalatoria de la misma dosis en pacientes con asma.
La exposición sistémica al tiotropio tras la administración inhalatoria mediante el inhalador RESPIMAT fue similar a la exposición sistémica al tiotropio tras la administración inhalatoria mediante el dispositivo HandiHaler.
Distribución. El fármaco se une a las proteínas plasmáticas en un 72 %; el volumen de distribución es de 32 l/kg. La concentración local en los pulmones es desconocida, pero, dada la vía de administración, se supone una alta concentración en los pulmones. Estudios en ratas mostraron que el tiotropio no atraviesa la barrera hematoencefálica de forma significativa.
Biotransformación. El grado de biotransformación es bajo. Esto se observa en la excreción urinaria del 74 % de la sustancia inalterada tras la administración intravenosa en jóvenes voluntarios sanos. El éter del bromuro de tiotropio se descompone no enzimáticamente en un alcohol (N-metilescopolina) y un ácido (ácido ditioglicólico), que no se unen a los receptores muscarínicos. Los resultados de estudios in vitro con microsomas hepáticos humanos y hepatocitos humanos indican que una parte del fármaco (< 20 % de la dosis tras administración intravenosa) se metaboliza mediante oxidación dependiente del citocromo P450 (CYP) y posterior conjugación con glutatión, formando diversos metabolitos en la fase II de la biotransformación.
Estudios in vitro con microsomas hepáticos mostraron que esta vía enzimática puede ser inhibida por inhibidores del CYP 2D6 (y 3A4) como la quinidina, el ketoconazol y el gestodeno. Por tanto, el CYP 2D6 y el 3A4 participan en la vía metabólica responsable de la eliminación de una fracción menor de la dosis.
El bromuro de tiotropio, incluso en concentraciones superiores a las terapéuticas, no inhibe el CYP 1A1, 1A2, 2B6, 2C9, 2C19, 2D6, 2E1 ni 3A en microsomas hepáticos humanos.
Eliminación. El periodo efectivo de semivida del tiotropio se sitúa entre 27 y 45 horas tras la inhalación en voluntarios sanos y pacientes con EPOC. El periodo efectivo de semivida del tiotropio fue de 34 horas tras la inhalación en pacientes con asma. Tras la administración intravenosa en jóvenes voluntarios sanos, el aclaramiento total fue de 880 ml/min. El tiotropio administrado por vía intravenosa se excreta principalmente por orina (74 %). Tras la inhalación de la solución en pacientes con EPOC hasta alcanzar el estado de equilibrio, la excreción urinaria fue del 18,6 % de la dosis (0,93 μg); el resto no fue absorbido por el intestino y se eliminó por heces.
Tras la inhalación de la solución en voluntarios sanos, la excreción urinaria fue del 20,1-29,4 % de la dosis; el resto no fue absorbido por el intestino y se eliminó por heces.
En pacientes con asma, el 11,9 % (0,595 μg) de la dosis se excreta inalterado por orina durante más de 24 horas tras la administración en estado de equilibrio.
El aclaramiento renal del tiotropio supera el aclaramiento de creatinina, lo que indica excreción urinaria.
Tras la administración diaria por inhalación durante un período prolongado en pacientes con EPOC, se alcanzó el estado de equilibrio farmacocinético en el día 7, sin acumulación adicional de tiotropio.
Linealidad/no linealidad. El tiotropio muestra propiedades farmacocinéticas lineales en el rango terapéutico, independientemente de la forma farmacéutica.
Pacientes de edad avanzada. Como ocurre con todos los demás medicamentos que se eliminan principalmente por vía renal, la administración de tiotropio en pacientes de edad avanzada se asocia con una reducción de su aclaramiento renal (347 ml/min en pacientes con EPOC < 65 años frente a 275 ml/min en pacientes con EPOC > 65 años). Esto no provocó un aumento correspondiente en los valores de AUC0-6,ss ni en Cmax,ss. En pacientes con asma no se identificaron diferencias en el efecto del tiotropio en función de la edad del paciente.
Pacientes con alteración de la función renal.
Tras la administración de tiotropio (una vez al día por inhalación) en pacientes con EPOC hasta alcanzar el estado de equilibrio, la insuficiencia renal leve (aclaramiento de creatinina de 50-80 ml/min) provocó un ligero aumento del parámetro AUC0-6,ss (1,8-30 %) y valores similares de Cmax,ss en comparación con pacientes con función renal normal (aclaramiento de creatinina >80 ml/min).
