Somavert
UkrainaSpis treści
INSTRUKCJA dotyczaca stosowania leku Somavert (Somavert)
Skład:
substancja czynna: pegwizomant;
1 fiolka zawiera 10 mg, 15 mg, 20 mg lub 30 mg pegwizomantu;
substancje pomocnicze: glicyna, manitol (E 421), fosforan sodu bezwodny, fosforan sodu dwusiarczony monohydrat.
1 strzykawka wstępnie napełniona z rozpuszczalnikiem zawiera wodę do wstrzykiwań.
Postać leku. Liofilizat i rozpuszczalnik do roztworu do wstrzykiwań.
Główne właściwości fizyko-chemiczne: masa liofilizowana od białej do prawie białej.
Grupa farmakoterapeutyczna. Inne hormony przysadki i ich analogi. Pegwizomant. Kod ATC H01A X01.
Właściwości farmakologiczne.
Opis
Somavert zawiera pegwisomant, analog ludzkiego hormonu wzrostu, który został strukturalnie zmodyfikowany, aby działać jako antagonist receptorów hormonu wzrostu.
Pegwisomant to białko wytworzone metodą rekombinowanego DNA, zawierające 191 reszt aminokwasowych, do których kowalencyjnie przyłączone są cząsteczki polimeru polietylenglikolu (PEG) (głównie 4–6 cząsteczek PEG/białko). Masa cząsteczkowa białka pegwisomantu wynosi 21 998 daltonów. Masa cząsteczkowa części PEG pegwisomantu wynosi około 5000 daltonów. W związku z tym ogólna masa cząsteczkowa pegwisomantu wynosi głównie około 42 000; 47 000 oraz 52 000 daltonów. Pegwisomant jest syntetyzowany przez specyficzny szczep bakterii Escherichia coli, który został zmodyfikowany genetycznie poprzez dodanie plazmidu niosącego gen antagonisty receptorów hormonu wzrostu. Aktywność biologiczną oznacza się za pomocą testu biologicznego proliferacji komórek. Wiązanie leku Somavert z receptorem hormonu wzrostu prowadzi do zaburzenia właściwego wiązania drugiego receptora hormonu wzrostu, hamując dimeryzację funkcjonalnego receptora oraz dalszą wewnątrzkomórkową drogę sygnalizacyjną.
Farmakodynamika.
Pegwisomant wiąże się selektywnie z receptorem hormonu wzrostu na powierzchni komórki i nie wykazuje reaktywności krzyżowej z 19 innymi badanymi receptorami cytookinowymi, w tym z prolaktyną. Pegwisomant powoduje zmniejszenie stężenia w surowicy insulinopodobnego czynnika wzrostu I (IGF-1), wolnego IGF-1, podjednostki kwasolabilnej (KLO) oraz białka wiążącego insulinopodobny czynnik wzrostu typu 3 (BZIGF-3) (patrz niżej „Badania kliniczne”, rys. 1).
Mechanizm działania
Pegwisomant wiąże się selektywnie z receptorami hormonu wzrostu na powierzchni komórki, gdzie blokuje wiązanie endogennego hormonu wzrostu i w ten sposób przeszkadza w przekazywaniu sygnału hormonu wzrostu.
Hamowanie działania hormonu wzrostu prowadzi do zmniejszenia stężenia w surowicy IGF-1 oraz innych białek surowicy wrażliwych na działanie hormonu wzrostu, takich jak wolny IGF-1, podjednostka kwasolabilna IGF-1 oraz BZIGF-3.
Badania kliniczne
W 12-tygodniowym, randomizowanym, podwójnie ślepych, wieloośrodkowym badaniu porównującym placebo i Somavert wzięło udział łącznie 112 pacjentów (63 mężczyzn i 49 kobiet) z akromegalią. Średni wiek ± odchylenie standardowe (SD) wynosił 48 ± 14 lat, a średnie trwanie akromegalii wynosiło 8 ± 8 lat. 93 pacjentom wcześniej przeprowadzono operację przysadki mózgu, z czego 57 osobom dodatkowo przeprowadzono standardową terapię promieniową. 6 pacjentom poddano napromienianie bez interwencji chirurgicznej, 9 osób otrzymywało jedynie leczenie farmakologiczne, a kolejnych 4 pacjentów wcześniej nie otrzymało żadnego rodzaju terapii. Na początku badania średnie wartości ± SD czasu, który upłynął od ostatniej operacji oraz/lub napromienienia, wynosiły odpowiednio 6,8 ± 0,93 roku (n = 63) oraz 5,6 ± 0,57 roku (n = 93).
Pacjentów włączono do badania, jeśli ich stężenie IGF-1 w surowicy, oznaczone po odpowiednim okresie wypłukania leku, było ≥ 1,3 razy wyższe od górnej granicy normy skorygowanej o wiek. W czasie wizyty kwalifikacyjnej pacjentów przydzielono w sposób losowy do jednej z czterech grup terapii: placebo (n = 32); Somavert podany podskórnie w dawce 10 mg/doba (n = 26), 15 mg/doba (n = 26) lub 20 mg/doba (n = 28). Głównym punktem końcowym skuteczności była procentowa zmiana stężenia IGF-1 od poziomu na wizycie kwalifikacyjnej do 12. tygodnia. W trzech grupach otrzymujących Somavert zaobserwowano istotne statystycznie (p < 0,01) obniżenie poziomu IGF-1 w surowicy krwi w porównaniu z grupą placebo (tab. 1).
Tabela 1. Średnia procentowa zmiana stężenia IGF-1 od poziomu na wizycie kwalifikacyjnej do 12. tygodnia w grupie „pacjentów leczonych”
| Wskaźniki |
Placebo n = 31 |
Somavert |
||
| 10 mg/doba n = 26 |
15 mg/doba n = 26 |
20 mg/doba n = 28 |
||
| Średnie stężenie IGF-1 w czasie wizyty inkluzji (ng/ml) (SW) |
670 (288) |
627 (251) |
649 (293) |
732 (205) |
| Średnia procentowa zmiana stężenia IGF-1 od wizyty inkluzji (SW) |
|
|
|
|
| Somavert minus placebo (95 % CI dla różnicy między lekami badawczymi) |
(–35; –11) |
(–56; –33) |
(–68; –49) |
|
* P < 0,01; n – liczba pacjentów; SD – odchylenie standardowe.
Dodatkowo w grupach otrzymujących Somavert obserwowano zmniejszenie stężenia wolnego IGF-1, IGFBP-3 oraz KLO w surowicy we krwi w porównaniu z grupą placebo w czasie wszystkich zaplanowanych wizyt badawczych (rys. 1).
| Somavert 15 mg/doba (n = 24–26) Somavert 20 mg/doba (n = 27–28) |
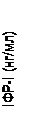
| 800 600 400 200 |
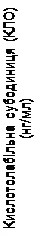
| Placebo (n = 31) Somavert 10 mg/doba (n = 25–26) |
| 0 2 4 8 12 Tydzień |
| 0 2 4 8 12 Tydzień |
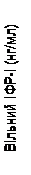
Rys. 1. Wpływ leku Somavert na markery w surowicy krwi (średnia ± standardowy błąd średniej).
Po 12 tygodniach terapii znormalizowane stężenia IGF-1 obserwowano u niżej wskazanej liczby pacjentów (w procentach) (rys. 2).
| Somavert 20 mg na dobę |
| Somavert 15 mg/dobu |
| Somavert 10 mg/dobę |
| Placebo |
Rys. 2. Odsetek pacjentów, u których stężenia IGF-1 uległy normalizacji w 12. tygodniu leczenia.
