Signifor lar
UkraineTable of Contents
INSTRUCTIONS for medical use of the medicinal product SIGNIFOR LAR (SIGNIFOR® LAR®)
Composition:
Active substance: pasireotide;
1 vial of powder for suspension for injection contains 20 mg, 40 mg, or 60 mg of pasireotide (as pasireotide pamoate).
Excipients:
powder: poly (D,L-lactic-co-glycolic acid) (50-60:40-50), poly (D,L-lactic-co-glycolic acid) (50:50);
solvent: 1 pre-filled syringe with 2 ml of solvent contains: sodium carmellose, mannitol (E421), poloxamer 188, water for injections.
Pharmaceutical form. Powder for suspension for injection.
Main physicochemical characteristics:
powder: powder from slightly yellowish to yellowish in color;
solvent: clear solution from colorless to yellowish or light brown in color.
Pharmacotherapeutic group. Pituitary, hypothalamic hormones and their analogues. Somatostatin and its analogues. ATC code H01C B05.
Pharmacological properties.
Pharmacodynamics.
Mechanism of action. Pasireotide is a cyclohexapeptide, an injectable somatostatin analog. Like the natural peptide hormones somatostatin-14 and somatostatin-28 (also known as somatotropin release-inhibiting factor [SRIF]) and other somatostatin analogs, pasireotide exerts its pharmacological effect by binding to somatostatin receptors. Five subtypes of human somatostatin receptors are known: hsst1, 2, 3, 4, and 5. Under normal physiological conditions, these receptor subtypes are expressed in various tissues. Somatostatin analogs bind to hsst receptors with varying potency (see table below). Pasireotide binds with high affinity to four of the five hsst receptors.
Binding affinity of somatostatin (SRIF-14), pasireotide, octreotide, and lanreotide to the five human somatostatin receptor subtypes (hsst1–5).
| Drug substance |
hsst1 |
hsst2 |
hsst3 |
hsst4 |
hsst5 |
| Somatostatin (SRIF-14) |
0.93 ± 0.12 |
0.15 ± 0.02 |
0.56 ± 0.17 |
1.5 ± 0.4 |
0.29 ± 0.04 |
| Pasireotide |
9.3 ± 0.1 |
1.0 ± 0.1 |
1.5 ± 0.3 |
> 100 |
0.16 ± 0.01 |
| Octreotide |
280 ± 80 |
0.38 ± 0.08 |
7.1 ± 1.4 |
>1000 |
6.3 ± 1.0 |
| Lanreotide |
180 ± 20 |
0.54 ± 0.08 |
14 ± 9 |
230 ± 40 |
17 ± 5 |
The results are presented as mean ± standard error of the mean (SEM) IC50, expressed in nmol/L.
Pharmacodynamic effects. Somatostatin receptors are expressed in many tissues, particularly in neuroendocrine tumors, where excessive hormone secretion occurs, including growth hormone (GH) in acromegaly and adrenocorticotropic hormone (ACTH) in Cushing's disease.
In vitro studies have shown that corticotroph tumor cells obtained from patients with Cushing's disease exhibit strong expression of hsst5, whereas other receptor subtypes are either not expressed or expressed to a minimal extent. Pasireotide binds and activates four of the five hsst subtypes, particularly hsst5, in corticotroph ACTH-producing adenomas, leading to inhibition of ACTH secretion.
Due to its broad somatostatin receptor binding profile, pasireotide is able to stimulate hsst2 and hsst5 receptor subtypes, which are necessary for inhibition of GH and insulin-like growth factor (IGF-1) secretion, making it potentially effective in the treatment of acromegaly.
Glucose metabolism. In a randomized, double-blind study in healthy volunteers, hyperglycemia observed after subcutaneous administration of pasireotide at doses of 0.6 mg and 0.9 mg twice daily was associated with a significant reduction in insulin hormone secretion, as well as in the secretion of incretins (specifically glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP)). Pasireotide did not affect insulin sensitivity.
Paediatric population. The European Medicines Agency has waived the obligation to submit the results of studies on the use of Signifor LAR in all paediatric subpopulations for acromegaly and pituitary gigantism, as well as for pituitary-dependent Cushing’s disease, hypersecretion of pituitary ACTH, and pituitary-dependent hypercortisolism (for information on use in children, see section “Posology and method of administration”).
Pharmacokinetics.