En pacientes con EPOC y alteración renal de moderada a grave (aclaramiento de creatinina < 50 ml/min), la administración intravenosa de una dosis única de tiotropio provocó una duplicación del efecto sistémico total (AUC0-4h un 82 % mayor y Cmax un 52 % mayor) en comparación con pacientes con EPOC y función renal normal, lo que fue confirmado por la concentración de tiotropio en plasma tras la inhalación de la forma farmacéutica en polvo seco. En pacientes con asma y alteración leve de la función renal (aclaramiento de creatinina de 50-80 ml/min), el tiotropio en forma de inhalación no provocó un aumento correspondiente de la exposición en comparación con pacientes con función renal normal.
Pacientes con alteración de la función hepática. La insuficiencia hepática no tiene un efecto clínicamente significativo sobre la farmacocinética del tiotropio. El tiotropio se elimina principalmente por vía renal (hasta el 74 % en jóvenes voluntarios sanos) y mediante una simple escisión no enzimática del éter en productos farmacológicamente inactivos.
Pacientes japoneses con EPOC. En comparaciones cruzadas, las concentraciones plasmáticas máximas medias de tiotropio 10 minutos tras la administración en estado de equilibrio fueron un 20-70 % más altas en pacientes japoneses que en europeos tras la inhalación de tiotropio, pero no hubo indicios de mayor mortalidad ni de mayor riesgo de complicaciones cardiovasculares en pacientes japoneses en comparación con europeos. No existen suficientes datos farmacocinéticos respecto a otras razas o grupos étnicos.
Pacientes pediátricos
Asma bronquial
La exposición máxima y total (AUC y excreción urinaria) del tiotropio en pacientes con asma de 6-11, 12-17 y ≥18 años fue comparable. En función de la excreción urinaria, la exposición total del tiotropio en pacientes de 1 a 5 años fue un 52-60 % menor que en otros grupos de edad. Tras la corrección por superficie corporal, los datos de exposición total en todos los grupos de edad fueron comparables. A los pacientes de 1 a 5 años se les administró SPIRIVA RESPIMAT utilizando un espaciador portátil con válvula unidireccional y una mascarilla médica.
EPOC
En los programas de EPOC no participaron pacientes pediátricos (véase la sección «Posología y forma de administración»).
Relación farmacocinética/farmacodinámica. No existe una relación directa entre farmacocinética y farmacodinámica.
Características clínicas.
Indicaciones.
EPOC
El medicamento SPIRIVA RESPIMAT está indicado como tratamiento broncodilatador de mantenimiento para aliviar los síntomas en pacientes con enfermedad pulmonar obstructiva crónica (EPOC).
Asma
El medicamento SPIRIVA RESPIMAT está indicado como tratamiento broncodilatador de mantenimiento adicional en adultos y niños a partir de 6 años con asma grave que hayan presentado uno o más episodios graves de exacerbación del asma durante el año anterior (véanse las secciones «Instrucciones de uso y dosis» y «Farmacodinamia»).
Contraindicaciones.
Hipersensibilidad al bromuro de tiotropio, a la atropina o a sus derivados, por ejemplo al ipratropio o al oxitropio, o a cualquiera de los excipientes del medicamento.
Interacción con otros medicamentos y otras formas de interacción.
Aunque no se han realizado estudios específicos sobre interacciones con otros medicamentos, el bromuro de tiotropio se ha utilizado en combinación con otros fármacos empleados en el tratamiento de la EPOC y del asma, incluyendo broncodilatadores simpaticomiméticos, metilxantinas, corticosteroides orales e inhalados, antihistamínicos, mucolíticos, modificadores de leucotrienos, cromonas e inmunoglobulina E anti-IgE, sin que se hayan observado datos clínicos de reacciones adversas.
La administración conjunta de agonistas de los receptores beta-adrenérgicos de acción prolongada y corticosteroides inhalados no ha mostrado tener efecto sobre el tiotropio.
No se ha estudiado la administración concomitante del bromuro de tiotropio con otros medicamentos anticolinérgicos y, por tanto, no se recomienda.