W tabeli 2 przedstawiono wpływ terapii lekiem Somavert na przykładzie rozmiaru pierścienia (standardowe rozmiary wyrobów jubilerskich przeliczone na skalę liczbową od 1 do 63 punktów), a także objawów i objawów akromegalii. Każdą pojedynczą cechę lub objaw akromegalii (obrzęk miękkich tkanek, artralgia, ból głowy, nadmierna potliwość i zwiększona zmęczalność) oceniano w dziewięciopunktowej skali porządkowej (0 – brak, 8 – ciężkie i niesprawność powodujące), a ogólny wynik oceny objawów lub objawów akromegalii uzyskiwano na podstawie sumy punktów poszczególnych skal. Średnie wyniki na poziomie wyjściowym były następujące: rozmiar pierścienia – 47,1; ogólny wynik oceny objawów i objawów – 15,2; obrzęk miękkich tkanek – 2,5; artralgia – 3,2; ból głowy – 2,4; nadmierna potliwość – 3,3 i zwiększona zmęczalność – 3,7.
Tabela 2. Średnia zmiana rozmiaru pierścienia oraz objawów i objawów akromegalii w 12. tygodniu w porównaniu z wizytą wprowadzenia do badania (SW).
| Wskaźniki |
Placebo n = 30 |
Somavert |
||
| 10 mg/dobę n = 26 |
15 mg/dobę n = 24–25 |
20 mg/dobę n = 26–27 |
||
| Rozmiar pierścienia |
|
|
|
|
| Całkowita liczba punktów w ocenie objawów i objawów akromegalii |
1,3 (6,0) |
|
|
|
| Obwód miękkich tkanek |
0,3 (2,3) |
|
|
|
| Artrodynia |
0,1 (1,8) |
|
|
|
| Ból głowy |
0,1 (1,7) |
|
|
|
| Potliwość |
0,1 (1,7) |
|
|
|
| Zwiększona zmęczalność |
0,7 (1,5) |
|
|
|
Stężenia surowiczego hormonu wzrostu mierzonych za pomocą metod ilościowych opracowanych do badań, z wykorzystaniem przeciwciał nie wykazujących reakcji krzyżowej z pegwizomantem, zwiększały się w ciągu dwóch tygodni od rozpoczęcia terapii lekiem Somavert. Największy wzrost stężenia hormonu wzrostu obserwowano u pacjentów otrzymujących Somavert w dawce 20 mg/dobę. Najprawdopodobniej efekt ten wynika ze zmniejszenia się hamowania sekrecji hormonu wzrostu przy obniżeniu poziomu IGF-1. Jak pokazano na rys. 3, gdy pacjenci z akromegalią otrzymywali dawkę załadunkową leku Somavert, a następnie ustalone codzienne dawki, wzrost stężenia hormonu wzrostu był odwrotnie proporcjonalny do spadku poziomu IGF-1 i zwykle ustabilizował się do drugiego tygodnia. Stężenie hormonu wzrostu w surowicy krwi pozostawało stabilne u pacjentów otrzymujących Somavert przez przeciętnie 43 tygodnie (zakres 0–82 tygodnie).
| Placebo |
| Tydzień Tydzień Tydzień Tydzień Tydzień 0 2 4 8 12 |
| 20 mg/d |
| 15 mg/d |
| 10 mg/d |

Rys. 3. Procentowa zmiana stężenia hormonu wzrostu oraz IGF-1 w surowicy krwi.
W otwartym etapie badania klinicznego uczestniczyło 109 pacjentów (w tym 6 nowych pacjentów) przy średnim czasie stosowania leku wynoszącym 42,6 tygodnia (zakres: 1 dzień – 82 tygodnie); u 93 (85,3%) pacjentów wystąpiły działania niepożądane; u 16 (14,7%) zaobserwowano poważne działania niepożądane (PZN), a u 4 (3,7%) osób leczenie zostało przerwane z powodu działań niepożądanych (ból głowy, podwyższenie poziomów biochemicznych wskaźników funkcji wątroby, rak trzustki oraz przyrost masy ciała). Ogółem 100 (92,6%) spośród 108 uczestników z dostępnymi danymi pomiarowymi IGF-1 miało stężenie IGF-1 w normie w czasie dowolnej wizyty podczas badania.
W różnych badaniach, a także w badaniu ACROSTUDY, pegwizomant normalizował stężenia IGF-1 u dużej części pacjentów (>70%) oraz istotnie obniżał stężenia glukozy we krwi na czczo (GPN) oraz insuliny we krwi na czczo (IPN).
Pegwizomant poprawia również wrażliwość na insulinę, co najprawdopodobniej wiąże się z blokadą receptorów GH w tkankach, głównie w wątrobie, a także w tkance tłuszczowej, nerkach i mięśniach szkieletowych, eliminując w ten sposób szkodliwy wpływ GH na szlaki sygnałowe insuliny, lipolizę i glukoneogenezę. Mechanizm działania tego wpływu nie jest jednak dokładnie znany. Pacjenci z cukrzycą i akromegalią mogą wymagać zmniejszenia dawek insuliny lub leków obniżających poziom glukozy we krwi (patrz sekcje „Szczególne ostrzeżenia i środki ostrożności stosowania”, „Współdziałanie z innymi lekami i inne formy interakcji”).
Farmakokinetyka.
Wchłanianie. Po podaniu podskórnie maksymalne stężenie pegwizomantu w surowicy krwi osiągane jest zazwyczaj nie wcześniej niż po 33–77 godzinach od momentu podania. Średnie wchłonięcie dawki podanej podskórnie w dawce 20 mg wynosi 57% w porównaniu do podania dożylnego dawki 10 mg.
Rozkład. Średnie widoczne objętości rozkładu pegwizomantu wynosi 7 l (współczynnik zmienności 12%), co wskazuje na niewielkie rozprowadzenie pegwizomantu do tkanek. Po jednorazowym podaniu podskórnie ekspozycja (Cmax, AUC) pegwizomantu wzrasta nieliniowo wraz ze zwiększaniem dawki. Średnie ± błąd standardowy średniej (BSS) stężenie pegwizomantu w surowicy krwi po 12 tygodniach stosowania dawek dziennych 10; 15 i 20 mg wynosiło odpowiednio 6600 ± 1330; 16 000 ± 2200 i 27 000 ± 3100 ng/ml.
Porównano względną biodostępność dawki 1 × 30 mg pegwizomantu z dawką 2 × 15 mg pegwizomantu w badaniu z jednorazowym podaniem. Wartości AUCinf i Cmax pegwizomantu po podaniu w postaci jednej iniekcji o mocy działania 30 mg były odpowiednio o około 6% i 4% wyższe niż po podaniu w postaci dwóch iniekcji po 15 mg.
Metabolizm i eliminacja. Cząsteczka pegwizomantu zawiera kowalencyjnie związane polimery polietylenglikolu, które mają na celu zmniejszenie szybkości klirensu. Klirens pegwizomantu po wielokrotnym podawaniu jest mniejszy niż klirens obserwowany po jednorazowym podaniu. Średni całkowity układowy klirens pegwizomantu w organizmie po wielokrotnym podawaniu szacuje się na poziomie od 36 do 28 ml/godz. dla dawek podskórnych w zakresie od 10 do 20 mg na dobę odpowiednio. Stwierdzono, że klirens pegwizomantu zwiększa się wraz z masą ciała. Pegwizomant jest wydalany z surowicy krwi ze średnią połową okresu półtrwania wynoszącą od 60 do 138 godzin po jednorazowym lub wielokrotnym podaniu. Mniej niż 1% podanej dawki wykrywa się w moczu w ciągu 96 godzin. Ścieżki eliminacji pegwizomantu u ludzi nie zostały zbadane.
Badania interakcji między lekami
W badaniach klinicznych pacjentom otrzymującym opioidy często wymagane były wyższe stężenia pegwizomantu w surowicy krwi w celu osiągnięcia odpowiedniego hamowania IGF-1 w porównaniu z pacjentami, którzy nie otrzymywali opioidów. Mechanizm tej interakcji jest nieznany (patrz sekcja „Współdziałanie z innymi lekami i inne formy interakcji”).