Pasireotide for intramuscular administration is formulated as prolonged-release microspheres. After a single injection, plasma concentrations of pasireotide show an initial "burst" release on the first day, followed by a decline in concentration between days 2–7. Subsequently, a gradual increase to peak concentration occurs around day 21, followed by a slow elimination phase over the following weeks, coinciding with the final degradation phase of the polymeric matrix of the formulation.
Absorption. The relative bioavailability of pasireotide after intramuscular administration compared to subcutaneous administration is complete. Studies to assess the absolute bioavailability of pasireotide in humans have not been conducted.
Distribution. In healthy volunteers, intramuscularly administered pasireotide showed extensive distribution with a large volume of distribution (Vz/F > 100 liters). The distribution between blood cells and plasma is concentration-independent and indicates that pasireotide is predominantly localized in plasma (91%). Protein binding to plasma proteins is moderate (88%) and concentration-independent.
Based on in vitro data, pasireotide is a substrate of the efflux transporter P-glycoprotein. Based on in vitro data, pasireotide is not a substrate of the efflux transporter BCRP (breast cancer resistance protein) or of the influx transporters OAT1 (organic anion transporter 1), OATP1B1, 1B3, or 2B1. At therapeutic doses, pasireotide is also not an inhibitor of UGT1A1, OATP1B1 or 1B3, OAT1 or OAT3, OCT1 or OCT2, P-gp, BCRp, MRP2, or BSEP.
Biological transformation. Pasireotide is highly metabolically stable. In vitro data indicate that pasireotide is not a substrate, inhibitor, or inducer of any major CYP450 enzymes. In healthy volunteers, pasireotide is predominantly found unchanged in plasma, urine, and feces.
Elimination. Pasireotide is primarily eliminated via hepatic clearance (biliary excretion), with minimal contribution from renal elimination. In an ADME (absorption, distribution, metabolism, excretion) study in humans, 55.9 ± 6.63% of the radioactive dose was recovered within the first 10 days after administration, including 48.3 ± 8.16% of radioactivity in feces and 7.63 ± 2.03% in urine.
The clearance (CL/F) of intramuscular pasireotide in healthy volunteers averages 4.5–8.5 L/h. Based on population pharmacokinetic analysis, measured CL/F ranged approximately from 4.8 to 6.5 L/h in patients with typical Cushing’s disease and from 5.6 to 8.2 L/h in patients with typical acromegaly.
Linearity and time dependency. Steady-state pharmacokinetics following intramuscular administration of pasireotide is achieved after 3 months. After multiple monthly doses ranging from 10 mg to 60 mg every 4 weeks, intramuscular pasireotide demonstrates approximately dose-proportional pharmacokinetics in patients.
Special patient populations.
Paediatric population. Studies in children have not been conducted.
Patients with renal impairment. Renal clearance plays a minimal role in the elimination of pasireotide in humans. In a clinical study, a single subcutaneous dose of pasireotide 900 mcg in patients with renal dysfunction, including mild, moderate, and severe renal impairment or end-stage renal disease (ESRD), did not have a significant effect on total plasma exposure to pasireotide. However, plasma exposure to unbound pasireotide (AUCinf,u) increased in patients with renal impairment (mild: 33%; moderate: 25%; severe: 99%; ESRD: 143%) compared to control subjects.
Patients with hepatic impairment. Clinical studies with intramuscular pasireotide in patients with hepatic impairment have not been conducted. In a clinical study administering a single subcutaneous dose of pasireotide to patients with hepatic impairment, statistically significant differences were observed in patients with moderate and severe hepatic impairment (Child-Pugh classes B and C). In these patients, AUCinf increased by 60% and 79%, Cmax increased by 67% and 69%, and CL/F decreased by 37% and 44%, respectively.
Elderly patients (≥ 65 years). Age was not a significant factor in the population pharmacokinetic analysis of patients.
Demographic characteristics. Population pharmacokinetic analysis of intramuscular pasireotide indicates that patient race and sex do not influence the pharmacokinetic parameters of the drug. Body weight had a minor effect on pharmacokinetics in a study of treatment-naïve patients but had no effect in a study of patients with inadequately controlled disease. Women with acromegaly showed greater exposure (32% and 51%) compared to men in studies of treatment-naïve patients and patients with inadequately controlled disease, respectively; however, these differences were not clinically relevant based on efficacy and safety data.
Clinical characteristics.
Indications.
Treatment of adult patients with acromegaly for whom surgery is not optimal or has been unsuccessful and who have not achieved adequate control with another somatostatin analogue.
Treatment of adult patients with Cushing’s disease for whom surgery is not optimal or has been unsuccessful.
The 60 mg dose is used only for the treatment of acromegaly.