Características de uso.
El bromuro de tiotropio es un broncodilatador que se administra una vez al día como tratamiento de mantenimiento. El medicamento no está indicado para el tratamiento inicial de los episodios agudos de broncoespasmo, es decir, para la asistencia de urgencia. En caso de un ataque agudo, deben utilizarse agonistas beta-2 de acción rápida.
El medicamento SPIRIVA RESPIMAT no debe usarse como monoterapia para el tratamiento del asma. Los pacientes con asma deben continuar tomando medicamentos antiinflamatorios, por ejemplo corticosteroides inhalados, sin modificar la dosis tras la introducción de SPIRIVA RESPIMAT, incluso si se produce una posible mejoría de los síntomas.
Pueden presentarse reacciones de hipersensibilidad de tipo inmediato tras la administración de la solución para inhalación de bromuro de tiotropio.
Debido a su actividad anticolinérgica, el bromuro de tiotropio debe administrarse con precaución a pacientes con glaucoma de ángulo cerrado, hiperplasia prostática benigna u obstrucción del cuello de la vejiga.
La administración de medicamentos inhalados puede provocar broncoespasmo provocado por la propia inhalación.
El tiotropio debe administrarse con precaución a pacientes con infarto de miocardio reciente (< 6 meses), cualquier arritmia cardíaca inestable o amenazante para la vida, o arritmia cardíaca que haya requerido intervención o cambio de tratamiento en el último año, o con hospitalización por insuficiencia cardíaca (clase NYHA III o IV) en el último año. Estos pacientes fueron excluidos de los ensayos clínicos, ya que podrían verse afectados por el efecto anticolinérgico del medicamento.
Dado que la concentración del medicamento en plasma aumenta con la disminución de la función renal, en pacientes con insuficiencia renal de moderada a grave (depuración de creatinina ≤ 50 ml/min), el bromuro de tiotropio solo debe administrarse si el beneficio esperado supera el riesgo potencial. No existe experiencia prolongada en el uso del medicamento en pacientes con insuficiencia renal grave (ver sección «Farmacocinética»).
Debe instruirse a los pacientes sobre la importancia de evitar que el aerosol entre en contacto con los ojos, ya que esto podría provocar un inicio más rápido o empeoramiento del glaucoma de ángulo cerrado, dolor o molestias oculares, visión borrosa temporal, sensación de halos o manchas coloreadas ante los ojos, combinado con enrojecimiento del ojo en forma de hiperemia conjuntival y edema corneal. Si aparecen estos síntomas en cualquier combinación, debe suspenderse el uso de bromuro de tiotropio y debe buscarse atención médica especializada.
La sequedad de la mucosa oral, observada durante la terapia anticolinérgica, puede estar asociada con caries dental en caso de tratamiento prolongado.
El bromuro de tiotropio no debe administrarse con más frecuencia de una vez al día (ver sección «Sobredosis»).
SPIRIVA RESPIMAT no se recomienda en pacientes con fibrosis quística. Cuando se administra a pacientes con fibrosis quística, el medicamento SPIRIVA RESPIMAT puede agravar las manifestaciones y síntomas de la enfermedad (por ejemplo, eventos adversos graves, exacerbaciones de enfermedades pulmonares, infecciones de las vías respiratorias).
Componentes auxiliares
El cloruro de benzalconio puede provocar obstrucción broncopulmonar y dificultad respiratoria. Los pacientes con asma tienen mayor riesgo de presentar estos efectos adversos. El medicamento contiene 0,0011 mg de cloruro de benzalconio por cada inhalación. La inhalación es la dosis disponible para el paciente tras pasar por la boquilla. 2 inhalaciones equivalen a una dosis terapéutica.
Uso durante el embarazo o la lactancia.
Embarazo
Existen muy pocos datos sobre el uso de tiotropio en mujeres embarazadas. Los estudios en animales no mostraron ausencia de efectos negativos directos o indirectos desde el punto de vista de la toxicidad reproductiva a dosis clínicamente relevantes.
Como medida de precaución, se recomienda evitar el uso de SPIRIVA RESPIMAT durante el embarazo.