Osobne grupy pacjentów
Nie przeprowadzono badań farmakokinetycznych z udziałem pacjentów z zaburzeniami funkcji nerek, zaburzeniami funkcji wątroby, pacjentów w wieku podeszłym ani dzieci. Nie badano wpływu przynależności rasowej pacjenta na farmakokinetykę pegwizomantu. W trakcie analizy populacyjnej farmakokinetyki nie stwierdzono różnic w farmakokinetyce pegwizomantu w zależności od płci pacjentów.
Immunogenność
Obserwowana częstość występowania przeciwciał w dużym stopniu zależy od czułości i specyficzności metody analizy. Różnice w metodach analizy wykluczają sensowne porównanie częstości występowania przeciwciał w badaniach opisanych poniżej z częstością występowania przeciwciał w innych badaniach, w tym w badaniach leku Somavert lub innych analogów hormonu wzrostu.
W badaniach klinicznych przed rejestracją u około 17% pacjentów otrzymujących Somavert rozwijały się niskie miana neutralizujących przeciwciał przeciwko hormonowi wzrostu. Chociaż obecność tych przeciwciał wydaje się nie wpływać na skuteczność leku Somavert, kliniczne znaczenie rozwoju tych przeciwciał w długoterminowej perspektywie jest nieznane. Obecnie nie są dostępne zestawy diagnostyczne do analizy przeciwciał przeciwko pegwizomantowi u pacjentów otrzymujących Somavert.
Dane podane powyżej odzwierciedlają odsetek pacjentów, u których wyniki analizy zostały uznane za pozytywne pod względem obecności przeciwciał przeciwko lekowi Somavert. Wykrycie powstawania przeciwciał w dużym stopniu zależy od czułości i specyficzności metody analizy. Ponadto na obserwowaną częstość pozytywnych wyników analizy przeciwciał mogły wpływać różne czynniki, w tym postępowanie z próbkami, czas pobrania próbek, leki współadministrowane oraz choroba podstawowa. Z tego powodu porównywanie częstości przypadków powstawania przeciwciał przeciwko lekowi Somavert z częstością powstawania przeciwciał przeciwko innym lekom może prowadzić do błędnych wniosków.
Dane kliniczne.
Wskazania.
Somavert jest wskazany w leczeniu akromegalii u pacjentów, u których zabiegi chirurgiczne lub radioterapia nie przyniosły wystarczającego efektu lub u których te metody leczenia są niewskazane. Celem leczenia jest znormalizowanie stężenia insulinopodobnego czynnika wzrostu 1 (IGF-1) w surowicy krwi.
Przeciwwskazania.
Nie należy stosować leku Somavert u pacjentów z uczuleniem na substancję czynną lub którykolwiek z pozostałych składników leku.
Interakcje z innymi lekami oraz inne formy interakcji.
Insluina oraz/lub doustne leki hipoglikemiczne
Pacjentom z akromegalią i cukrzycą, którzy otrzymują insulinę i/lub doustne leki hipoglikemiczne, może być wymagane zmniejszenie dawki insuliny i/lub doustnych środków hipoglikemicznych po rozpoczęciu terapii lekiem Somavert (patrz sekcja „Szczególne środki ostrożności podczas stosowania”).
Alkohole
W trakcie badań klinicznych u pacjentów przyjmujących opony często wymagane były wyższe dawki leku Somavert w celu znormalizowania stężenia IGF-1 w porównaniu z pacjentami nieprzyjmującymi opioidów. Mechanizm tej interakcji jest nieznany.
Szczególne środki ostrożności.
Hipoglikemia związana ze zmniejszeniem stężenia hormonu wzrostu u pacjentów z cukrzycą
Hormon wzrostu przeciwdziała działaniu insuliny na metabolizm węglowodanów poprzez zmniejszenie wrażliwości na insulinę. W związku z tym u niektórych pacjentów stosujących Somavert może poprawić się tolerancja glukozy. U pacjentów z cukrzycą należy dokładnie monitorować stan, a w razie potrzeby zmniejszyć dawki leków przeciwcukrzycowych, aby uniknąć hipoglikemii.
Toxyczność wątroby
Przed rozpoczęciem terapii lekiem Somavert należy ocenić wyjściowe stężenia alaninotransaminazy (ALT), asparaginianotransaminazy (AST), bilirubiny ogólnej oraz fosfatazy alkalicznej w surowicy. W tabeli 3 przedstawiono zalecenia dotyczące rozpoczęcia terapii lekiem Somavert w zależności od wyników oceny czynności wątroby (LFT).
Bezobjawowe, przemijające podwyższenie stężenia transaminaz, przekraczające do 15 razy górny limit normy (ULN), obserwowano u < 2 % pacjentów w trakcie dwóch otwartych badań (łącznie z udziałem 147 pacjentów). Wysokie stężenia transaminaz nie były związane ze wzrostem stężenia bilirubiny. Podwyższone stężenia transaminaz wracały do normy z czasem, najczęściej po tymczasowym wstrzymaniu terapii. Doniesienia po rejestracji wykazały podwyższenie stężenia transaminaz wątrobowych w surowicy powyżej 20 razy w stosunku do ULN, związane ze wzrostem bilirubiny ogólnej powyżej 2 razy w stosunku do ULN. W wielu z tych przypadków przerwanie terapii lekiem Somavert prowadziło do zmniejszenia się lub zniknięcia odchyleń od normy wskaźników wątrobowych.
Podczas terapii lek Somavert należy stosować zgodnie z informacjami dotyczącymi odchyleń biochemicznych wskaźników czynności wątroby, przedstawionymi w tabeli 4.
Tabela 3. Zalecenia dotyczące rozpoczęcia terapii lekiem Somavert w zależności od biochemicznych wskaźników czynności wątroby (LFT) na poziomie wyjściowym oraz dotyczące okresowego monitorowania LFT w czasie terapii lekiem Somavert
| Poziom wyjściowy PFP |
Zalecenia |
| Normalny |
|
| Zwiększony, ale mniejszy lub równy 3 × WGN |
Można stosować Somavert, jednak należy kontrolować poziom PFP miesięcznie przez co najmniej rok od rozpoczęcia terapii, a następnie 2 razy w roku przez następny rok. |
| Więcej niż 3 razy przekracza WGN |
|
Jeśli podczas leczenia lekiem Somavert u pacjenta występuje wzrost stężenia PFP lub pojawiają się inne objawy lub oznaki zaburzeń funkcji wątroby, zaleca się leczenie przedstawione poniżej (tabela 4).
Tabela 4. Rekomendacje kliniczne oparte na wynikach oceny biochemicznych wskaźników funkcji wątroby podczas leczenia lekiem Somavert
| Poziom enzymów wątrobowych i objawy kliniczne/symptomy |
Zalecenia |
| Co najmniej 3-krotne, ale mniej niż 5-krotne przekroczenie GGP (bez objawów/objawów zapalenia wątroby lub innego uszkodzenia wątroby lub bez podwyższenia stężenia bilirubiny całkowitej w surowicy) |
|
| Co najmniej 5-krotne przekroczenie GGP lub podwyższenie transaminaz co najmniej 3-krotnie w porównaniu z GGP, towarzyszące dowolnemu wzrostowi stężenia bilirubiny całkowitej w surowicy (z objawami/objawami zapalenia wątroby lub innego uszkodzenia wątroby lub bez nich) |
|
| Objawy lub symptomy wskazujące na zapalenie wątroby lub inne uszkodzenie wątroby (np. żółtaczka, bilirubinuria, nasilone zmęczenie, nudności, wymioty, ból w prawym górnym kwadrancie brzucha, wodobrzusze, obrzęki o nieznanej etiologii, skłonność do powstawania siniaków) |
|
Pacjenci powinni oddawać krew w celu analizy poziomu IGF-1 oraz biochemicznych wskaźników funkcji wątroby przed i podczas leczenia lekiem Somavert; dawka leku może zostać zmieniona na podstawie wyników tych badań.
