Signifor Lar
UkrainaSpis treści
INSTRUKCJA DOTYCZĄCA STOSOWANIA LEKU SIGNIFOR® LAR®
Skład:
substancja czynna: pasireotyd;
1 buteleczka z proszkiem do zawiesiny do wstrzykiwań zawiera 20 mg, 40 mg lub 60 mg pasireotydu (w postaci pasireotydu pamoatu).
substancje pomocnicze:
proszek: poli(D,L-laktyd-ko-glikolid) (50-60:40-50), poli(D,L-laktyd-ko-glikolid) (50:50);
rozpuszczalnik: 1 wstępnie wypełniony strzykawka z rozpuszczalnikiem o objętości 2 ml zawiera: karboksymetylocelulozę sodową, manitol (E421), poloksymer 188, wodę do wstrzykiwań.
Postać farmaceutyczna. Proszek do zawiesiny do wstrzykiwań.
Główne właściwości fizykochemiczne:
proszek: proszek od lekko żółtawego do żółtawego;
rozpuszczalnik: klarowny roztwór od bezbarwnego do żółtawego lub jasnobrunatnego.
Grupa farmakoterapeutyczna. Hormony przysadki, podwzgórza i ich analogi. Somatostatyna i jej analogi. Kod ATC H01C B05.
Właściwości farmakologiczne.
Farmakodynamika.
Mechanizm działania. Pasireotyd – cykloheksapeptyd, lek do wstrzykiwań analog somatostatyny. Podobnie jak naturalne peptydowe hormony somatostatyna-14 i somatostatyna-28 (znane również jako czynnik hamujący wydzielanie hormonu wzrostu [SRIF]) oraz inne analogi somatostatyny, pasireotyd wykazuje swoje działanie farmakologiczne poprzez wiązanie się z receptorami somatostatyny. Znanych jest pięć podtypów ludzkich receptorów somatostatyny: hsst1, 2, 3, 4 i 5. W normalnych warunkach fizjologicznych różne podtypy receptorów są eksprymowane w różnych tkankach. Analogi somatostatyny wiążą się z różną siłą z receptorami hsst (patrz tabela poniżej). Pasireotyd wiąże się z wysokim powinowactwem z czterema z pięciu receptorów hsst.
Powinowactwo wiązania somatostatyny (SRIF-14), pasireotydu, oktreotydu i lanreotydu do pięciu ludzkich podtypów receptorów sst (hsst1–5).
| Lek |
hsst1 |
hsst2 |
hsst3 |
hsst4 |
hsst5 |
| Somatostatyna (SRIF-14) |
0,93 ± 0,12 |
0,15 ± 0,02 |
0,56 ± 0,17 |
1,5 ± 0,4 |
0,29 ± 0,04 |
| Pasireotyd |
9,3 ± 0,1 |
1,0 ± 0,1 |
1,5 ± 0,3 |
> 100 |
0,16 ± 0,01 |
| Oktreotyd |
280 ± 80 |
0,38 ± 0,08 |
7,1 ± 1,4 |
>1000 |
6,3 ± 1,0 |
| Lantreotyd |
180 ± 20 |
0,54 ± 0,08 |
14 ± 9 |
230 ± 40 |
17 ± 5 |
Wyniki przedstawiono jako średnią wartość ± błąd standardowy średniej (SEM) wartości IC50 wyrażonej w nmol/l.
Efekty farmakodynamiczne. Receptory somatostatyny są obecne w wielu tkankach, szczególnie w nowotworach neuroendokrynnych, u których występuje nadmierna sekrecja hormonów, w tym hormonu wzrostu (GH) w akromegalii oraz hormonu adrenokortykotropowego (ACTH) w chorobie Cushinga.
Badania in vitro wykazały, że komórki guzów kortykotropowych pobrane od pacjentów z chorobą Cushinga wykazują silną ekspresję receptora hsst5, podczas gdy receptory innych podtypów są nieobecne lub wykazują niewielką ekspresję. Pasireotyd wiąże się i aktywuje cztery z pięciu receptora hsst, szczególnie hsst5, w adenomach kortykotropowych produkujących ACTH, co prowadzi do hamowania sekrecji ACTH.
Dzięki swojemu szerokiemu profilowi wiązania z receptorami somatostatyny pasireotyd może stymulować podtypy receptora hsst2 i hsst5, które są niezbędne do hamowania sekrecji GH oraz insulinopodobnego czynnika wzrostu (IGF-1), dlatego może być skuteczny w leczeniu akromegalii.
Metabolizm glukozy. W przebiegu randomizowanego, podwójnie ślepego badania z udziałem zdrowych ochotników stwierdzono, że rozwój hiperglikemii po podskórnej aplikacji pasireotydu w dawkach 0,6 mg i 0,9 mg dwa razy dziennie był związany ze znaczącym zmniejszeniem sekrecji insuliny oraz incretinów (a mianowicie glukagonopodobnego peptydu-1 (GLP-1) i glukozozależnego insulino-tropowego polipeptydu (GIP)). Pasireotyd nie wpływał na wrażliwość na insulinę.
Populacja pediatryczna. Europejska Agencja Leków zwolniła z obowiązku przedstawienia wyników badań dotyczących stosowania leku Signifor Lar u dzieci we wszystkich podkategoriiach w przypadku akromegalii i gigantyzmu przysadkowego, a także przysadkowego zespołu Cushinga, nadprodukcji ACTH pochodzenia przysadkowego i przysadkowego hiperkortykoidyzmu (informacje dotyczące stosowania u dzieci znajdują się w sekcji „Sposób stosowania i dawki”).
Farmakokinetyka.
Pasireotyd do wstrzykiwania w formie wstrzykiwalnych mikrosfer o przedłużonym działaniu. Po pojedynczej dawce wstrzykniętej do mięśnia stwierdzono początkowe „eksplozyjne” uwolnienie leku w pierwszym dniu wstrzyknięcia, po którym nastąpiło zmniejszenie stężenia w dniach 2–7. Następnie obserwowano stopniowe osiągnięcie maksymalnego stężenia w przybliżeniu w 21. dniu i fazę stopniowego zmniejszania się stężenia w kolejnych tygodniach, wraz z końcową fazą rozpadu polimerowej matrycy postaci leku.
Wchłanianie. Względna biodostępność pasireotydu po wstrzyknięciu do mięśnia w porównaniu z podskórnym wstrzyknięciem jest pełna. Nie przeprowadzono badań mających na celu ocenę absolutnej biodostępności pasireotydu u ludzi.
Rozkład. U zdrowych ochotników pasireotyd po wstrzyknięciu do mięśnia szeroko się rozkładał, charakteryzując się dużym objętością rozkładu (Vz/F > 100 litrów). Rozkład między komórkami krwi a osoczem nie zależy od stężenia i wskazuje, że pasireotyd znajduje się głównie w osoczu krwi (91%). Wiązanie z białkami osocza jest umiarkowane (88%) i nie zależy od stężenia.