Contraindications.
Hypersensitivity to the active substance or to any of the excipients of the medicinal product. Severe hepatic impairment (Child-Pugh class C).
Interaction with other medicinal products and other forms of interaction.
Expected pharmacokinetic interactions affecting pasireotide. The effect of the P-glycoprotein inhibitor verapamil on the pharmacokinetics of pasireotide following subcutaneous administration was evaluated in a drug interaction study involving healthy volunteers. No changes in the pharmacokinetics (rate or extent of exposure) of pasireotide were observed.
Expected pharmacokinetic interactions affecting other medicinal products. Pasireotide may reduce the relative bioavailability of cyclosporine. Dose adjustment of cyclosporine may be required when administered concomitantly with pasireotide to maintain therapeutic concentrations.
Expected pharmacodynamic interactions. Medicinal products prolonging the QT interval.
Pasireotide should be used with caution in patients who are concurrently receiving medicinal products that prolong the QT interval, such as class Ia antiarrhythmics (e.g., quinidine, procainamide, disopyramide), class III antiarrhythmics (e.g., amiodarone, dronedarone, sotalol, dofetilide, ibutilide), certain antibacterial agents (intravenous erythromycin, pentamidine injections, clarithromycin, moxifloxacin), certain antipsychotics (e.g., chlorpromazine, thioridazine, fluphenazine, pimozide, haloperidol, tiapride, amisulpride, sertindole, methadone), certain antihistamines (e.g., terfenadine, astemizole, mizolastine), antimalarials (e.g., chloroquine, halofantrine, lumefantrine), and certain antifungal agents (ketoconazole, except shampoo).
Medicinal products causing bradycardia. Clinical monitoring of heart rate, especially at the beginning of treatment, is recommended for patients receiving pasireotide concomitantly with medicinal products that cause bradycardia, such as beta-blockers (e.g., metoprolol, carteolol, propranolol, sotalol), acetylcholinesterase inhibitors (e.g., rivastigmine, physostigmine), certain calcium channel blockers (e.g., verapamil, diltiazem, bepridil), and certain antiarrhythmic agents.
Insulin and antidiabetic medicinal products. Dose adjustment (reduction or increase) of insulin and antidiabetic medicinal products (e.g., metformin, liraglutide, vildagliptin, nateglinide) may be required when administered concomitantly with pasireotide.
Special precautions for use.
Disorders of glucose metabolism. Changes in blood glucose levels were frequently observed in healthy volunteers and patients after treatment with pasireotide. Hyperglycemia and, less frequently, hypoglycemia were observed in patients participating in clinical trials with pasireotide (see section "Adverse reactions").
Patients who developed hyperglycemia generally responded to antidiabetic therapy. Dose reduction or discontinuation of pasireotide treatment due to hyperglycemia were infrequent during clinical trials with pasireotide.
The development of hyperglycemia is associated with reduced secretion of insulin and incretin hormones (specifically glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP)).
Glycemic status (fasting plasma glucose/hemoglobin A1c (FPG/HbA1c)) should be assessed prior to initiating pasireotide therapy. Monitoring of FPG/HbA1c during treatment should be performed according to current recommendations. Self-monitoring of blood glucose levels and/or assessment of FPG should be performed weekly during the first 3 months and then periodically according to clinical practice, as well as during the first 4–6 weeks after any dose increase. Additionally, monitoring of FPG should be performed 4 weeks after completion of treatment and hemoglobin A1c 3 months after completion of treatment.
If hyperglycemia develops in a patient receiving Signifor LAR, initiation or adjustment of antidiabetic therapy should be considered according to current recommendations to control hyperglycemia. If uncontrolled hyperglycemia persists despite appropriate medical management, the dose of Signifor LAR should be reduced or treatment discontinued (see section "Interaction with other medicinal products and other forms of interaction").
Post-marketing cases of ketoacidosis have been reported with the use of Signifor LAR in patients with and without a history of diabetes mellitus. Patients who develop signs and symptoms consistent with severe metabolic acidosis should be evaluated for the development of ketoacidosis regardless of a history of diabetes mellitus.
In patients with poor glycemic control (defined as HbA1c > 8% despite antidiabetic therapy), intensified monitoring and management of diabetes should be implemented prior to and during pasireotide therapy.
Liver tests. In patients receiving pasireotide, mild transient increases in aminotransferase levels were generally observed. Rare cases of concurrent elevation of ALT (alanine aminotransferase) above 3 × ULN (upper limit of normal) and bilirubin above 2 × ULN have also been reported (see section "Adverse reactions").