Reakcja krzyżowa w oznaczeniach ilościowych hormonu wzrostu
Somavert wykazuje istotne podobieństwo strukturalne do hormonu wzrostu, wskutek czego występuje jego reakcja krzyżowa przy stosowaniu zestawów do ilościowego oznaczania hormonu wzrostu. Ponieważ stężenie leku Somavert w surowicy krwi przy zastosowaniu dawek terapeutycznie skutecznych jest zazwyczaj 100–1000 razy wyższe niż rzeczywiste stężenie hormonu wzrostu w surowicy krwi u pacjentów z akromegalią, oznaczenie stężenia hormonu wzrostu w surowicy krwi może prowadzić do fałszywie podwyższonych wyników.
Wzrost guzów produkujących hormon wzrostu
Guzów przysadki produkujących hormon wzrostu czasem mogą się powiększać, powodując poważne powikłania (np. zaburzenia pola widzenia), dlatego ważne jest, aby wszyscy pacjenci byli dokładnie kontrolowani. W przypadku pojawienia się objawów wzrostu guza mogą być zalecane alternatywne procedury.
Lipohypertrofia
Zarejestrowano przypadki lipohypertrofii u pacjentów stosujących Somavert. W trakcie podwójnie ślepego, 12-tygodniowego, placebo-kontrolowanego badania zanotowano jeden przypadek (1,3%) lipohypertrofii w miejscu wstrzyknięcia u pacjenta otrzymującego dawki 10 mg/dobę. Zjawisko ustąpiło mimo kontynuacji terapii. W dwóch otwartych badaniach (łącznie z udziałem 147 pacjentów) u dwóch pacjentów (oba otrzymujące dawki 10 mg/dobę) rozwinęła się lipohypertrofia. W jednym przypadku zjawisko ustąpiło podczas terapii, w drugim doprowadziło do przerwania leczenia. Aby zapobiec lipohypertrofii, należy codziennie zmieniać miejsce wstrzyknięcia (czyli wstrzykiwać lek w inne miejsce niż poprzednio).
Naruszenia funkcji nerek
Leku Somavert nie badano pod kątem stosowania u pacjentów z zaburzeniami funkcji nerek, a bezpieczeństwo i skuteczność stosowania u tych pacjentów są nieznane.
Stosowanie u pacjentów w wieku podeszłym
Liczba uczestników w wieku 65 lat i starszych w badaniach klinicznych leku Somavert nie była wystarczająca, aby ocenić różnicę w odpowiedzi pacjentów z tej grupy wiekowej w porównaniu z młodszych pacjentami. Ogólnie dawkę dla pacjentów w wieku podeszłym należy dobierać ostrożnie, zazwyczaj rozpoczynając od dolnej granicy zakresu dawek, ze względu na częstsze występowanie u pacjentów tej grupy wiekowej obniżonej funkcji wątroby, nerek lub serca, a także częstsze występowanie chorób współistniejących lub stosowanie innych leków.
Zawartość sodu
Ten lek zawiera mniej niż 1 mmol sodu (23 mg) w jednej dawce. Pacjentom przestrzegającym diety ubogosolnej można przekazać, że ten lek może być uznany za niezawierający sodu.
Stosowanie w okresie ciąży lub karmienia piersią.
Ciąża
Przegląd informacji dotyczących ryzyka
Dane z badań po wprowadzeniu na rynek dotyczące stosowania leku Somavert u kobiet w ciąży są niewystarczające, aby ustalić związek z ryzykiem wystąpienia poważnych wad wrodzonych, poronienia lub niekorzystnych skutków dla matki i płodu związanymi ze stosowaniem leku. Kliniczny przebieg akromegalii może się poprawić w czasie ciąży (patrz sekcja „Przegląd danych klinicznych”). W badaniach rozrodu na zwierzętach fetotoksyczność obserwowano przy dawce 6-krotnie przekraczającej maksymalną zalecaną dawkę dla człowieka, obliczoną na podstawie powierzchni ciała, po podskórnej aplikacji pegwisomantu podczas organogenezy lub w okresie przed implantacją (patrz sekcja „Dane”). Szacowane tło ryzyka wystąpienia poważnych wad wrodzonych i poronienia dla populacji docelowej jest nieznane. W ogólnej populacji USA szacowane tło ryzyka wystąpienia poważnych wad wrodzonych i poronienia w czasie klinicznie potwierdzonych ciąż wynosi odpowiednio 2–4% i 15–20%.
Przegląd danych klinicznych
Związane z chorobą ryzyko matki i/lub embrionalne i płodowe
Opublikowane dane z przypadków klinicznych, serii przypadków i małego badania interwencyjnego z udziałem ciężarnych kobiet z akromegalią wskazują, że kliniczny przebieg akromegalii może się poprawić lub ustabilizować bez leczenia w czasie ciąży, szczególnie jeśli akromegalię leczono przed ciążą. W rzadkich przypadkach akromegalia może się pogorszyć w czasie ciąży. Ponieważ poziomy hormonów mogą się fizjologicznie zmieniać w czasie ciąży, a interpretacja poziomu hormonu wzrostu u ciężarnych kobiet z akromegalią może być niepewna, zaleca się monitorowanie kliniczne. Stosowanie leku u ciężarnych kobiet nie było badane, dlatego pacjentki powinny natychmiast poinformować swojego lekarza, jeśli dowiedzą się o ciąży.
Dane
Dane z badań na zwierzętach
Wpływ pegwisomantu na wczesny rozwój embrionalny i rozwój embrionalny i płodowy oceniano w dwóch oddzielnych badaniach na ciężarnych królikach, którym podawano pegwisomant podskórnie w dawkach 1, 3 i 10 mg/kg/doba. Nie stwierdzono żadnych dowodów wpływu teratogennego związanego ze stosowaniem pegwisomantu w okresie organogenezy. Przy dawce 10 mg/kg/doba (6-krotnie przekraczającej maksymalną dawkę terapeutyczną dla człowieka, obliczoną na podstawie powierzchni ciała) w obu badaniach obserwowano powtarzalne, niewielkie zwiększenie liczby poronień po implantacji.
Karmienie piersią
Przegląd informacji dotyczących ryzyka
Zgodnie z ograniczoną informacją z przypadków klinicznych, w publikacjach podano, że poziom pegwisomantu w mleku kobiecym był niższy niż granica wykrywalności. Nie ma dostępnych informacji o wpływie leku na niemowlęta karmione piersią ani o wpływie leku na produkcję mleka. Należy wziąć pod uwagę korzyści karmienia piersią dla rozwoju i zdrowia dziecka, kliniczną potrzebę matki w leku Somavert oraz wszelki potencjalny niekorzystny wpływ leku Somavert na karmiące piersią dzieci lub na podstawową chorobę matki.
Pacjentka powinna poinformować swojego lekarza, jeśli planuje karmienie piersią.
Kobiety i mężczyźni w wieku rozrodczym
Należy omówić możliwość nieplanowanej ciąży z pacjentkami w okresie przedmenopauzalnym, ponieważ korzyści terapeutyczne wynikające ze zmniejszenia poziomu hormonu wzrostu (GH) i normalizacji stężenia insulinopodobnego czynnika wzrostu 1 u kobiet z akromegalią otrzymujących pegwisomant mogą prowadzić do poprawy płodności.
Wpływ na zdolność do prowadzenia pojazdów i obsługiwania maszyn.
Nie przeprowadzono badań wpływu leku na zdolność prowadzenia pojazdów i obsługiwanie maszyn.
Sposób stosowania i dawki.
Informacja dotycząca dawkowania
Zalecana dawka ładująca leku Somavert wynosi 80 mg podana do podskórnej iniekcji pod nadzorem lekarza. Konieczne jest odpowiednie przeszkolenie w technice wykonywania iniekcji podskórnych. Od następnego dnia po podaniu dawki ładującej pacjent powinien otrzymywać codziennie podskórne iniekcje leku Somavert w dawce 10 mg.