Na podstawie danych badań in vitro pasireotyd jest substratem białka transportowego eflluksowego P-glikoproteiny. Dane badań in vitro wskazują, że pasireotyd nie jest substratem białka transportowego eflluksowego BCRP (białko oporności nowotworu piersi) ani transporterów influxowych OCT1 (transporter organicznych kationów 1), OATP (polipeptyd transportujący organiczne aniony) 1B1, 1B3 ani 2B1. W dawkach terapeutycznych pasireotyd nie jest również inhibitorem UGT1A1, OATP1B1 ani 1B3, OAT1 ani OAT3, OCT1 ani OCT2, P-gp, BCRp, MRP2 ani BSEP.
Biotransformacja. Pasireotyd jest metabolicznie bardzo stabilny. Dane badań in vitro wskazują, że pasireotyd nie jest substratem, inhibitorem ani induktorem żadnych głównych enzymów CYP450. U zdrowych ochotników pasireotyd występuje głównie w niezmienionej formie w osoczu, moczu i kale.
Eliminacja. Pasireotyd jest usuwany głównie drogą wątrobową (wydzielanie żółciowe) przy niewielkim udziale nerek. W badaniu ADME (wchłanianie, rozkład, metabolizm, wydalanie) u ludzi 55,9 ± 6,63% dawki promieniotwórczej odzyskano w ciągu pierwszych 10 dni po podaniu, w tym 48,3 ± 8,16% radioaktywności w kale i 7,63 ± 2,03% w moczu.
Klirens (CL/F) pasireotydu wstrzykiwanego do mięśnia u zdrowych ochotników wynosi średnio 4,5–8,5 l/h. Na podstawie wyników analizy populacyjnej farmakokinetyki zmierzony CL/F wynosił około 4,8–6,5 l/h u pacjentów z typową chorobą Cushinga oraz około 5,6–8,2 l/h u pacjentów z typową akromegalią.
Linowość i zależność od czasu. Stan stacjonarny farmakokinetyczny po wstrzyknięciu do mięśnia osiągany jest po 3 miesiącach. Po kilku dawkach miesięcznych w zakresie od 10 mg do 60 mg co 4 tygodnie pasireotyd wstrzykiwany do mięśnia wykazuje u pacjentów przybliżoną proporcjonalność dawkowo-kinetyczną.
Osobliwe grupy pacjentów.
Populacja pediatryczna. Badania u dzieci nie były prowadzone.
Pacjenci z zaburzeniem funkcji nerek. Klirens nerkowy odgrywa minimalną rolę w eliminacji pasireotydu u ludzi. W badaniu klinicznym pojedyncza dawka podskórna pasireotydu 900 μg u pacjentów z dysfunkcją nerek, z łagodnym, umiarkowanym i ciężkim zaburzeniem czynności nerek lub z chorobą nerek w stadium końcowym (ESRD) nie wywierała istotnego wpływu na całkowite narażenie osoczowe na pasireotyd. Narażenie osoczowe na niezwiązany pasireotyd (AUCinf,u) wzrastało u pacjentów z zaburzeniem czynności nerek (łagodne zaburzenie – 33%; umiarkowane – 25%, ciężkie – 99%, ESRD – 143%) w porównaniu z pacjentami z grupy kontrolnej.
Pacjenci z zaburzeniem funkcji wątroby. Badania kliniczne z zastosowaniem pasireotydu wstrzykiwanego do mięśnia u pacjentów z zaburzoną czynnością wątroby nie były prowadzone. W badaniu klinicznym z podaniem pojedynczej dawki podskórnej pasireotydu pacjentom z zaburzoną czynnością wątroby stwierdzono istotne statystycznie różnice u pacjentów z umiarkowanym i ciężkim zaburzeniem czynności wątroby (klasy B i C wg skali Childa-Pugha). U pacjentów z umiarkowanym i ciężkim zaburzeniem czynności wątroby AUCinf wzrastało odpowiednio o 60% i 79%, Cmax – o 67% i 69%, a CL/F zmniejszał się o 37% i 44%.
Pacjenci starsi (≥ 65 lat). Wiek nie jest istotnym parametrem w analizie populacyjnej farmakokinetyki u pacjentów.
Cechy demograficzne. Analiza populacyjnej farmakokinetyki pasireotydu wstrzykiwanego do mięśnia wskazuje, że przynależność rasowa i płeć pacjenta nie wpływają na parametry farmakokinetyczne leku. Masa ciała miała niewielki wpływ na farmakokinetykę w badaniu z udziałem pacjentów wcześniej nieleczonych, ale nie miała wpływu w badaniu z udziałem pacjentów z niewystarczającym kontrolowaniem choroby. Kobiety z akromegalią wykazywały większe narażenie (o 32% i 51%) niż mężczyźni odpowiednio w badaniach pacjentów nieleczonych i pacjentów z niewystarczającym kontrolowaniem choroby; różnice te nie były klinicznie istotne na podstawie danych dotyczących skuteczności i bezpieczeństwa.
Charakterystyki kliniczne.
Wskazania.
Leczenie dorosłych pacjentów z akromegalią, u których zabieg chirurgiczny nie jest optymalny lub okazał się nieskuteczny oraz u których nie osiągnięto odpowiedniej kontroli za pomocą innego analogu somatostatyny.
Leczenie dorosłych pacjentów z chorobą Cushinga, u których zabieg chirurgiczny nie jest optymalny lub okazał się nieskuteczny.
Dawka 60 mg stosowana jest wyłącznie w leczeniu akromegalii.
Przeciwwskazania.
Podwyższona wrażliwość na substancję czynną lub na którąkolwiek ze substancji pomocniczych leku. Ciężkie zaburzenia funkcji wątroby (klasa C wg skali Childa-Pugha).
Interakcje z innymi lekami oraz inne rodzaje interakcji.
Oczekiwane interakcje farmakokinetyczne wpływające na pasireotyd. Wpływ inhibitora białka P-glikoproteiny – werapamilu – na farmakokinetykę pasireotydu po podaniu podskórnie badano w badaniu interakcji leków z udziałem zdrowych ochotników. Nie zaobserwowano żadnych zmian farmakokinetyki (szybkości lub stopnia działania) pasireotydu.
Oczekiwane interakcje farmakokinetyczne wpływające na inne leki. Pasireotyd może obniżać względną biodostępność cyklosporyny. W przypadku jednoczesnego stosowania pasireotydu i cyklosporyny może być konieczna korekta dawki cyklosporyny w celu utrzymania stężenia terapeutycznego.
Oczekiwane interakcje farmakodynamiczne. Leki wydłużające interwał QT.
Pasireotyd należy stosować z ostrożnością u pacjentów przyjmujących jednocześnie leki wydłużające interwał QT, takie jak leki przeciwnadżerkowe klasy Ia (np. chinidyna, prokainamid, disopyramid), leki przeciwnadżerkowe klasy III (np. amiodaron, dronedaron, sotalol, dofetylid, ibutilid), niektóre leki przeciwbakteryjne (erytromycyna dożylne, wstrzyknięcia pentamidyny, klaritromycyna, moxifloksacyna), niektóre leki przeciwpsychotyczne (np. chloropromazyna, tiorydazyna, fluorfenazyna, pimocydyna, haloperidol, tiapryd, amisulpryd, sertindol, metadon), niektóre leki przeciwhistaminowe (np. terfenadyna, astemizol, mizolastyna), leki przeciwmalaryczne (np. chlorochina, halofantryna, lumefantryna), niektóre leki przeciwgrzybicze (ketoconazol, z wyjątkiem szamponu).