Liver function monitoring should subsequently be performed based on clinical indications. Liver function should be monitored prior to initiation of intramuscular pasireotide administration and 2–3 weeks after starting therapy, followed by monthly monitoring for 3 months after initiation of treatment.
Patients with elevated transaminase levels require frequent monitoring of liver function until values return to pre-treatment levels. Pasireotide therapy should be discontinued if a patient develops jaundice or other signs of clinically significant liver dysfunction, persistent elevation of AST (aspartate aminotransferase) or ALT ≥ 5 × ULN, or ALT or AST > 3 × ULN concurrent with bilirubin elevation > 2 × ULN. Patients should be monitored after discontinuation of pasireotide until symptoms resolve. Reinitiation of treatment should not be considered if there is suspicion that liver dysfunction is related to pasireotide.
Cardiovascular effects. Bradycardia has been reported during pasireotide use. Careful monitoring is recommended in patients with cardiac conditions and/or risk factors for bradycardia, such as clinically significant bradycardia or history of acute myocardial infarction, high-grade cardiac conduction blocks, congestive heart failure (NYHA class III or IV), unstable angina, sustained ventricular tachycardia, or ventricular fibrillation. Dose adjustments of concomitant medications such as beta-blockers, calcium channel blockers, or drugs used to manage electrolyte balance may be required.
In two studies involving healthy volunteers, pasireotide was shown to prolong the QT interval on ECG. The clinical significance of this prolongation is unknown. In phase III clinical trials in patients with acromegaly, no clinically significant differences in QT interval prolongation were observed between intramuscular pasireotide and somatostatin analogs used as active comparators. All QT interval prolongation events were transient and resolved without therapeutic intervention.
No episodes of torsades de pointes tachycardia were observed in any clinical trial of pasireotide.
Pasireotide should be used with caution and with careful consideration of benefit/risk in patients with risk factors for QT interval prolongation, such as:
- congenital long QT syndrome;
- uncontrolled or significant cardiac disease, including recent myocardial infarction, congestive heart failure, unstable angina, or clinically significant bradycardia;
- concomitant use of antiarrhythmic drugs or other medicinal products known to prolong the QT interval;
- hypokalemia and/or hypomagnesemia.
An initial ECG is recommended before starting treatment with Signifor LAR. Monitoring for effects on QTc interval is advisable 21 days after initiation of treatment and subsequently as clinically indicated. Hypokalemia and/or hypomagnesemia should be corrected before starting Signifor LAR and appropriate periodic monitoring should be conducted during treatment.
Hypocortisolism. Suppression of ACTH (adrenocorticotropic hormone) secretion may lead to hypocortisolism in patients receiving Signifor LAR. Therefore, monitoring and patient education regarding signs and symptoms associated with hypocortisolism (e.g., weakness, fatigue, anorexia, nausea, vomiting, hypotension, hyperkalemia, hyponatremia, hypoglycemia) are necessary. In confirmed cases of hypocortisolism, temporary replacement therapy with exogenous steroids (glucocorticoids) and/or dose reduction or interruption of Signifor LAR treatment may be required. Rapid reduction in cortisol levels may be associated with decreased white blood cell count.
Gallbladder and related effects. Cholelithiasis (gallstone disease) is a known adverse reaction associated with somatostatin analogs and was frequently reported in clinical trials with pasireotide. Post-marketing cases of cholangitis have been reported with Signifor LAR, most of which were reported as complications of gallstone disease. Therefore, ultrasonographic examination of the gallbladder is recommended before and at 6- and 12-month intervals during treatment with Signifor LAR. Gallstones in patients receiving Signifor LAR are mostly asymptomatic; symptomatic gallstones should be managed according to standard clinical practice.
Pituitary hormones. Since the pharmacological action of pasireotide mimics that of somatostatin, inhibition of pituitary hormones other than GH and/or IGF-1 in patients with acromegaly and of ACTH/cortisol in patients with Cushing's disease cannot be excluded. Therefore, monitoring of pituitary function (e.g., TSH/free T4, growth hormone levels) should be performed prior to and periodically during treatment with Signifor LAR according to clinical standards.
Effect on reproductive function in women. The therapeutic effect of reducing growth hormone levels and normalizing insulin-like growth factor 1 (IGF-1) concentrations in women with acromegaly, and reducing or normalizing serum cortisol levels in women with Cushing's disease, may potentially restore reproductive function. Women of reproductive potential should be advised to use appropriate contraceptive methods, if necessary, during treatment with Signifor LAR.