Następnie dawkę należy modyfikować aż do osiągnięcia normy stężenia IGF-1 w surowicy (stężenie IGF-1 w surowicy należy mierzyć co 4–6 tygodni). Wybór dawki nie powinien opierać się na stężeniu hormonu wzrostu ani na objawach i objawach akromegalii. Nie wiadomo, czy pacjenci, u których mimo osiągnięcia normy stężenia IGF-1 nadal występują objawy, odnieśli korzyść z zwiększenia dawki leku Somavert.
-
Jeśli stężenie IGF-1 jest podwyższone, dawkę leku należy zwiększać o 5 mg co 4–6 tygodni.
-
Jeśli stężenie IGF-1 jest poniżej zakresu normy, dawkę leku należy zmniejszać o 5 mg co 4–6 tygodni.
-
Poziom IGF-1 należy również monitorować, gdy pacjent jest przechodzony z codziennego trybu podawania wymaganej dawki leku Somavert w postaci kilku iniekcji na codzienny tryb podawania wymaganej dawki w postaci jednej iniekcji (patrz sekcja „Właściwości farmakologiczne”).
Zalecany zakres dawkowania to od 10 do 30 mg podskórnie 1 raz na dobę, a maksymalna dawka dzienna wynosi 30 mg podskórnie 1 raz na dobę.
Ocena biochemicznych wskaźników funkcji wątroby przed rozpoczęciem terapii lekiem Somavert
Przed rozpoczęciem terapii lekiem Somavert należy ocenić wyjściowy poziom wskaźników funkcji wątroby pacjenta (surowiczy poziom alaninotransferazy (ALT), asparaginianotransferazy (AST), bilirubiny ogólnej surowicy (BOS) oraz fosfatazy alkalicznej). Zalecenia dotyczące rozpoczęcia terapii lekiem Somavert na podstawie wyjściowego poziomu wskaźników funkcji wątroby oraz zalecenia dotyczące monitorowania wskaźników funkcji wątroby podczas terapii lekiem Somavert przedstawiono w tabeli 3 (patrz sekcja „Szczególne ostrzeżenia i środki ostrożności”).
Procedura wykonania iniekcji z dawką ładującą
Poniższe instrukcje dotyczące przygotowania roztworu i przygotowania do podania dawki ładującej 80 mg przeznaczone są dla personelu medycznego. Personel medyczny powinien rozpuścić liofilizowany proszek w 4 fiolkach leku Somavert, z których każda zawiera 20 mg pegwisomantu, za pomocą rozpuszczalnika dostarczonego w zestawie (do przygotowania dawki ładującej 80 mg potrzebne są 4 fiolki liofilizowanego proszku oraz 4 strzykawki o objętości 2,25 ml zawierające rozpuszczalnik (woda do wstrzykiwań sterylna)). Personel medyczny powinien również wykonać 4 iniekcje odtworzonego roztworu leku Somavert w dowolne miejsce: ramię, górna część uda, brzuch lub pośladki pacjenta (każda iniekcja w inne miejsce).
- Przed podaniem dawki ładującej wyjmij z lodówki pierwszy zestaw (1 fiolka liofilizowanego proszku Somavert zawierająca 20 mg pegwisomantu oraz 1 strzykawka o objętości 2,25 ml zawierająca rozpuszczalnik) około 10 minut przed zaplanowanym czasem iniekcji.
- Rozpuść liofilizowany proszek leku Somavert zawierający 20 mg pegwisomantu w pierwszej fiolce za pomocą rozpuszczalnika. Podczas stosowania rozpuszczalnika w strzykawce o objętości 2,25 ml należy powoli wprowadzić zawartość strzykawki po ścianie fiolki z liofilizowanym proszkiem leku Somavert. Nie kieruj rozpuszczalnika bezpośrednio na proszek.
- Nie odwracaj fiolki ani nie wstrząsaj roztworem, ponieważ może to spowodować denaturację białka pegwisomantu. Powoli wymieszaj roztwór ruchami okrężnymi, aby cały liofilizowany proszek rozpuścił się w roztworze. Jeśli w odtworzonym roztworze leku Somavert występuje pienienie, roztwór może być uszkodzony i dlatego nie nadaje się do podania.
- Przed zastosowaniem wizualnie sprawdź odtworzony roztwór leku Somavert pod kątem obecności zanieczyszczeń i zmiany koloru. Odtworzony roztwór powinien być klarowny. Jeśli roztwór jest mętny, nie należy go stosować. Po odtworzeniu roztwór zawiera 20 mg pegwisomantu w 1 ml roztworu.
- Odbierz 1 ml odtworzonego roztworu leku Somavert. Roztworu należy użyć nie później niż w ciągu 6 godzin od momentu odtworzenia.
- Wykonaj pierwszą iniekcję podskórną odtworzonego roztworu leku Somavert (20 mg/ml) w ramię lub górną część uda, lub brzuch, lub pośladki pacjenta pod kątem 90 stopni.
- Powtórz kroki od (a) do (d), aby odtworzyć roztwór dla drugiej oraz kolejnych 3 i 4 dawki leku Somavert 20 mg.
- Następnie wykonaj drugą (a następnie 3 i 4) iniekcję podskórną odtworzonego roztworu leku Somavert (20 mg/ml) w ramię lub górną część uda, lub brzuch, lub pośladki pacjenta (wybierz inne miejsce niż dla poprzednich iniekcji) pod kątem 90 stopni.
- Powtórz kroki od (a) do (d), aby odtworzyć roztwór dla drugiej oraz kolejnych 3 i 4 dawki leku Somavert 20 mg.
- Wykonaj pierwszą iniekcję podskórną odtworzonego roztworu leku Somavert (20 mg/ml) w ramię lub górną część uda, lub brzuch, lub pośladki pacjenta pod kątem 90 stopni.
- Odbierz 1 ml odtworzonego roztworu leku Somavert. Roztworu należy użyć nie później niż w ciągu 6 godzin od momentu odtworzenia.
- Przed zastosowaniem wizualnie sprawdź odtworzony roztwór leku Somavert pod kątem obecności zanieczyszczeń i zmiany koloru. Odtworzony roztwór powinien być klarowny. Jeśli roztwór jest mętny, nie należy go stosować. Po odtworzeniu roztwór zawiera 20 mg pegwisomantu w 1 ml roztworu.
- Nie odwracaj fiolki ani nie wstrząsaj roztworem, ponieważ może to spowodować denaturację białka pegwisomantu. Powoli wymieszaj roztwór ruchami okrężnymi, aby cały liofilizowany proszek rozpuścił się w roztworze. Jeśli w odtworzonym roztworze leku Somavert występuje pienienie, roztwór może być uszkodzony i dlatego nie nadaje się do podania.
- Rozpuść liofilizowany proszek leku Somavert zawierający 20 mg pegwisomantu w pierwszej fiolce za pomocą rozpuszczalnika. Podczas stosowania rozpuszczalnika w strzykawce o objętości 2,25 ml należy powoli wprowadzić zawartość strzykawki po ścianie fiolki z liofilizowanym proszkiem leku Somavert. Nie kieruj rozpuszczalnika bezpośrednio na proszek.
Procedura wykonania iniekcji z dawką utrzymującą
Poniżej przedstawiono zalecenia dla pacjenta lub osoby opiekującej się pacjentem dotyczące przygotowania roztworu i podania codziennych dawek (od 10 do 30 mg) w „Krok po kroku instrukcji dotyczącej podania leku Somavert”.
- Przed podaniem dawki wyjmij z lodówki 1 fiolkę liofilizowanego proszku Somavert zawierającą 10 mg, 15 mg, 20 mg lub 30 mg pegwisomantu oraz jedną strzykawkę o objętości 2,25 ml zawierającą rozpuszczalnik około 10 minut przed zaplanowanym czasem iniekcji.
- Rozpuść liofilizowany proszek leku Somavert za pomocą rozpuszczalnika. Podczas stosowania rozpuszczalnika w strzykawce o objętości 2,25 ml należy powoli wprowadzić zawartość strzykawki po ścianie fiolki z liofilizowanym proszkiem leku Somavert. Nie kieruj rozpuszczalnika bezpośrednio na proszek.