Leki powodujące bradykardię. Zaleca się kliniczne monitorowanie rytmu serca, szczególnie na początku leczenia, u pacjentów przyjmujących pasireotyd jednocześnie z lekami powodującymi bradykardię, takimi jak beta-blokery (np. metoprolol, carteolol, propranolol, sotalol), inhibitory acetylocholinesterazy (np. rywastygmina, fizioostygmina), niektóre blokery kanałów wapniowych (np. werapamil, dyltiazem, beprydyl), niektóre leki przeciwnadżerkowe.
Insulina oraz leki przeciwcukrzycowe. Może być konieczna korekta dawki (obniżenie lub zwiększenie) insuliny oraz leków przeciwcukrzycowych (np. metforminy, liraglutydu, wildagliptyny, nateglinidu) w przypadku jednoczesnego stosowania z pasireotydem.
Szczególne wskazania dotyczące stosowania.
Zaburzenia gospodarki glukozą. Zmiany poziomu glukozy we krwi były często obserwowane u zdrowych ochotników i pacjentów po leczeniu pasireotydem. Hiperglikemia, a rzadziej hipoglikemia, występowały u pacjentów uczestniczących w badaniach klinicznych z zastosowaniem pasireotydu (patrz sekcja „Efekty niepożądane”).
Pacjenci, u których rozwijała się hiperglikemia, ogólnie odpowiadali na leczenie przeciwcukrzycowe. Zmniejszenie dawki lub przerwanie leczenia pasireotydem z powodu hiperglikemii było rzadkie w trakcie badań klinicznych z zastosowaniem pasireotydu.
Rozwój hiperglikemii jest związany ze zmniejszeniem sekrecji insuliny oraz hormonów inkretynowych (a mianowicie peptydu podobnego do glukagonu-1 (GLP-1) oraz glukozozależnego insulino-tropowego polipeptydu (GIP)).
Stan glikemii (poziom glukozy we krwi na czczo / hemoglobina A1c (PGN/HA1c)) należy ocenić przed rozpoczęciem leczenia pasireotydem. Monitorowanie PGN/A1c w trakcie leczenia należy prowadzić zgodnie z obowiązującymi zaleceniami. Samokontrola poziomu glukozy we krwi oraz/lub ocena PGN powinny być wykonywane co tydzień przez pierwsze 3 miesiące, a następnie okresowo zgodnie z praktyką kliniczną, a także w ciągu pierwszych 4–6 tygodni po każdej zmianie dawki. Ponadto, należy wykonać monitorowanie PGN po 4 tygodniach oraz hemoglobiny A1c po 3 miesiącach od zakończenia leczenia.
Jeśli u pacjenta podczas stosowania leku Signifor Lar rozwija się hiperglikemia, zaleca się rozpoczęcie lub dostosowanie leczenia przeciwcukrzycowego zgodnie z obowiązującymi zaleceniami w celu kontrolowania hiperglikemii. Jeśli hiperglikemia nie ulega kontrolowaniu mimo odpowiedniego leczenia farmakologicznego, należy rozważyć zmniejszenie dawki Signifor Lar lub przerwanie leczenia (patrz sekcja „Interakcje z innymi lekami i inne rodzaje interakcji”).
Zarejestrowano przypadki ketoacydozy w okresie postmarketingowym podczas stosowania leku Signifor Lar u pacjentów z wywiadem cukrzycy i bez niej. Pacjentów, u których pojawiają się objawy i znaki odpowiadające ciężkiemu kwasici metabolicznej, należy przebadać pod kątem rozwoju ketoacydozy niezależnie od obecności cukrzycy w wywiadzie.
U pacjentów z niewłaściwą kontrolą glikemii (określoną jako wartość HbA1c > 8% mimo terapii przeciwcukrzycowej) należy wzmocnić kontrolę cukrzycy i monitorowanie przed rozpoczęciem oraz w trakcie terapii pasireotydem.
Próby funkcji wątroby. U pacjentów przyjmujących pasireotyd obserwowano zazwyczaj niewielkie, przejściowe podwyższenie poziomu aminotransferaz. Zarejestrowano również rzadkie przypadki jednoczesnego wzrostu ALT (alaninotransaminazy) powyżej 3 × ULN (górna granica normy) i bilirubiny powyżej 2 × ULN (patrz sekcja „Efekty niepożądane”).
Monitorowanie funkcji wątroby należy dalej prowadzić zgodnie z wskazaniami klinicznymi. Zaleca się kontrolę funkcji wątroby przed rozpoczęciem wstrzykiwania wewnątrzmięśniowego pasireotydu oraz po 2–3 tygodniach terapii, a następnie co miesiąc przez 3 miesiące po rozpoczęciu leczenia.
Pacjenci, u których stwierdzono podwyższenie poziomu transaminaz, wymagają częstszego monitorowania funkcji wątroby aż do powrotu wartości do poziomów sprzed leczenia. Leczenie pasireotydem należy przerwać, jeśli u pacjenta rozwinie się żółtaczka lub inne objawy klinicznie istotnej dysfunkcji wątroby, przy trwałym wzroście AST (aspartaminotransferazy) lub ALT powyżej 5 × ULN lub przy wzroście ALT lub AST powyżej 3 × ULN jednocześnie ze wzrostem bilirubiny powyżej 2 × ULN. Po przerwaniu leczenia pasireotydem należy kontynuować kontrolę pacjentów aż do ustąpienia objawów. Nie należy wznowić leczenia, jeśli podejrzewa się, że zaburzenia funkcji wątroby są związane z pasireotydem.
Zjawiska związane z układem sercowo-naczyniowym. W trakcie stosowania pasireotydu zgłaszano przypadki bradykardii. Pacjentom z chorobami serca i/lub czynnikami ryzyka bradykardii, takimi jak klinicznie istotna bradykardia lub niedawny zawał mięśnia sercowego w wywiadzie, bloki serca wysokiego stopnia, przewlekła niewydolność serca (klasa III lub IV według klasyfikacji NYHA), niestabilna dławica piersiowa, trwała tachykardia komorowa, migotanie komór, zaleca się staranne monitorowanie. Może istnieć potrzeba dostosowania dawek leków, np. beta-blokerów, blokerów kanałów wapniowych lub leków stosowanych do kontroli równowagi elektrolitowej.