Lactancia
No se sabe si el bromuro de tiotropio se excreta en la leche materna. Aunque estudios en roedores han demostrado que el bromuro de tiotropio se excreta en la leche materna solo en pequeñas cantidades, no se recomienda el uso de SPIRIVA RESPIMAT durante la lactancia. El bromuro de tiotropio es un compuesto de acción prolongada. La decisión sobre continuar o interrumpir la lactancia o continuar o interrumpir el tratamiento con SPIRIVA RESPIMAT debe tomarse considerando los beneficios de la lactancia para el niño y los beneficios del tratamiento para la madre.
Fertilidad
No existen datos clínicos sobre el efecto del tiotropio sobre la fertilidad. Los estudios preclínicos con tiotropio no mostraron ningún efecto negativo sobre la fertilidad.
Capacidad para afectar la velocidad de reacción al conducir vehículos o manejar mecanismos.
No se han realizado estudios sobre la capacidad de afectar la velocidad de reacción al conducir vehículos o al trabajar con otros mecanismos.
El vértigo o la visión borrosa pueden afectar la velocidad de reacción al conducir vehículos o al trabajar con otros mecanismos.
Vía de administración y dosis.
Vía de administración
El medicamento está indicado únicamente para uso por inhalación. El cartucho solo puede utilizarse con el dispositivo inhalador RESPIMAT.
Dos inhalaciones con el inhalador RESPIMAT contienen una dosis.
La dosis recomendada para adultos es de 5 mcg de tiotropio en forma de dos inhalaciones con el inhalador RESPIMAT, una vez al día a la misma hora.
No se debe superar la dosis recomendada.
En el tratamiento del asma, el beneficio del medicamento se manifiesta plenamente tras varias dosis. A los pacientes adultos con asma grave se debe administrar tiotropio además de corticosteroides inhalados (CIS) (≥ 800 mcg de budesonida/día o equivalente) y al menos un medicamento de terapia de mantenimiento.
Categorías especiales de pacientes
Los pacientes de edad avanzada pueden utilizar el medicamento SPIRIVA RESPIMAT en las dosis recomendadas.
Los pacientes con insuficiencia renal pueden utilizar el medicamento SPIRIVA RESPIMAT en las dosis recomendadas. En cuanto al uso en pacientes con insuficiencia renal de moderada a grave (depuración de creatinina ≤ 50 ml/min, véanse las secciones «Precauciones de uso» y «Propiedades farmacológicas»).
Los pacientes con insuficiencia hepática pueden utilizar el medicamento en las dosis recomendadas (véase la sección «Propiedades farmacológicas»).
Niños
Asma
La dosis recomendada para pacientes de 6 a 17 años es de 5 mcg de tiotropio en forma de dos inhalaciones con el inhalador RESPIMAT, una vez al día a la misma hora.
A los adolescentes (12─17 años) con asma grave se debe administrar tiotropio además de corticosteroides inhalados (> 800─1600 mcg de budesonida/día o equivalente) y un medicamento de terapia de mantenimiento, o además de corticosteroides inhalados (400─800 mcg de budesonida/día o equivalente) con dos medicamentos de terapia de mantenimiento.
A los niños (6─11 años) con asma grave se debe administrar tiotropio además de corticosteroides inhalados (> 400 mcg de budesonida/día o equivalente) y un medicamento de terapia de mantenimiento, o además de corticosteroides inhalados (200─400 mcg de budesonida/día o equivalente) con dos medicamentos de terapia de mantenimiento.
No se ha establecido la seguridad y eficacia del medicamento SPIRIVA RESPIMAT en niños de 6─17 años con asma moderada. No se ha establecido la seguridad y eficacia del medicamento SPIRIVA RESPIMAT en niños menores de 6 años. La información disponible se presenta en las secciones «Farmacodinamia» y «Farmacocinética», pero no pueden darse recomendaciones sobre la dosificación.
EPOC
No existe experiencia en el uso del medicamento SPIRIVA RESPIMAT en niños (menores de 18 años).
Fibrosis quística
No se ha establecido la eficacia ni la seguridad del uso del medicamento SPIRIVA RESPIMAT (véase la sección «Precauciones de uso»).
Vía de administración
Este medicamento está indicado únicamente para uso por inhalación. RESPIMAT es un inhalador que genera un aerosol en spray para inhalación. Está diseñado para uso por un solo paciente y para la administración de múltiples dosis a partir de un único cartucho.