- Nie odwracaj fiolki ani nie wstrząsaj roztworem, ponieważ może to spowodować denaturację białka pegwisomantu. Powoli wymieszaj roztwór ruchami okrężnymi, aby cały liofilizowany proszek rozpuścił się w roztworze. Jeśli w odtworzonym roztworze leku Somavert występuje pienienie, roztwór może być uszkodzony i dlatego nie nadaje się do podania.
- Przed zastosowaniem wizualnie sprawdź odtworzony roztwór leku Somavert pod kątem obecności zanieczyszczeń i zmiany koloru. Odtworzony roztwór powinien być klarowny. Jeśli roztwór jest mętny, nie należy go stosować. Po odtworzeniu roztwór zawiera 10 mg, 15 mg, 20 mg lub 30 mg pegwisomantu w 1 ml roztworu.
- Odbierz 1 ml odtworzonego roztworu leku Somavert. Roztworu należy użyć nie później niż w ciągu 6 godzin od momentu odtworzenia.
Wykonaj iniekcję podskórną odtworzonego roztworu leku Somavert w ramię lub górną część uda, lub brzuch, lub pośladki pod kątem 90 stopni.
Instrukcja krok po kroku dotycząca podania leku Somavert
Krok 1. Potrzebne materiały
Jeden opakowanie leku Somavert zawiera fiolkę z proszkiem leku Somavert, strzykawkę wstępnie wypełnioną rozpuszczalnikiem, bezpieczną igłę.
Będziesz również potrzebować: waty, chusteczki alkoholowej, pojemnika na przedmioty ostry.
| podparcie palca na strzykawce |
| zatka fiolki (ze zdjętą pokrywką fiolki) |
| nakrywka igły |
| Bezpieczna igła |
| Szpryc |
| osłona ochronna igły |
| trzpień tłoka strzykawki |
| nakrętka strzykawki |
| cylindra strzykawki |
| cylinderek strzykawki |
| data końcowa ważności |
| nakrętka fiolki |
| otwór w zatyczce |
| Fiolka |
Krok 2. Przygotowanie
Przed rozpoczęciem pracy:
- Mieszaj Somavert i płyn tylko wtedy, gdy jesteś gotowy podać dawkę.
- Wyjmij z lodówki jedną opakowanie Somavert i pozostaw lek, aby ogrzał się do temperatury pokojowej w bezpiecznym miejscu przez co najmniej 10 minut, zanim zaczniesz go stosować.
- Nie należy ogrzewać opakowania z lekiem Somavert za pomocą zewnętrznego źródła ciepła, takiego jak woda ciepła lub kuchenka mikrofalowa. Lek powinien ogrzać się samodzielnie.
- Umij ręce wodą z mydłem i dokładnie je osusz.
- Otwórz opakowanie strzykawki i bezpiecznej igły, aby mieć wygodny dostęp do każdego niezbędnego przedmiotu podczas przygotowania do iniekcji.
- Nie należy stosować strzykawek ani fiolków, jeśli:
- są uszkodzone lub wadliwe;
- upłynął ich termin ważności;
- zostały zamrożone, nawet jeśli obecnie już odmroziły (dotyczy tylko strzykawek).
Krok 3. Wybór miejsca iniekcji
| Wybór miejsca na wstrzyknięcie |
| Brzuch Oddalaj się o co najmniej 5 cm od pępka |
| Uda |
| Ramiona lub dolna część pleców Tylna część górnej części ramienia (tylko dla lekarza lub opiekuna) |
- Do każdej iniekcji wybieraj inne miejsce w obrębie obszarów przeznaczonych na zastrzyki.
- Unikaj obszarów w pobliżu kości lub miejsc z siniakami, zaczerwienieniami, uczuciem bólu lub zgrubieniami, a także obszarów ze strzępiami lub chorobami skóry.
- Przetrzyj miejsce iniekcji gazikiem alkoholowym.
- Pozwól, aby miejsce iniekcji wyschnęło.
Krok 4. Zdejmowanie nakładki z fiolki
| Ściąganie pokrywki z fiolki |
- Zdejmij pokrywkę z fiolki.
- Wyrzuć pokrywkę. Nie będzie Ci jej już potrzebna.
Uwaga! Nie dotykaj niczym do wkładu fiolki.
Krok 5. Zdjęcie nakrywki z strzykawki
| Zdjęcie czapki ze strzykawki |
| Kliknij |
- Zdejmij osłonkę ze strzykawki, pozostawiając nakrętkę strzykawki na swoim miejscu. Aby zdjąć osłonkę, może być wymagane więcej wysiłku, niż się wydaje na pierwszy rzut oka.
- Wyrzuć osłonkę strzykawki. Nie będzie ona już Ci potrzebna.
- Trzymaj strzykawkę pionowo, aby zapobiec wyciekaniu cieczy.
Uwaga! Nie dotykaj końcówki strzykawki żadnych przedmiotów po zdjęciu osłonki.
Krok 6. Przyłączenie bezpiecznej igły
| Dołączanie |
- Naciśnij i zakręć do oporu bezpieczną igłą na strzykawce.
Krok 7. Zdjęcie nakrywki z igły
| Ściąganie nakrywki igły |
- Usuń osłonę igły z kierunku nakrywki igły.
- Ostrożnie zdejmij nakrywkę z igły.
- Wyrzuć nakrywkę igły. Nie będzie ona już potrzebna.
Uwaga! Nie dotykaj niczego igłą.
Krok 8. Wprowadzenie igły
| Wprowadzenie igły |
- Nakłuj środkową część przeciwcienia fiolki igłą, jak pokazano na rysunku.
- Trzymaj strzykawkę nieruchomo, gdy igła znajduje się w przeciwcieniu fiolki, aby zapobiec jej zgięciu.
Krok 9. Dodanie cieczy
| Dodawanie cieczy |
- Nachyl butelkę i strzykawkę pod kątem, jak pokazano na rysunku.
- Powoli naciskaj na tłoczek strzykawki, aż cała ciecz przejdzie do butelki.
- Uwaga! Nie skierowuj strumienia cieczy bezpośrednio na proszek. Może to spowodować powstawanie piany, co uczyni lek nieprzydatnym do stosowania.
- Nie wyjmuj jeszcze igły.
Krok 10. Mieszanie roztworu ruchami kołowymi
| Mieszanie roztworu ruchami okrężnymi |
- Trzymaj strzykawkę i fiolkę w jednej ręce, jak pokazano na rysunku.
- Ostrożnie i powoli zmieszaj roztwór, poruszając fiolką w ruchu kołowym na płaskiej powierzchni.
- Kontynuuj mieszanie cieczy ruchami kołowymi, aż cały proszek całkowicie się rozpuści.
Uwaga. Może to potrwać do 5 minut. Nie wstrząsać.
Krok 11. Sprawdzenie roztworu
| Sprawdzanie roztworu |
- Trzymając igłę w fiolce, dokładnie obejrzyj roztwór. Powinien być klarowny i nie zawierać żadnych zanieczyszczeń.
- Nie należy stosować leku, jeśli:
- roztwór jest mętny lub nieprzezroczysty;
- roztwór ma zabarwienie w dowolnym kolorze;
- w fiolce znajdują się jakiekolwiek cząstki lub piana.
Krok 12. Przeniesienie igły
| Przesunięcie igły |
- Odwróć fiolkę tak, aby można było zobaczyć otwór w korku, jak pokazano na rysunku.
- Umieść igłę do góry nogami, tak aby koniec igły znajdował się w najniższym punkcie cieczy. Dzięki temu uda Ci się pobrać jak największą ilość cieczy z fiolki.
- Upewnij się, że trzpień strzykawki nie przesunął się. Jeśli trzpień strzykawki został przesunięty, wsuń go z powrotem do strzykawki. Pozwoli to usunąć całe powietrze ze strzykawki przed rozpoczęciem pobierania dawki.