W dwóch badaniach z udziałem zdrowych ochotników wykazano, że pasireotyd wydłużał odcinek QT na EKG. Kliniczne znaczenie tego wydłużenia jest nieznane. W badaniach klinicznych fazy III u pacjentów z akromegalią nie stwierdzono żadnych klinicznie istotnych różnic w wydłużeniu odcinka QT między wstrzykiwaniem wewnątrzmięśniowym pasireotydu a analogami somatostatyny stosowanymi jako aktywny lek porównawczy. Wszystkie zjawiska związane z wydłużeniem odcinka QT były tymczasowe i ustępowały bez interwencji terapeutycznej.
Epizody tachykardii typu „pistolet” nie były obserwowane w żadnym z badań klinicznych z zastosowaniem pasireotydu.
Pasireotyd należy stosować z ostrożnością i po ocenie stosunku korzyści do ryzyka u pacjentów z czynnikami ryzyka wydłużenia odcinka QT, takimi jak:
- wrodzony zespół wydłużonego odcinka QT;
- niekontrolowane lub istotne choroby serca, w tym niedawny zawał mięśnia sercowego, przewlekła niewydolność serca, niestabilna dławica piersiowa lub klinicznie istotna bradykardia;
- jednoczesne stosowanie leków przeciwarytmicznych lub innych leków znanych z możliwości wydłużania odcinka QT;
- hipokaliemia i/lub hipomagnezemia.
Przed rozpoczęciem terapii lekiem Signifor Lar zaleca się wykonanie wstępnego EKG. Wskazane jest monitorowanie wpływu na odcinek QTc po 21 dniach od rozpoczęcia leczenia oraz dalej w przypadku wskazań klinicznych. Hipokaliemię i/lub hipomagnezemię należy skorygować przed rozpoczęciem przyjmowania leku Signifor Lar, a następnie prowadzić odpowiednie okresowe monitorowanie w trakcie leczenia.
Hipokortyzolemia. Hamowanie sekrecji ACTH (hormonu adrenokortykotropowego) może prowadzić do hipokortyzolemii u pacjentów przyjmujących Signifor Lar. Dlatego konieczne jest monitorowanie i instruowanie pacjentów w zakresie objawów i symptomów związanych z hipokortyzememią (np. osłabienie, zmęczenie, anoreksja, nudności, wymioty, hipotensja tętnicza, hiperkaliemia, hiponatremia, hipoglikemia). W przypadku potwierdzonej hipokortyzolemii może być konieczna tymczasowa terapia zastępcza egzogennymi steroidami (glukokortykosteroidami) i/lub zmniejszenie dawki lub przerwanie terapii lekiem Signifor Lar. Szybkie obniżenie poziomu kortyzolu może być związane ze spadkiem liczby leukocytów.
Pęcherz żółciowy i zjawiska z nim związane. Kamica żółciowa (choroba kamieni w pęcherzu żółciowym) jest ustaloną niepożądaną reakcją związaną ze stosowaniem analogów somatostatyny i często zgłaszana w badaniach klinicznych z zastosowaniem pasireotydu. Zarejestrowano przypadki cholangitów w okresie postmarketingowym podczas stosowania leku Signifor Lar, z których większość była raportowana jako powikłanie kamicy żółciowej. Dlatego zaleca się wykonanie USG pęcherza żółciowego przed rozpoczęciem terapii oraz w odstępach 6- i 12-miesięcznych podczas leczenia lekiem Signifor Lar. Obecność kamieni żółciowych u pacjentów przyjmujących Signifor Lar ma zazwyczaj przebieg bezobjawowy; kamica żółciowa z objawami klinicznymi powinna być leczona zgodnie z obowiązującymi standardami klinicznymi.
Hormony przysadki. Ponieważ działanie farmakologiczne pasireotydu naśladuje działanie somatostatyny, nie można wykluczyć możliwości hamowania wydzielania hormonów przysadki, poza GH i/lub IGF-1 u pacjentów z akromegalią oraz ACTH/kortyzolem u pacjentów z zespołem Cushinga. Dlatego konieczne jest monitorowanie funkcji przysadki (np. poziom TSH/wolnego T4, hormonu wzrostu) przed rozpoczęciem i okresowo w trakcie terapii lekiem Signifor Lar zgodnie z obowiązującymi standardami klinicznymi.
Wpływ na funkcję rozrodczą u kobiet. Terapeutyczny efekt polegający na zmniejszeniu poziomu hormonu wzrostu i normalizacji stężenia insulinopodobnego czynnika wzrostu 1 (IGF-1) u kobiet z akromegalią oraz zmniejszenie lub normalizacja poziomu kortyzolu we krwi u kobiet z zespołem Cushinga może potencjalnie przywrócić funkcję rozrodczą. Kobietom w wieku rozrodczym należy zalecić stosowanie odpowiednich środków antykoncepcyjnych, jeśli jest to konieczne, w trakcie leczenia lekiem Signifor Lar.
Zaburzenia krzepnięcia krwi. Pacjenci z istotnie podwyższonym czasem protrombinowym (PT) i częściowym czasem tromboplastynowym (PTT) lub pacjenci przyjmujący pochodne kumaryny lub antykoagulancy pochodne heparyny byli wykluczani z badań klinicznych z pasireotydem, ponieważ bezpieczeństwo połączenia z takimi antykoagulantami nie zostało ustalone. Jeśli jednoczesnego stosowania pochodnych kumaryny lub antykoagulantów pochodnych heparyny podczas wstrzykiwania wewnątrzmięśniowego leku Signifor Lar nie można uniknąć, pacjentom należy zapewnić regularną kontrolę zmian parametrów krzepnięcia (PT i PTT), a dawki antykoagulantu należy odpowiednio skorygować.
Zaburzenia funkcji nerek. Ze względu na zwiększoną ekspozycję na niezwiązany lek Signifor Lar należy stosować z ostrożnością u pacjentów z ciężkim zaburzeniem funkcji nerek lub chorobą nerek w stadium końcowym.
Zawartość sodu. Ten lek zawiera mniej niż 1 mmol sodu (23 mg) na jedną zalecaną dawkę, co oznacza praktycznie brak sodu.
Stosowanie w okresie ciąży lub karmienia piersią.
Ciąża.
Dane dotyczące stosowania pasireotydu u kobiet w ciąży są ograniczone. Badania podskórnej aplikacji pasireotydu u zwierząt wykazały toksyczność rozrodczą. Pasireotyd nie jest zalecany do stosowania u kobiet w ciąży oraz u kobiet w wieku rozrodczym, które nie stosują środków antykoncepcyjnych.
Karmienie piersią.
Nie wiadomo, czy pasireotyd wydzielany jest z mlekiem matki. Dostępne dane z badań podskórnej aplikacji pasireotydu u szczurów wykazały, że pasireotyd przenika do mleka. W trakcie leczenia lekiem Signifor Lar należy przerwać karmienie piersią.
Plodność.
Badania podskórnej aplikacji pasireotydu u szczurów wykazały wpływ na parametry rozrodcze u samic. Kliniczne znaczenie tych efektów dla ludzi jest nieznane.
Wpływ na zdolność prowadzenia pojazdów i innych maszyn.