Los pacientes deben leer las instrucciones de uso del inhalador RESPIMAT antes de comenzar a utilizar el medicamento SPIRIVA RESPIMAT.
Para garantizar la correcta administración del medicamento, el médico u otro profesional sanitario debe demostrar al paciente cómo utilizar el inhalador.
Instrucciones para el uso y mantenimiento del inhalador RESPIMAT
Los niños deben utilizar el medicamento SPIRIVA RESPIMAT bajo supervisión de un adulto.
El paciente debe utilizar el inhalador UNA VEZ AL DÍA.
Cada vez que se utilice, debe realizar DOS INHALACIONES.
**

**
- Si no se ha utilizado el medicamento SPIRIVA RESPIMAT durante más de 7 días, libere una inhalación dirigiendo el inhalador hacia abajo.
- Si no se ha utilizado el medicamento SPIRIVA RESPIMAT durante más de 21 días, repita los pasos 4─6 de «Preparación para el primer uso», hasta que aparezca una nube de aerosol. Luego repita los pasos 4─6 otras tres veces.
Cómo cuidar el inhalador RESPIMAT
- Limpiar la boquilla, incluida la parte metálica en su interior, únicamente con una toallita húmeda o tela, al menos una vez por semana.
- Cualquier pequeño cambio de color en la boquilla no afecta al funcionamiento del inhalador RESPIMAT.
- Si es necesario, limpie el exterior del inhalador RESPIMAT con una toallita húmeda o tela.
Cuándo debe adquirirse un nuevo medicamento SPIRIVA RESPIMAT

-
El inhalador con el medicamento SPIRIVA RESPIMAT contiene 60 inhalaciones (30 dosis) cuando se utiliza según las indicaciones (dos inhalaciones una vez al día).
-
El indicador de dosis muestra aproximadamente la cantidad restante de medicamento.
-
Cuando el indicador llega a la zona roja de la escala, significa que queda aproximadamente medicamento para 7 días (14 inhalaciones).
-
Tan pronto como el indicador de dosis alcance el final de la escala roja, el inhalador con el medicamento SPIRIVA RESPIMAT se bloqueará automáticamente: no habrá más dosis disponibles para liberar. A partir de ese momento, no será posible girar la base transparente.
-
No más tarde de tres meses después del primer uso, el inhalador con el medicamento SPIRIVA RESPIMAT debe desecharse, incluso si no se ha utilizado todo el líquido.
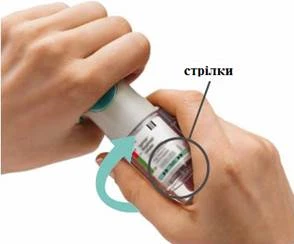
Preparación para el primer uso
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
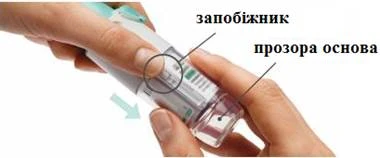
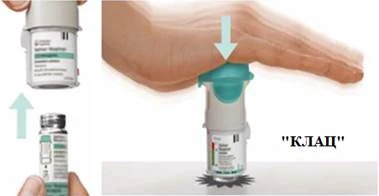
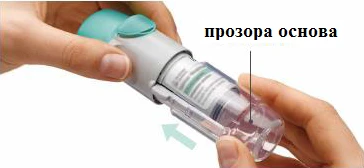
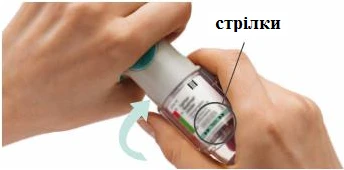


 PASOS 4-6
PASOS 4-6
Uso diario
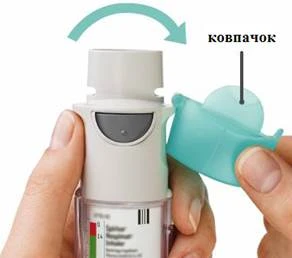
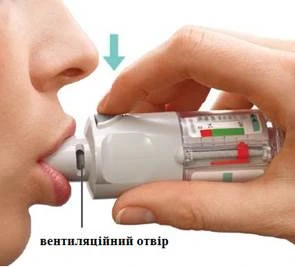
| GIRE
|
|
| ABRA
|
|
| PULSE
|
2 INHALACIONES UNA VEZ AL DÍA |
Niños.