Coagulation disorders. Patients with markedly prolonged prothrombin time (PT) and partial thromboplastin time (PTT), or patients receiving coumarin derivatives or heparin-derived anticoagulants, were excluded from pasireotide clinical trials, as the safety of concomitant use with such anticoagulants has not been established. If concomitant use of coumarin derivatives or heparin-derived anticoagulants with intramuscular Signifor LAR cannot be avoided, patients should be regularly monitored for changes in coagulation parameters (PT and PTT), and anticoagulant doses should be adjusted accordingly.
Renal function. Due to increased exposure to unbound Signifor LAR, caution should be exercised when administering the drug to patients with severe renal impairment or end-stage renal disease.
Sodium content. This medicinal product contains less than 1 mmol sodium (23 mg) per recommended dose, i.e., essentially "sodium-free".
Use during pregnancy or breastfeeding.
Pregnancy.
Data on the use of pasireotide in pregnant women are limited. Animal studies with subcutaneous pasireotide showed reproductive toxicity. Pasireotide is not recommended for use in pregnant women or in women of reproductive potential who are not using contraception.
Breastfeeding.
It is unknown whether pasireotide is excreted in human milk. Available data from subcutaneous pasireotide studies in rats show that pasireotide passes into milk. Breastfeeding should be discontinued during treatment with Signifor LAR.
Fertility.
Subcutaneous pasireotide studies in rats showed effects on reproductive parameters in females. The clinical relevance of these effects in humans is unknown.
Ability to influence reaction speed when driving or operating machinery.
Signifor LAR may have a minor influence on the ability to drive or operate machinery. Patients should be advised to exercise caution when driving or operating machinery if they experience fatigue, dizziness, or headache during treatment with Signifor LAR.
Method of administration and dosage.
Dosage.
Acromegaly.
The recommended starting dose for the treatment of acromegaly is 40 mg of pasireotide every 4 weeks.
The dose may be increased up to a maximum of 60 mg in patients whose growth hormone (GH) and/or insulin-like growth factor-1 (IGF-1) levels are not adequately controlled after 3 months of treatment with the 40 mg dose.
To manage suspected adverse reactions or an excessive response to treatment (IGF-1 < lower limit of normal), temporary dose reduction may be required. The dose may be temporarily or permanently reduced.
Cushing’s disease.
The recommended starting dose for the treatment of Cushing’s disease is 10 mg of pasireotide administered as a deep intramuscular injection every 4 weeks.
Patients should be evaluated after the first month of treatment and periodically thereafter to assess clinical benefit. Dose titration should be performed every 2–4 months based on treatment efficacy and tolerability. The maximum dose of Signifor LAR in Cushing’s disease is 40 mg every 4 weeks. If no clinical benefit is observed, discontinuation of Signifor LAR should be considered.
Management of suspected adverse reactions or an excessive treatment effect (cortisol levels < lower limit of normal) may require dose reduction, temporary interruption, or discontinuation of treatment with Signifor LAR.
Transition from subcutaneous to intramuscular administration in Cushing’s disease.
Clinical data on switching from subcutaneous to intramuscular pasireotide administration are lacking. If such a transition is necessary, the recommended starting dose for the treatment of Cushing’s disease is 10 mg of pasireotide administered as a deep intramuscular injection every 4 weeks. Patients should be monitored to evaluate treatment response and tolerability; additional dose adjustments may also be required.
Missed dose.
If a dose of Signifor LAR is missed, the missed injection should be administered as soon as possible. The next dose should be given 4 weeks after the administration of the missed dose to re-establish the every-4-week dosing schedule.
Special patient populations.
Elderly patients (≥ 65 years of age). Data on the use of Signifor LAR in patients aged 65 years and older are limited; however, there is no evidence to suggest that dose adjustment is required in these patients.
Renal impairment. Dose adjustment is not required in patients with renal impairment.
Hepatic impairment. Dose adjustment is not required in patients with mild hepatic impairment (Child–Pugh class A).
The recommended starting dose in acromegaly for patients with moderate hepatic impairment (Child–Pugh class B) is 20 mg every 4 weeks. The maximum recommended dose for these patients is 40 mg every 4 weeks.
Cushing’s disease: the recommended starting dose in Cushing’s disease for patients with moderate hepatic impairment (Child–Pugh class B) is 10 mg every 4 weeks, and the maximum recommended dose for these patients is 20 mg every 4 weeks.
Signifor LAR is contraindicated in patients with severe hepatic impairment (Child–Pugh class C).