Signifor Lar może mieć nieznaczny wpływ na zdolność prowadzenia pojazdów i pracy z innymi maszynami. Pacjentom należy zalecić zachowanie ostrożności podczas prowadzenia pojazdów lub pracy z innymi maszynami, jeśli odczuwają zmęczenie, zawroty głowy lub ból głowy podczas leczenia lekiem Signifor Lar.
Sposób stosowania i dawki.
Dawkowanie.
Akromegalia.
Zalecana dawka początkowa w leczeniu akromegalii to 40 mg pasireotydu co 4 tygodnie.
Dawkę można zwiększyć maksymalnie do 60 mg u pacjentów, u których stężenia hormonu wzrostu (GH) i/lub insulinopodobnego czynnika wzrostu 1 (IGF-1) nie są wystarczająco kontrolowane po 3 miesiącach leczenia lekiem w dawce 40 mg.
W przypadku podejrzenia działań niepożądanych lub nadmiernej reakcji na leczenie (IGF-1 < dolnej granicy normy) może być konieczne tymczasowe zmniejszenie dawki. Dawka może być zmniejszana tymczasowo lub trwale.
Choroba Cushinga.
Zalecana dawka początkowa w leczeniu choroby Cushinga to 10 mg pasireotydu w postaci głębokiej iniekcji wewnątrzmięśniowej co 4 tygodnie.
Po pierwszym miesiącu leczenia oraz okresowo później pacjentów należy badać, aby ocenić korzyści kliniczne. Dawkę należy dostosowywać co 2–4 miesiące w zależności od efektu leczenia i jego tolerancji. Maksymalna dawka Signifor Lar w chorobie Cushinga wynosi 40 mg co 4 tygodnie. Jeśli nie stwierdza się korzyści klinicznych, należy rozważyć możliwość przerwania leczenia lekiem Signifor Lar.
Leczenie podejrzewanych działań niepożądanych lub nadmiernej reakcji na leczenie (stężenia kortyzolu < dolnej granicy normy) może wymagać zmniejszenia dawki, tymczasowego przerwania lub odstawienia leku Signifor Lar.
Przejście z podskórnej na wewnątrzmięśniową aplikację w chorobie Cushinga.
Brak danych z badań klinicznych dotyczących przejścia z podskórnej na wewnątrzmięśniową aplikację pasireotydu. Jeśli taka zmiana jest konieczna, zalecana dawka początkowa w leczeniu choroby Cushinga to 10 mg pasireotydu w postaci głębokiej iniekcji wewnątrzmięśniowej co 4 tygodnie. Należy obserwować stan pacjenta w celu oceny efektu leczenia i jego tolerancji; może być również konieczna dodatkowa korekta dawki.
Pominięcie dawki.
Jeśli dawkę Signifor Lar pominięto, zaleca się podanie pominiętej iniekcji jak najszybciej. Następną dawkę należy podać 4 tygodnie po tej iniekcji, aby przywrócić harmonogram dawkowania co 4 tygodnie.
Osoby z grup szczególnych.
Pacjenci w wieku podeszłym (≥ 65 lat). Dane dotyczące stosowania Signifor Lar u pacjentów w wieku 65 lat i starszych są ograniczone, jednak nie ma dowodów wskazujących na konieczność korekty dawki u tych pacjentów.
Naruszenie funkcji nerek. Korekty dawki u pacjentów z zaburzeniem funkcji nerek nie wymaga się.
Naruszenie funkcji wątroby. Korekty dawki u pacjentów z łagodnym zaburzeniem funkcji wątroby (klasa A wg skali Childa-Pugha) nie wymaga się.
Zalecana dawka początkowa w przypadku akromegalii u pacjentów z umiarkowanym zaburzeniem funkcji wątroby (klasa B wg skali Childa-Pugha) wynosi 20 mg co 4 tygodnie. Maksymalna zalecana dawka u tych pacjentów to 40 mg co 4 tygodnie.
Choroba Cushinga: zalecana dawka początkowa w chorobie Cushinga u pacjentów z umiarkowanym zaburzeniem funkcji wątroby (klasa B wg skali Childa-Pugha) wynosi 10 mg co 4 tygodnie, a maksymalna zalecana dawka u tych pacjentów to 20 mg co 4 tygodnie.
Signifor Lar jest przeciwwskazany u pacjentów z ciężkim zaburzeniem funkcji wątroby (klasa C wg skali Childa-Pugha).
Sposób stosowania.
Signifor Lar należy podawać w postaci głębokiej iniekcji wewnątrzmięśniowej przez specjalnie przygotowanego personelu medycznego. Zawiesinę leku należy przygotowywać tuż przed podaniem.
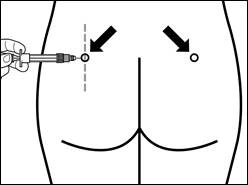
Miejsca powtarzanych iniekcji wewnątrzmięśniowych należy naprzemiennie zmieniać: lewy i prawy mięsień pośladkowy.
Składniki zestawu do iniekcji:
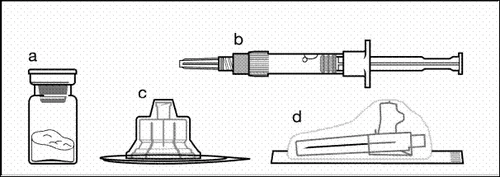
a jeden fiolka zawierająca proszek;
b jeden wstępnie napełniony strzykawka z rozpuszczalnikiem;
c jeden adapter do fiolki do przygotowania zawiesiny;
d jedna bezpieczna igła do iniekcji (20G × 1,5ʺ).
W celu prawidłowego przygotowania zawiesiny leku Signifor Lar należy dokładnie przestrzegać poniższych instrukcji.