Krok 13. Pobieranie dawki
| Dozowanie |
- Powoli wyciągnij tłok strzykawki, aby pobrać jak największą ilość roztworu z fiolki.
Uwaga. Jeśli widzisz pęcherzyki powietrza w strzykawce, lekko postukaj w cylinder strzykawki, aby pęcherzyki uniosły się do góry, a następnie ostrożnie usuń je z powrotem do fiolki.
- Wyjmij igłę z fiolki.
Krok 14. Wprowadzenie igły
| Wprowadzenie igły |
- Ostrożnie uchwycić fałd skóry w miejscu wstrzyknięcia.
- Wprowadzić igłę na pełną długość w fałd skóry.
Krok 15. Wykonanie wstrzyknięcia leku
| Wykonywanie wstrzyknięcia leku |
- Powoli przesuwaj tłok strzykawki w dół, aż do całkowitego opróżnienia cylinderka strzykawki.
Uwaga. Upewnij się, że wprowadziłeś igłę na pełną długość.
- Puść skладkę skóry i wyciągnij igłę pod kątem prostym.
Krok 16. Bezpieczeństwo obsługi igły
| Kliknij! |
| Bezpieczeństwo użytkowania igły |
- Opuść osłonę ochronną na igłę.
Uważnie naciśnij na osłonę, używając twardej powierzchni, aby przywrócić ją do pozycji wyjściowej.
- Uwaga. Usłyszysz kliknięcie, gdy osłona ochronna zaskoczy na miejsce.
Krok 17. Unieszkodliwienie
- Umieść użyte strzykawki w pojemniku na przedmioty ostry.
- Nie wyrzucaj (nie unieszkodliwiaj) strzykawek wraz z odpadami komunalnymi.
Krok 18. Po wstrzyknięciu
- W razie potrzeby weź czysty wacik i delikatnie przyciśnij go do miejsca wstrzyknięcia.
- Nie należy pocierać miejsca wstrzyknięcia.
Dzieci.
Bezpieczeństwo i skuteczność stosowania leku Somavert u dzieci nie zostały ustalone.
Przedawkowanie.
Podczas klinicznych badań po wprowadzeniu na rynek zgłoszono jeden przypadek ostrego przedawkowania leku Somavert: pacjent samodzielnie wykonywał wstrzyknięcia w dawce 80 mg/dobę (co było 2,7 razy wyższe niż maksymalna zalecana dawka utrzymująca) przez siedem dni. U pacjenta zaobserwowano niewielkie zwiększenie zmęczenia bez innych dolegliwości oraz istotnych odchyleń w wynikach badań laboratoryjnych.
W przypadku przedawkowania należy przerwać stosowanie leku Somavert i nie wznawiać go, dopóki poziom IGF-1 nie powróci do normy.
Niepożądane reakcje.
Niepożądane reakcje klinicznie istotne, wymienione w sekcji „Szczególne ostrzeżenia i środki ostrożności podczas stosowania”, to:
- hipoglikemia związana ze zmniejszeniem stężenia hormonu wzrostu u pacjentów z cukrzycą;
- hepatotoksyczność;
- reakcje krzyżowe w ilościowym oznaczaniu hormonu wzrostu;
- lipohipertrofia;
- układowe reakcje nadwrażliwości.
W trakcie badań klinicznych prowadzonych przed rejestracją, stężenie w surowicy ALAT i ASAT wzrosło ponad 10-krotnie w stosunku do GWN u dwóch pacjentów (0,8%), którzy otrzymywali Somavert. U jednego z pacjentów przeprowadzono ponowną próbę prowokacyjną z zastosowaniem Somavertu, po której zaobserwowano nawrót podwyższenia poziomu transaminaz, co wskazuje na prawdopodobny związek przyczynowo-skutkowy między podaniem leku a wzrostem enzymów wątrobowych. Wyniki biopsji wątroby przeprowadzonej u drugiego pacjenta były zgodne z przewlekłym zapaleniem wątroby o nieznanej etiologii. U obu pacjentów poziom transaminaz powrócił do normy po zaprzestaniu stosowania leku.
Wzrost stężenia ALAT i ASAT nie był towarzyszone zwiększeniem stężenia bilirubiny ogólnej ani fosfatazy alkalicznej, z wyjątkiem dwóch pacjentów, u których stwierdzono minimalny wzrost poziomu fosfatazy alkalicznej (tj. mniej niż 3 × GWN). Wzrost poziomu transaminaz najprawdopodobniej nie zależy od dawki podanego Somavertu i zwykle występuje w ciągu 4–12 tygodni od rozpoczęcia terapii. Nie wykryto żadnych czynników prognostycznych biochemicznych, fenotypowych ani genetycznych związanych z tym zjawiskiem.
Doświadczenie z badań klinicznych
Ponieważ badania kliniczne przeprowadza się w bardzo różnych warunkach, częstość występowania niepożądanych reakcji w badaniach klinicznych jednego leku nie może być bezpośrednio porównywana z częstością uzyskaną w badaniach klinicznych innego leku, ani nie można oczekiwać takiej samej częstości w praktyce klinicznej.
W 12-tygodniowym, randomizowanym, placebo-kontrolowanym, podwójnie ślepym badaniu dawek ustalonych Somavertu u pacjentów z akromegalią, 32 pacjentów otrzymywało placebo, a 80 pacjentów otrzymywało Somavert raz na dobę (patrz sekcja „Właściwości farmakologiczne” („Badania kliniczne”)). Ogółem 108 pacjentów (30 w grupie placebo, 78 w grupie Somavertu) ukończyło 12 tygodni leczenia.
Ogółem z powodu niepożądanych reakcji z badań klinicznych prowadzonych przed rejestracją wycofano ośmioro pacjentów z akromegalią (5,3%), w tym dwóch pacjentów z wyraźnym wzrostem poziomu transaminaz, jednego pacjenta z lipohipertrofią w miejscach wstrzyknięć oraz jednego pacjenta z istotnym przyrostem masy ciała. Większość niepożądanych reakcji wydaje się nie być zależna od dawki. W tabeli 5 przedstawiono częstość niepożądanych reakcji zgłoszonych u co najmniej dwóch pacjentów otrzymujących Somavert, których częstość była wyższa niż w grupie placebo, w 12-tygodniowym, placebo-kontrolowanym badaniu.
Tabela 5. Niepożądane reakcje obserwowane w 12-tygodniowym, placebo-kontrolowanym badaniu u pacjentów z akromegalią*
Poświadczenia niepożądane |
Placebo n = 32 |
Somavert |
||
| 10 mg na dobę n = 26 |
15 mg na dobę n = 26 |
20 mg na dobę N = 28 |
||
| Infekcja† |
2 (6 %) |
6 (23 %) |
0 |
0 |
| Ból |
2 (6 %) |
2 (8 %) |
1 (4 %) |
4 (14 %) |
| Nudności |
1 (3 %) |
0 |
2 (8 %) |
4 (14 %) |
| Diaree |
1 (3 %) |
1 (4 %) |
0 |
4 (14 %) |
| Odchylenie od normy biochemicznych wskaźników funkcji wątroby |
1 (3 %) |
3 (12 %) |
1 (4 %) |
1 (4 %) |
| Zespół grypopodobny |
0 |
1 (4 %) |
3 (12 %) |
2 (7 %) |
| Reakcja w miejscu iniekcji |
0 |
2 (8 %) |
1 (4 %) |
3 (11 %) |
| Zawroty głowy |
2 (6 %) |
2 (8 %) |
1 (4 %) |
1 (4 %) |
| Przypadkowa uraz |
1 (3 %) |
2 (8 %) |
1 (4 %) |
0 |
| Ból pleców |
1 (3 %) |
2 (8 %) |
0 |
1 (4 %) |
| Zatokowe zapalenie |
1 (3 %) |
2 (8 %) |
0 |
1 (4 %) |
| Ból w klatce piersiowej |
0 |
1 (4 %) |
2 (8 %) |
0 |
| Obwódowe obrzęki |
0 |
2 (8 %) |
0 |
1 (4 %) |
| Choroba nadciśnieniowa |
0 |
0 |
2 (8 %) |
0 |
| Parastezja |
2 (6 %) |
0 |
0 |
2 (7 %) |
* Tabela zawiera tylko reakcje, które zostały zarejestrowane u co najmniej 2 pacjentów i które występowały częściej u pacjentów otrzymujących Somavert niż u pacjentów z grupy placebo.