Method of administration.
Signifor LAR must be administered by deep intramuscular injection by a trained healthcare professional. The medicinal product suspension should be prepared immediately before administration.
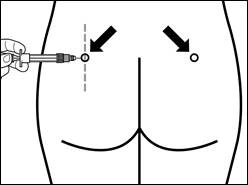
The site of repeated intramuscular injections should be alternated: left and right gluteal muscles.
Components of the injection kit:
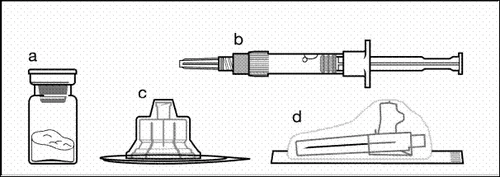
a one vial containing powder;
b one pre-filled syringe with solvent;
c one vial adapter for suspension preparation;
d one safety needle for injection (20G × 1.5ʺ).
To ensure proper preparation of the Signifor LAR suspension, the instructions below must be strictly followed.
| Step 1 Remove Signifor LA from the refrigerator. Caution The injection kit must reach room temperature. Leave the injection kit at room temperature for at least 30 minutes (but no longer than 24 hours). Note: An injection kit not used within 24 hours may be returned to the refrigerator. |
|
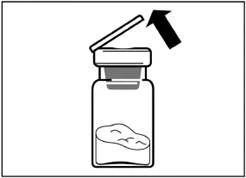
| Step 2 Remove the plastic cap from the vial and wipe the vial's rubber stopper with an alcohol swab. |
|
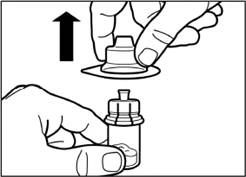
| Remove the film from the packaging containing the vial adapter, but do not remove the adapter from this packaging. Holding the vial adapter by the packaging, place it onto the top of the vial and push the adapter firmly downwards until it clicks into place. |
|
| Remove the plastic cover from the vial adapter with a vertical upward movement. |
|
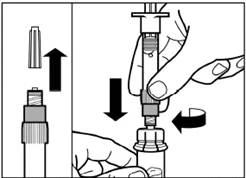
| Step 3 Remove the cap from the pre-filled solvent syringe and screw the syringe into the vial adapter. |
|
| Slowly push the plunger down completely to transfer all the solvent into the vial. |
|
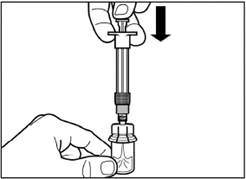
| Step 4 Caution: while keeping the plunger depressed, gently move the vial horizontally for at least 30 seconds until a uniform suspension is formed. If the powder has not been completely suspended, gently mix the contents again by moving the vial horizontally for 30 seconds. |
|
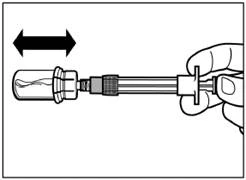
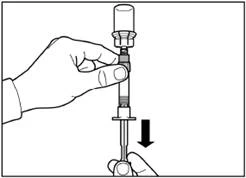
| Step 5 Turn the vial with the attached syringe upside down, slowly pull the plunger down to transfer the contents from the vial into the syringe. |
|
| Unscrew the syringe from the vial adapter. |
|
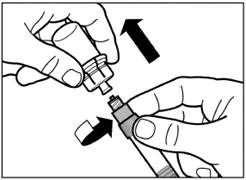
| Step 6 Screw the safety injection needle onto the syringe. |
|
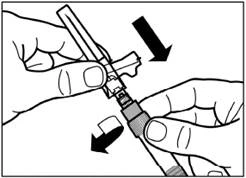
| Remove the protective cap from the needle by pulling it straight off along the line of the needle. To prevent sedimentation, the suspension can be kept homogeneous by gently shaking the syringe. Gently tap the syringe so that any visible air bubbles rise to the top, then remove them by carefully pressing the plunger. The suspension is now ready for immediate administration. |
|
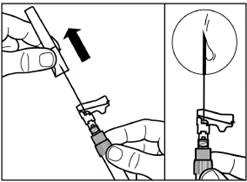
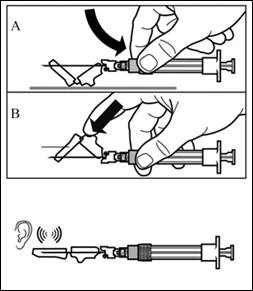
| Step 7 SIGNIFOR LAR must only be administered by deep intramuscular injection. Prepare the injection site by wiping it with an alcohol swab. Insert the needle fully into the right or left gluteal muscle at a 90° angle to the skin surface. Pull back slightly on the plunger to make sure the needle has not entered a blood vessel (if blood appears, withdraw the needle and inject at another site). Slowly depress the plunger until the syringe is empty. Remove the needle from the injection site and activate the safety mechanism (as shown in the figure below). |
|
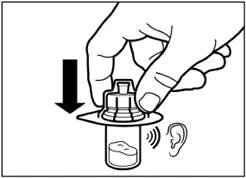
| Step 8 Activate the needle safety mechanism by one of the following methods:
A clicking sound confirms correct activation of the safety mechanism. Immediately dispose of the vial, syringe, and needle into a sharps container. |
|
Children.