| Krok 1 Wyjmij Signifor Lar z lodówki. Uwaga Zestaw do wstrzyknięcia musi osiągnąć temperaturę pokojową. Należy pozostawić zestaw do wstrzyknięcia w temperaturze pokojowej co najmniej na 30 minut (ale nie dłużej niż na 24 godziny). Uwaga: nieużywany zestaw do wstrzyknięcia, który był w temperaturze pokojowej nie dłużej niż 24 godziny, można ponownie odstawić do lodówki. |
|
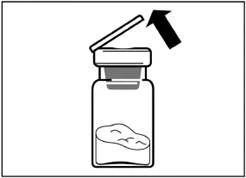
| Krok 2 Ściągnij z fiolki plastikowy korek i przetrzyj gumową septum fiolki gazikiem alkoholowym. |
|
| Usuń folię z opakowania, w którym znajduje się adapter do fiolki, ale nie wyciągaj go z tego opakowania. Trzymając adapter do fiolki za opakowanie, umieść go na wierzchu fiolki i wciśnij całkowicie w dół, aż do zatrzaśnięcia (charakterystyczny dźwięk kliknięcia). |
|
| Połóż plastikowy pojemnik w górę, aby zdjąć go z adaptera do fiolki. |
|
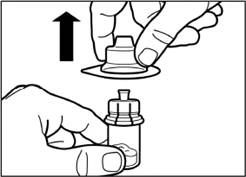
| Krok 3 Ściągnij nakrywkę z wstępnie wypełnionego strzykawki z rozpuszczalnikiem i wkręć strzykawkę w adapter do fiolki. |
|
| Wolno naciśnij tłok w dół do końcowej pozycji, aby przenieść cały rozpuszczalnik do fiolki. |
|
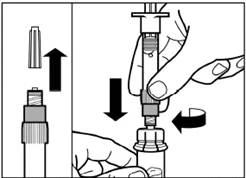
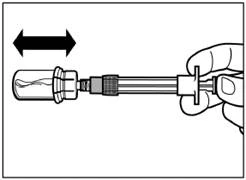
| Krok 4 Uwaga: trzymając tłok naciśniętym, ostrożnie przesuwaj fiolkę w płaszczyźnie poziomej przez co najmniej 30 sekund, aż do powstania jednolitej zawiesiny. Jeśli proszek nie został całkowicie zawieszony, ponownie ostrożnie wymieszaj zawartość poprzez przesuwanie fiolki w płaszczyźnie poziomej przez 30 sekund. |
|
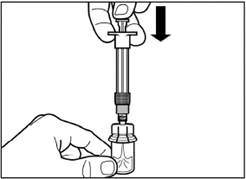
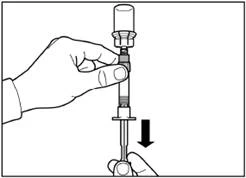
| Krok 5 Odwróć fiolkę z dołączoną strzykawką do góry nogami, powoli wyciągnij tłoczek, aby przenieść zawartość z fiolki do strzykawki. |
|
| Odkręć strzykawkę od adaptera fiolki. |
|
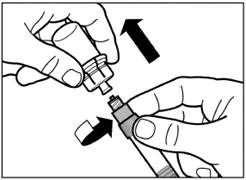
| Krok 6 Nakręć bezpieczną igłę do wstrzykiwania na strzykawkę. |
|
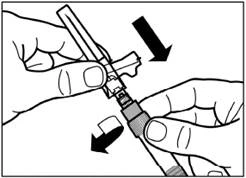
| Usuń ochronny kaptur z igły, ciągnąc go w górę wzdłuż linii igły. Aby zapobiec sedymentacji, można utrzymać jednorodność zawiesiny poprzez delikatne kołysanie strzykawką. Delikatnie postukaj w strzykawkę, aby widoczne pęcherzyki powietrza uniosły się do góry, a następnie usuń je, ostrożnie naciskając na tłoczek. Teraz zawiesina jest gotowa do natychmiastowego zastosowania. |
|
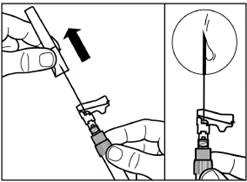
| Krok 7 Signifor Lar można wstrzykiwać wyłącznie głęboko do wewnętrznej tkanki mięśniowej. Przygotuj miejsce wstrzyknięcia, przemywając je watą nasączoną alkoholem. Wprowadź igłę do końca do mięśnia pośladkowego prawego lub lewego pod kątem 90° do powierzchni skóry. Delikatnie przyciągnij tłok do siebie, aby upewnić się, że igła nie trafiła do naczynia krwionośnego (jeśli igła trafiła do naczynia krwionośnego, wprowadź ją w inne miejsce). Wolno naciskaj na tłok, aż szpryt będzie pusty. Wyciągnij igłę z miejsca wstrzyknięcia i aktywuj mechanizm ochronny (jak pokazano na rysunku poniżej). |
|
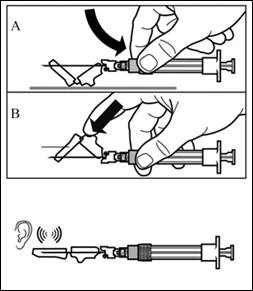
| Krok 8 Aktywuj mechanizm ochronny igły w jeden z poniższych sposobów:
Dźwięk kliknięcia potwierdza poprawną aktywację mechanizmu ochronnego. Natychmiast zutylizuj fiolkę oraz strzykawkę z igłą w pojemniku na ostre przedmioty. |
|
Dzieci.
Bezpieczeństwo i skuteczność stosowania leku Signifor Lar u dzieci i nastolatków (w wieku od 0 do 18 roku życia) nie były oceniane. Brak danych.
Przedawkowanie.
W przypadku przedawkowania zaleca się rozpoczęcie odpowiedniego leczenia wspomagającego, które należy dostosować do stanu klinicznego pacjenta i kontynuować do ustąpienia objawów.
Działania niepożądane.
Podsumowanie profilu bezpieczeństwa. Profil bezpieczeństwa po wstrzykiwaniu do mięśnia pasireotydu odpowiada profilowi bezpieczeństwa leków z grupy analogów somatostatyny, z wyjątkiem zwiększonej częstości i nasilenia hiperglikemii obserwowanej po wstrzykiwaniu do mięśnia pasireotydu. Profil bezpieczeństwa po wstrzykiwaniu do mięśnia pasireotydu jest w dużej mierze podobny przy stosowaniu w różnych wskazaniach – akromegalii i chorobie Cushinga.
Akromegalia. Ocena bezpieczeństwa w akromegalii była przeprowadzona u 491 pacjentów, którzy otrzymywali pasireotyd (419 pacjentów otrzymywało pasireotyd wstrzykiwany do mięśnia, a 72 podskórnie) podczas badań faz I, II i III.
Najczęstsze działania niepożądane (częstość ≥ 1/10) na podstawie połączonych danych bezpieczeństwa z badań faz III C2305 i C2402 to były (w kolejności malejącej): biegunka (najczęściej obserwowana w badaniu C2305), kamica żółciowa, hiperglikemia (najczęściej obserwowana w badaniu C2402) i cukrzyca. Działania niepożądane według Ogólnych Kryteriów Toksyczności (ZKT) stopnia 3 i 4 były głównie związane z hiperglikemią.
Choroba Cushinga. Ocena bezpieczeństwa leków wstrzykiwanych do mięśnia w chorobie Cushinga była przeprowadzona na podstawie danych zebranych u 150 pacjentów, którzy otrzymywali pasireotyd, podczas badania fazy III G2304 (średnia długość ekspozycji 57 tygodni). Pacjenci byli losowani w stosunku 1:1 w celu otrzymania dawek początkowych 10 mg lub 30 mg pasireotydu z możliwością zwiększenia dawki do maksymalnej dawki 40 mg co 28 dni. Najczęstsze działania niepożądane (częstość ≥ 1/10) podczas badania fazy III G2304 to były hiperglikemia, biegunka, kamica żółciowa i cukrzyca. Częstość i nasilenie działań niepożądanych wzrastały wraz ze zwiększeniem dawki początkowej do 30 mg, ale nie były jednakowe dla wszystkich działań niepożądanych.