Asma
No se ha establecido la seguridad y eficacia del medicamento SPIRIVA RESPIMAT en niños menores de 6 años (ver secciones «Propiedades farmacológicas» y «Instrucciones de uso y dosis»).
EPOC
No existe experiencia en el uso del medicamento SPIRIVA RESPIMAT en niños (menores de 18 años).
Sobredosificación.
Dosis elevadas del medicamento pueden provocar signos y síntomas anticolinérgicos.
Sin embargo, no se han observado efectos adversos sistémicos anticolinérgicos en voluntarios sanos tras una inhalación única de hasta 340 mcg de bromuro de tiotropio. Adicionalmente, tras 14 días de tratamiento con dosis de hasta 40 mcg de tiotropio, solución para inhalación, en voluntarios sanos, no se observaron otros efectos adversos significativos más allá de sequedad de las mucosas de la boca/garganta y de la cavidad nasal, excepto una disminución evidente en la secreción salival tras 7 días de tratamiento.
Reacciones adversas.
Información resumida sobre la seguridad del medicamento
Muchos de los efectos adversos indicados a continuación pueden atribuirse a las propiedades anticolinérgicas del bromuro de tiotropio.
Tabla resumida de reacciones adversas
La frecuencia de aparición de las reacciones adversas indicadas a continuación, observadas en el grupo de pacientes que recibieron bromuro de tiotropio, se calculó sobre la base de datos combinados procedentes de 7 ensayos clínicos controlados con placebo en pacientes con EPOC (3 282 pacientes) y 12 ensayos clínicos controlados con placebo en adultos y pacientes pediátricos con asma (1 930 pacientes), con periodos de tratamiento de entre cuatro semanas y un año.
Criterios de evaluación de la frecuencia de reacciones adversas:
muy frecuentes (≥1/10); frecuentes (≥1/100 - <1/10); poco frecuentes (≥1/1 000 - <1/100); raras (≥1/10 000 - <1/1 000); muy raras (<1/10 000); frecuencia desconocida (no puede determinarse a partir de los datos disponibles).
Tabla 4
| Clasificación por órganos y sistemas/término preferente según la clasificación MedDRA |
Frecuencia |
|
| EPOC |
Asma |
|
| Alteraciones del metabolismo y de la nutrición |
||
| Deshidratación |
No conocido |
No conocido |
| Trastornos del sistema nervioso |
||
| Vertigo |
Infrecuente |
Infrecuente |
| Cefalea |
Infrecuente |
Infrecuente |
| Alteraciones del sueño |
Isolado |
Infrecuente |
| Trastornos oculares |
||
| Glaucoma |
Isolado |
No conocido |
| Aumento de la presión intraocular |
Isolado |
No conocido |
| Visión borrosa |
Isolado |
No conocido |
| Trastornos del sistema cardiovascular |
||
| Fibrilación auricular |
Isolado |
No conocido |
| Sensación de palpitaciones |
Isolado |
Infrecuente |
| Taquicardia supraventricular |
Isolado |
No conocido |
| Taquicardia |
Isolado |
No conocido |
| Trastornos del aparato respiratorio, del tórax y del mediastino |
||
| Tos |
Infrecuente |
Infrecuente |
| Faringitis |
Infrecuente |
Infrecuente |
| Disfonía |
Infrecuente |
Infrecuente |
| Hemorragias nasales |
Isolado |
Isolado |
| Broncoespasmo |
Isolado |
Infrecuente |
| Laringitis |
Isolado |
No conocido |
| Sinusitis |
No conocido |
No conocido |
| Trastornos gastrointestinales |
||
| Sequedad de boca |
Frecuente |
Infrecuente |
| Estreñimiento |
Infrecuente |
Isolado |
| Candidiasis orofaríngea |
Infrecuente |
Infrecuente |
| Disfagia |
Isolado |
No conocido |
| Enfermedad por reflujo gastroesofágico |
Isolado |
No conocido |
| Caries dental |
Isolado |
No conocido |
| Gingivitis |
Isolado |
Isolado |
| Glositis |
Isolado |
No conocido |
| Estomatitis |
No conocido |
Isolado |
| Obstrucción intestinal, incluyendo obstrucción intestinal paralítica |
No conocido |
No conocido |
| Náuseas |
No conocido |
No conocido |