The safety and efficacy of Signifor LAR in children and adolescents (aged 0 to 18 years) have not been established. Data are lacking.
Overdose.
In case of overdose, appropriate supportive treatment should be initiated, based on the patient's clinical condition, and maintained until symptoms resolve.
Side effects.
Summary of safety profile. The safety profile following intramuscular administration of pasireotide is consistent with that of drugs in the somatostatin analog class, except for increased frequency and severity of hyperglycemia observed with intramuscular pasireotide. The safety profile following intramuscular administration of pasireotide is largely similar across different indications – acromegaly and Cushing’s disease.
Acromegaly. Safety was evaluated in 491 patients who received pasireotide (419 patients received intramuscular pasireotide and 72 received subcutaneous pasireotide) during Phase I, II, and III studies.
The most common adverse reactions (frequency ≥ 1/10), based on pooled safety data from Phase III studies C2305 and C2402, in descending order, were: diarrhea (most frequently observed in study C2305), cholelithiasis, hyperglycemia (most frequently observed in study C2402), and diabetes mellitus. Adverse reactions graded as severity level 3 and 4 according to the Common Terminology Criteria for Toxicity (CTC) were predominantly related to hyperglycemia.
Cushing’s disease. For Cushing’s disease, safety assessment of the intramuscular formulation was based on data collected from 150 patients who received pasireotide in the Phase III study G2304 (mean exposure duration 57 weeks). Patients were randomized in a 1:1 ratio to receive initial doses of 10 mg or 30 mg pasireotide, with dose escalation permitted up to a maximum dose of 40 mg every 28 days. The most common adverse reactions (frequency ≥ 1/10) during Phase III study G2304 were hyperglycemia, diarrhea, cholelithiasis, and diabetes mellitus. The frequency and severity of adverse reactions increased with higher initial doses (30 mg), although this was not consistent across all adverse reactions.
Adverse reactions include events reported during core studies following administration of the intramuscular formulation to patients with acromegaly and Cushing’s disease. Adverse reactions are listed according to MedDRA primary system organ classes. Within each organ system class, adverse reactions are categorized by frequency. Within each frequency group, adverse reactions are listed in order of decreasing severity. Frequency is defined as follows: very common (≥ 1/10); common (≥ 1/100 to < 1/10); uncommon (≥ 1/1000 to < 1/100); unknown (cannot be estimated due to limited available data).
Adverse reactions (by preferred terms) with intramuscular administration of pasireotide
Blood and lymphatic system disorders: common – anemia.
Endocrine disorders: common – adrenal insufficiency*.
Metabolism and nutrition disorders: very common – hyperglycemia, diabetes mellitus; common – type 2 diabetes mellitus, impaired glucose tolerance, decreased appetite; unknown – diabetic ketoacidosis.
Nervous system disorders: common – headache, dizziness.
Cardiac disorders: common – sinus bradycardia**, QT interval prolongation.
Gastrointestinal disorders: very common – diarrhea, nausea, abdominal pain***; common – abdominal distension, vomiting; unknown – steatorrhea, fecal discoloration.
Hepatobiliary disorders: very common – cholelithiasis; common – cholecystitis***, cholestasis.
Skin and subcutaneous tissue disorders: common – alopecia, pruritus.
General disorders and administration site conditions: very common – fatigue***; common – injection site reactions***.
Investigations: common – increased glycated hemoglobin, increased alanine aminotransferase, increased aspartate aminotransferase, increased gamma-glutamyl transferase, increased blood glucose, increased blood glucose and creatine phosphokinase, increased lipase; uncommon – increased amylase, prolonged prothrombin time.
* Adrenal insufficiency includes adrenal insufficiency and decreased blood cortisol levels.