† W grupie otrzymującej Somavert 10 mg zgłoszono 6 reakcji zakodowanych terminem „infekcja”: objawy przeziębienia (3), infekcja dróg oddechowych górnych (1), pęcherze (1) oraz infekcja ucha (1). W grupie otrzymującej placebo zarejestrowano 2 takie reakcje: objawy przeziębienia (1) oraz infekcja klatki piersiowej (1).
Doświadczenie po rejestracji leku
Reakcje niepożądane zgłoszone spontanicznie w okresie po rejestracji
W okresie po rejestracji leku Somavert zaobserwowano reakcje niepożądane wymienione poniżej. Ponieważ zgłoszenia te pochodzą z dobrowolnych doniesień w populacji o nieznanej liczebności, nie zawsze możliwe jest wiarygodne oszacowanie ich częstości ani ustalenie związku przyczynowo-skutkowego z zastosowaniem leku.
W okresie po rejestracji zgłaszano o systemowe reakcje nadwrażliwości, w tym reakcje anafilaktyczne, laryngospazm, obrzęk naczynioruchowy, ogólnoustrojowe reakcje skórne (osutka, zaczerwienienie, świąd, pokrzywka). Niektórzy pacjenci wymagali hospitalizacji. Po ponownym włączeniu terapii u żadnego z pacjentów objawy się nie powtarzały (patrz sekcja „Szczególne ostrzeżenia i środki ostrożności stosowania” („Reakcje nadwrażliwości systemowe”)).
Reakcje niepożądane zgłoszone w badaniu obserwacyjnym
ACROSTUDY – międzynarodowy rejestr obserwacyjny, w którym zebrano dane dotyczące długoterminowej bezpieczeństwa u 2221 pacjentów z akromegalią leczonych Somavertem przy średnim czasie leczenia wynoszącym 8,5 roku. Pacjenci mogli również otrzymywać inne leczenie akromegalii w trakcie okresu rejestracji. Dawkę i schemat terapii dobierał na własną odpowiedzialność każdy lekarz przepisujący leczenie. Chociaż monitorowanie bezpieczeństwa zgodnie z zalecanym schematem było obowiązkowe, nie wszystkie oceny zostały wykonane we wszystkich punktach czasowych u każdego pacjenta. Z tego powodu porównanie częstości występowania reakcji niepożądanych z wartościami częstości obserwowanymi w pierwotnym badaniu klinicznym nie jest możliwe.
Spośród 1327 pacjentów, którzy na początku badania mieli normalne stężenie AST i ALT, u 20 (1,5%) pacjentów zaobserwowano wzrost tych parametrów powyżej 3–5-krotnej wartości górnej granicy normy, a u 22 (1,7%) pacjentów wzrost przekraczał 5-krotność wartości górnej granicy normy. Lipohipertrofię zarejestrowano u 35 (1,6%) pacjentów.
Spośród 1795 uczestników badania, u których dostępne były wyniki rezonansu magnetycznego (MRI) wykonanego na początku oraz co najmniej raz w trakcie dalszej obserwacji, wyniki MRI wykazały u 128 (7,1%) – zwiększenie, u 310 (17,3%) – zmniejszenie, u 81 (4,5%) – zarówno zwiększenie, jak i zmniejszenie, oraz u 1276 (71,1%) – brak zmian.
Należy poinformować pacjentów (i/lub osoby opiekujące się nimi) o możliwym wystąpieniu następujących reakcji niepożądanych.
- Najczęściej zgłaszano reakcje niepożądane w postaci reakcji w miejscu wstrzyknięcia, podwyższenia stężenia biochemicznych markerów funkcji wątroby, bólu, nudności i biegunki.
- Jeżeli u pacjentów zostanie stwierdzone podwyższenie stężenia biochemicznych markerów funkcji wątroby, może być konieczne częstsze oddawanie krwi w celu oceny markerów funkcji wątroby i/lub przerwania leczenia lekiem Somavert. Należy poinformować pacjentów, że w przypadku wystąpienia żółtaczki muszą natychmiast przerwać terapię i skontaktować się z lekarzem.
- U pacjentów z akromegalią guzy wydzielające hormon wzrostu mogą się zwiększać, dlatego należy je starannie monitorować za pomocą rezonansu magnetycznego (MRI).
- W miejscu podskórnej iniekcji może pojawić się zgrubienie, które może prowadzić do powstania guła; wykonywanie iniekcji w nowe miejsce za każdym razem może pomóc w zapobieganiu temu zjawisku lub jego minimalizacji.
- U pacjentów z cukrzycą może być konieczna dokładna kontrola stanu oraz zmniejszenie dawki insuliny i/lub doustnych leków hipoglikemizujących w trakcie leczenia lekiem Somavert.
- Jeżeli pacjenci otrzymują opioidy, mogą wymagać wyższych dawek leku Somavert w celu osiągnięcia odpowiedniego hamowania IGF-1.
Zgłaszanie podejrzewanych reakcji niepożądanych
Zgłaszanie reakcji niepożądanych po rejestracji leku ma istotne znaczenie. Umożliwia to monitorowanie stosunku korzyści do ryzyka związanego z zastosowaniem tego leku. Osoby medyczne i farmaceutyczne, a także pacjenci lub ich prawni opiekunowie, powinni zgłaszać wszystkie przypadki podejrzewanych reakcji niepożądanych oraz brak skuteczności leku poprzez zautomatyzowany system informacyjny nadzoru farmakologicznego pod adresem: https://aisf.dec.gov.ua.
Okres ważności.
3 lata.
Warunki przechowywania.
Fiolki z liofilizatem należy przechowywać w oryginalnym opakowaniu w celu ochrony przed światłem, w temperaturze od 2 do 8 °C w lodówce. Nie mrozić.
Lek w fiolkach w oryginalnym opakowaniu można przechowywać w temperaturze nie wyższej niż 25 °C jednorazowo przez okres 30 dni. Na tekturowej puszce należy zaznaczyć datę, do której lek należy zużyć (do 30 dni od daty wyjęcia z lodówki). Fiolki należy chronić przed światłem i nie wolno ich ponownie przechowywać w lodówce. Lek należy zutylizować, jeśli nie został wykorzystany w ciągu 30 dni przechowywania w temperaturze pokojowej lub upłynął okres ważności wskazany na opakowaniu, w zależności od tego, co nastąpi wcześniej.
Rozpuszczalnik w strzykawkach wstępnie napełnionych należy przechowywać w temperaturze nie wyższej niż 30 °C lub w temperaturze od 2 do 8 °C w lodówce. Nie mrozić.
Odtworzony roztwór należy użyć natychmiast lub w ciągu 6 godzin od przygotowania.
Przechowywać w miejscu niedostępnym dla dzieci.
Opakowanie.
10 fiolki z liofilizatem w tekturowej puszce środkowej; 3 puszki środkowe, 30 strzykawek wstępnie napełnionych rozpuszczalnikiem po 1 ml, 30 bezpiecznych igieł w tekturowej puszce.
Kategoria wydawania.
Na receptę.
Producent.
Pfizer Manufacturing Belgium NV / Pfizer Manufacturing Belgium NV.
Adres producenta i miejsce prowadzenia działalności.
Rijksweg 12, Puurs-Sint-Amands, 2870, Belgia / Rijksweg 12, Puurs-Sint-Amands, 2870, Belgium.