| Trastornos de la piel, tejidos subcutáneos e inmunológico |
||
| Erupción cutánea |
Infrecuente |
Infrecuente |
| Prurito |
Infrecuente |
Isolado |
| Angioedema |
Isolado |
Isolado |
| Urticaria |
Isolado |
Isolado |
| Infección de la piel/úlcera de la piel |
Isolado |
No conocido |
| Sequedad de la piel |
Isolado |
No conocido |
| Hipersensibilidad (incluyendo reacciones alérgicas de tipo inmediato) |
No conocido |
Isolado |
| Reacción anafiláctica |
No conocido |
No conocido |
| Trastornos del sistema musculoesquelético y del tejido conectivo |
||
| Edema articular |
No conocido |
No conocido |
| Trastornos renales y urinarios |
||
| Retención urinaria |
Infrecuente |
No conocido |
| Alteraciones miccionales |
Infrecuente |
No conocido |
| Infección del tracto urinario |
Isolado |
Isolado |
Descripción de reacciones adversas individuales
Durante estudios clínicos controlados con participación de pacientes con EPOC, las reacciones adversas frecuentes asociadas con el efecto anticolinérgico fueron sequedad de boca. Esta se presentó en aproximadamente el 2,9 % de los pacientes. En pacientes asmáticos, la frecuencia de sequedad de boca fue del 0,83 %.
En el transcurso de 7 ensayos clínicos con pacientes con EPOC, la sequedad de boca fue la causa de abandono prematuro del estudio en 3 de 3282 pacientes tratados con tiotropio (0,1 %). En el transcurso de 12 ensayos clínicos con pacientes asmáticos, no se registró ningún caso de abandono prematuro del estudio debido a sequedad de boca (1930 pacientes).
Las reacciones adversas graves asociadas con el efecto anticolinérgico incluyen glaucoma, estreñimiento, obstrucción intestinal, incluyendo obstrucción intestinal paralítica, y retención urinaria.
Pacientes pediátricos
La base de datos de seguridad incluye a 560 pacientes pediátricos (296 pacientes de 1*─11 años y 264 pacientes de 12─*17 años) que participaron en 5 ensayos clínicos controlados con placebo, en los que los periodos de tratamiento variaron entre 12 semanas y un año. La frecuencia, tipo y gravedad de las reacciones adversas en pacientes pediátricos fueron similares a las observadas en adultos.
Otras categorías de pacientes.
Puede desarrollarse un aumento del efecto anticolinérgico del medicamento con el incremento de la edad del paciente.
Notificación de reacciones adversas sospechosas
La notificación de reacciones adversas sospechosas tras la autorización del medicamento es importante. Permite continuar con el seguimiento de la relación beneficio-riesgo del medicamento. Los profesionales médicos y farmacéuticos, así como los pacientes o sus representantes legales, deben informar sobre todos los casos de reacciones adversas sospechosas y sobre la falta de eficacia del medicamento a través del Sistema de Información Automatizado de Farmacovigilancia, disponible en el siguiente enlace: https://aisf.dec.gov.ua».
Período de validez. 3 años.
Período de validez tras la primera utilización: 3 meses.
Condiciones de conservación.
Conservar a una temperatura no superior a 25 °C. ¡No congelar! Conservar en un lugar fuera del alcance de los niños.
Envase.
4 ml en un cartucho (60 inhalaciones); 1 cartucho junto con 1 inhalador RESPIMAT en una caja de cartón.
Categoría de dispensación.
Bajo receta médica.
Fabricante.
Boehringer Ingelheim Pharma GmbH & Co. KG
o
Boehringer Ingelheim Espana, S.A.
Domicilio del fabricante y dirección del lugar de actividad.
Binger Strasse 173, 55216 Ingelheim am Rhein, Alemania / Binger Strasse 173, 55216 Ingelheim am Rhein, Germany.
o
Prat de la Riba, 50, 08174 Sant Cugat del Valles (Barcelona), España / Prat de la Riba, 50, 08174 Sant Cugat del Valles (Barcelona), Spain.