Działania niepożądane obejmują zdarzenia zgłaszane podczas podstawowych badań po podaniu leków wstrzykiwanych do mięśnia pacjentom z akromegalią i chorobą Cushinga. Działania niepożądane są wymienione według głównych klas układów narządów według Medycznego Słownika do Działań Regulacyjnych (MedDRA). W ramach każdej klasy układów narządów niepożądane reakcje są pogrupowane według częstości. W ramach każdej grupy według częstości niepożądane reakcje są wymienione w kolejności malejącej ciężkości. Częstość określono następująco: bardzo często (≥ 1/10); często (od ≥ 1/100 do < 1/10); rzadko (od ≥ 1/1000 do < 1/100); nieznana (nie można oszacować z powodu ograniczonej dostępności danych).
Działania niepożądane (zgodnie z dominującymi terminami) po wstrzykiwaniu do mięśnia pasireotydu
Z boku krwi i układu limfatycznego: często – anemia.
Z boku układu endokrynnego: często – niewydolność nadnerczy*.
Zaburzenia przemiany materii i odżywiania: bardzo często – hiperglikemia, cukrzyca; często – cukrzyca typu II, zaburzenia tolerancji glukozy, zmniejszenie apetytu; nieznana – kwasica ketonowa cukrzycowa.
Z boku układu nerwowego: często – ból głowy, zawroty głowy.
Z boku serca: często – bradykardia zatokowa**, wydłużenie odcinka QT.
Z boku przewodu pokarmowego: bardzo często – biegunka, nudności, ból brzucha***; często – wzdęcia, wymioty; nieznana – steatorea, odbarwienie stolca.
Z boku wątroby i dróg żółciowych: bardzo często – kamica żółciowa; często – zapalenie pęcherzyka żółciowego***, cholestaza.
Z boku skóry i tkanek podskórnych: często – łysienie, świąd.
Ogólne zaburzenia i reakcje w miejscu wstrzyknięcia: bardzo często – zmęczenie***; często – reakcje w miejscu wstrzyknięcia***.
Badania: często – podwyższenie poziomu hemoglobiny glikowanej, podwyższenie poziomu alaninotransferazy, podwyższenie poziomu asparaginianotransferazy, podwyższenie poziomu gamma-glutamylotranspeptydazy, podwyższenie poziomu glukozy we krwi, podwyższenie poziomu glukozy we krwi i kinazy fosfokreatynowej we krwi, podwyższenie poziomu lipazy; rzadko – podwyższenie poziomu amylazy, wydłużenie czasu protrombinowego.
* Niewydolność nadnerczy obejmuje niewydolność nadnerczy i obniżenie poziomu kortyzolu we krwi.
** Bradykardia zatokowa obejmuje bradykardię i bradykardię zatokową.
*** Ból brzucha obejmuje ból brzucha i ból w górnej części brzucha. Reakcje w miejscu wstrzyknięcia obejmują ból w miejscu wstrzyknięcia, powstawanie guzka w miejscu wstrzyknięcia, dyskomfort w miejscu wstrzyknięcia, powstawanie siniaków w miejscu wstrzyknięcia, świąd w miejscu wstrzyknięcia, reakcje w miejscu wstrzyknięcia, zwiększona wrażliwość w miejscu wstrzyknięcia i obrzęk w miejscu wstrzyknięcia. Zapalenie pęcherzyka żółciowego obejmuje ostre i przewlekłe zapalenie pęcherzyka żółciowego. Zmęczenie obejmuje zmęczenie i osłabienie.
Opis poszczególnych działań niepożądanych.
Zaburzenia przemiany glukozy.
Akromegalia. U pacjentów z akromegalią podwyższenie poziomu glukozy na czczo było najczęstszym odchyleniem wyników badań laboratoryjnych stopnia 3/4 w dwóch badaniach fazy III. W badaniu C2305 podwyższenia poziomu glukozy na czczo stopnia 3 obserwowano u 9,7 % i 0,6 %, a stopnia 4 – u 0,6 % i 0 % pacjentów z akromegalią, którzy otrzymywali odpowiednio pasireotyd wstrzykiwany do mięśnia i oktreotyd wstrzykiwany do mięśnia. W badaniu C2402 o podwyższeniach poziomu glukozy na czczo stopnia 3 zgłaszano u 14,3 % i 17,7 % pacjentów z akromegalią, którzy otrzymywali odpowiednio pasireotyd 40 mg i 60 mg wstrzykiwany do mięśnia, oraz u 0 % pacjentów w grupie aktywnego kontroli. Dwa przypadki nagłych zdarzeń związanych z hiperglikemią (kwasica ketonowa cukrzycowa i hiperglikemiczne śpiączki cukrzycowe) zostały zarejestrowane po zwiększeniu dawki pasireotydu do 60 mg u pacjentów, którzy wcześniej nie otrzymywali leczenia: jeden u pacjenta z nieleczoną hiperglikemią i HbA1c > 8 % przed rozpoczęciem stosowania pasireotydu oraz jeden u pacjenta z nieleczoną hiperglikemią i poziomem glukozy we krwi na czczo 359 mg/dl. W obu badaniach poziomy FPG i HbA1c wzrosły w ciągu pierwszych trzech miesięcy wstrzykiwania do mięśnia pasireotydu. U pacjentów, którzy wcześniej nie otrzymywali leczenia (badanie C2305), średnie bezwzględne zwiększenie FPG i HbA1c było zasadniczo podobne u wszystkich pacjentów, którzy otrzymywali pasireotyd wstrzykiwany do mięśnia, niezależnie od wartości wyjściowych.
Stopień i częstość hiperglikemii obserwowane w dwóch głównych badaniach z udziałem pacjentów z akromegalią były wyższe w grupie wstrzykiwania do mięśnia Signifor Lar niż w grupie aktywnego kontroli (oktreotyd do wstrzykiwania do mięśnia lub lanreotyd w postaci głębokiego wstrzykiwania podskórnego). W analizie uogólnionej dwóch głównych badań ogólna częstość niepożądanych działań niepożądanych związanych z hiperglikemią wynosiła 58,6 % (wszystkie stopnie) i 9,9 % (kryteria ogólnej toksyczności stopnia 3 i 4) w grupie wstrzykiwania do mięśnia Signifor Lar w porównaniu z 18,0 % (wszystkie stopnie) i 1,1 % (KZT stopnia 3 i 4) w grupie aktywnego kontroli. W badaniu referencyjnym z udziałem pacjentów, u których nie zapewniono wystarczającego kontroli za pomocą innego analogu somatostatyny, odsetek pacjentów, którzy wcześniej nie otrzymywali antydiabetycznych środków i wymagali terapii antydiabetycznej w trakcie badania, wynosił 17,5 % i 16,1 % w grupach leku Signifor Lar 40 mg i 60 mg w porównaniu z 1,5 % w grupie aktywnego kontroli; w badaniu referencyjnym z udziałem pacjentów, którzy nie otrzymywali wcześniejszego leczenia, odsetek pacjentów, którzy wymagali terapii antydiabetycznej w trakcie badania, wynosił 36 % w grupie leku Signifor Lar w porównaniu z 4,4 % w grupie aktywnego kontroli.
Choroba Cushinga. U pacjentów z chorobą Cushinga podwyższony poziom GPN był najczęstszym odchyleniem wyników badań laboratoryjnych stopnia 3 według KZT (14,7 % pacjentów) podczas badania fazy III G2304; przypadki odchyleń laboratoryjnych stopnia 4 nie były obserwowane. Średnie zwiększenie HbA1c było mniej wyrażone u pacjentów z normalnym poziomem glikemii w momencie włączenia do badania w porównaniu z pacjentami z przedcukrzycą lub cukrzycą. Średnie poziomy GPN często wzrastały w ciągu pierwszego miesiąca leczenia, a następnie w kolejnych miesiącach spadały i się stabilizowały. Zwiększenie poziomów GPN i HbA1c zależało od dawki, a wartości zazwyczaj spadały po przerwaniu wstrzykiwania do mięśnia pasireotydu, ale pozostawały wyższe niż wartości wyjściowe. Ogólna częstość działań niepożądanych związanych z hiperglikemią wynosiła 75,3 % (wszystkie stopnie) i 22,7 % (stopień 3 według KZT). Takie działania niepożądane, jak hiperglikemia i cukrzyca, wymagały przerwania uczestnictwa w badaniu u 3 (2,0 %) i 4 pacjentów (2,7 %).
Zwiększenie poziomów glukozy i HbA1c we krwi na czczo obserwowane po wstrzykiwaniu do mięśnia pasireotydu były odwracalne po odstawieniu leku.
Zaleca się monitorowanie poziomu glukozy we krwi u pacjentów stosujących Signifor Lar.
Zaburzenia ze strony przewodu pokarmowego. Podczas leczenia lekiem Signifor Lar często zgłaszano zaburzenia ze strony przewodu pokarmowego. Zjawiska te były zazwyczaj niskiego stopnia nasilenia, nie wymagały interwencji i ustępowały przy kontynuacji leczenia. U pacjentów z akromegalią zaburzenia przewodu pokarmowego występowały rzadziej u chorych, u których nie zapewniono wystarczającego kontroli, w porównaniu z chorymi, którzy wcześniej nie otrzymywali leczenia.
Reakcje w miejscu wstrzyknięcia. Podczas badań fazy III reakcje w miejscu wstrzyknięcia (np. ból w miejscu wstrzyknięcia, dyskomfort w miejscu wstrzyknięcia) miały głównie nasilenie stopnia 1 lub 2. Częstość takich zjawisk była najwyższa w pierwszych 3 miesiącach leczenia. Podczas badań z udziałem pacjentów z akromegalią reakcje w miejscu wstrzyknięcia były podobne przy wstrzykiwaniu do mięśnia pasireotydu i wstrzykiwaniu do mięśnia oktreotydu i rzadziej obserwowane u pacjentów, u których nie zapewniono wystarczającego kontroli, w porównaniu z pacjentami, którzy wcześniej nie otrzymywali leczenia.
Wydłużenie odcinka QT. W badaniu C2305 z udziałem pacjentów z akromegalią odsetek pacjentów z nowo wykrytymi istotnymi odcinkami QT/QTc był porównywalny w grupach otrzymujących pasireotyd wstrzykiwany do mięśnia i oktreotyd wstrzykiwany do mięśnia do fazy krzyżowej, z kilkoma istotnymi nieprawidłowymi wartościami. Wartości QTcF > 480 ms obserwowano u 3 pacjentów w porównaniu z 2 pacjentami w grupie wstrzykiwania do mięśnia pasireotydu i wstrzykiwania do mięśnia oktreotydu odpowiednio, a wydłużenie QTcF > 60 ms w porównaniu z wartością wyjściową zgłaszano u 2 pacjentów w porównaniu z 1 pacjentem w odpowiednich grupach. W badaniu C2402 jedynym istotnym odchyleniem od normy była wartość QTcF > 480 ms u 1 pacjenta w grupie wstrzykiwania do mięśnia pasireotydu 40 mg. Podczas badania G2304 z udziałem pacjentów z chorobą Cushinga wartość QTcF > 480 ms obserwowano u 2 pacjentów. Wartości QTcF > 500 ms nie były obserwowane w żadnym z głównych badań.
Enzymy wątrobowe. Zgłaszano przejściowe podwyższenie poziomu enzymów wątrobowych na tle stosowania analogów somatostatyny, co również obserwowano u pacjentów, którzy przyjmowali pasireotyd w trakcie badań klinicznych. Podwyższenie było głównie bezobjawowe, niskiego stopnia i odwracalne przy kontynuacji leczenia. Kilka przypadków jednoczesnego podwyższenia ALP > 3 × WNN i bilirubiny > 2 × WNN obserwowano przy podanym podskórnie, jednak nie u pacjentów, którzy otrzymywali pasireotyd wstrzykiwany do mięśnia. Wszystkie przypadki jednoczesnych podwyższeń obserwowano w ciągu 10 dni od rozpoczęcia leczenia. Pacjenci wyzdrowieli bez następstw klinicznych, a wyniki badań funkcji wątroby powróciły do wartości wyjściowych po odstawieniu leczenia.
Zaleca się monitorowanie poziomu enzymów wątrobowych przed i podczas leczenia lekiem Signifor Lar zgodnie z zasadami klinicznymi.
Enzymy trzustki. U pacjentów, którzy przyjmowali pasireotyd w trakcie badań klinicznych, obserwowano bezobjawowe podwyższenie poziomu lipazy i amylazy. Podwyższenie było głównie niskiego stopnia i miało charakter odwracalny przy kontynuacji leczenia. Zapalenie trzustki jest potencjalnym niepożądaniem związanym ze stosowaniem analogów somatostatyny, wynikającym ze związku między kamica żółciową a ostrym zapaleniem trzustki.
Niezgodność.
Ze względu na brak badań zgodności ten lek nie powinien być mieszany z innymi lekami.
Okres ważności. 3 lata.
Warunki przechowywania. Przechowywać w oryginalnym opakowaniu w temperaturze 2–8 ºC. Nie zamrażać. Przechowywać w miejscu niedostępnym dla dzieci.
Opakowanie. Proszek w fiolce z brąszowego szkła o pojemności 6 ml, zatkanej gumową korką szarego koloru pod aluminiową pokrywką systemu flip-off szarego (do dawkowania 20 mg), czerwonego (do dawkowania 40 mg) lub pomarańczowego (do dawkowania 60 mg) koloru w zestawie z: rozpuszczalnikiem w wstępnie wypełnionym strzykawce o pojemności 3 ml z bezbarwnego szkła z 2 gumowymi korkami szarego koloru, oparciem dla palców, tłokiem i pokrywką; jedną igłą i jednym adapterem w tekturowym pudełku.
Kategoria wydania. Na receptę.
Producent. Recordati Rare Diseases.
Miejsce produkcji i adres miejsca prowadzenia działalności.
Eco River Park, 30 Rue des Peupliers, Nanterre, 92000, Francja;
Immeuble Le Wilson, 70 Avenue du Général de Gaulle, Puteaux, 92800, Francja.