** Sinus bradycardia includes bradycardia and sinus bradycardia.
*** Abdominal pain includes abdominal pain and upper abdominal pain. Injection site reactions include injection site pain, injection site nodule, injection site discomfort, injection site bruising, injection site pruritus, injection site reaction, increased injection site sensitivity, and injection site swelling. Cholecystitis includes acute cholecystitis and chronic cholecystitis. Fatigue includes fatigue and asthenia.
Description of selected adverse reactions.
Glucose metabolism disorders.
Acromegaly. In patients with acromegaly, elevated fasting plasma glucose levels were the most common Grade 3/4 laboratory abnormality in both Phase III studies. In study C2305, Grade 3 elevations in fasting glucose levels occurred in 9.7% and 0.6%, and Grade 4 elevations in 0.6% and 0% of acromegaly patients receiving intramuscular pasireotide and intramuscular octreotide, respectively. In study C2402, Grade 3 elevations in fasting glucose levels were reported in 14.3% and 17.7% of acromegaly patients receiving intramuscular pasireotide 40 mg and 60 mg, respectively, compared to 0% in the active control group. Two serious hyperglycemia-related events (diabetic ketoacidosis and diabetic hyperglycemic coma) occurred after dose escalation to 60 mg in previously untreated patients: one in a patient with untreated hyperglycemia and HbA1c > 8% prior to starting pasireotide, and one in a patient with untreated hyperglycemia and a fasting plasma glucose level of 359 mg/dL. In both studies, FPG and HbA1c levels increased during the first three months of intramuscular pasireotide administration. In treatment-naïve patients (study C2305), mean absolute increases in FPG and HbA1c were generally similar across all patients receiving intramuscular pasireotide, regardless of baseline values.
The degree and frequency of hyperglycemia observed in the two main studies involving acromegaly patients were higher in the intramuscular Signifor LAR group compared to the active control group (intramuscular octreotide or deep subcutaneous lanreotide). In a pooled analysis of the two main studies, the overall incidence of adverse reactions related to hyperglycemia was 58.6% (all grades) and 9.9% (CTC Grade 3 and 4) in the intramuscular Signifor LAR group, compared to 18.0% (all grades) and 1.1% (CTC Grade 3 and 4) in the active control group. In the pivotal study involving patients inadequately controlled on another somatostatin analog, the proportion of patients who were previously not on antidiabetic therapy and required antidiabetic treatment during the study was 17.5% and 16.1% in the Signifor LAR 40 mg and 60 mg groups, respectively, compared to 1.5% in the active control group. In the pivotal study involving treatment-naïve patients, the proportion requiring antidiabetic therapy during the study was 36% in the Signifor LAR group compared to 4.4% in the active control group.
Cushing’s disease. In patients with Cushing’s disease, elevated fasting plasma glucose (FPG) was the most common Grade 3 laboratory abnormality according to CTC criteria (14.7% of patients) during Phase III study G2304; no Grade 4 laboratory abnormalities were observed. Mean increases in HbA1c were less pronounced in patients with normal glycemia at baseline compared to those with prediabetes or diabetes. Mean FPG levels often increased during the first month of treatment, then decreased and stabilized in subsequent months. Increases in FPG and HbA1c were dose-dependent, and values generally decreased after discontinuation of intramuscular pasireotide, although they remained above baseline. The overall incidence of adverse reactions related to hyperglycemia was 75.3% (all grades) and 22.7% (Grade 3 CTC). Adverse reactions such as hyperglycemia and diabetes mellitus led to study discontinuation in 3 (2.0%) and 4 patients (2.7%), respectively.
Elevations in fasting plasma glucose and HbA1c levels observed after intramuscular pasireotide administration were reversible upon discontinuation of the drug.
Monitoring of blood glucose levels is recommended in patients receiving Signifor LAR.
Gastrointestinal disorders. Gastrointestinal disorders were commonly reported during treatment with Signifor LAR. These events were generally mild in severity, did not require intervention, and resolved with continued treatment. In acromegaly patients, gastrointestinal disorders were less frequent in those inadequately controlled compared to treatment-naïve patients.
Injection site reactions. During Phase III studies, injection site reactions (e.g., injection site pain, injection site discomfort) were predominantly Grade 1 or 2 in severity. The frequency of these events was highest during the first 3 months of treatment. In studies involving acromegaly patients, injection site reactions were similar between intramuscular pasireotide and intramuscular octreotide, and were less frequent in inadequately controlled patients compared to treatment-naïve patients.
QT interval prolongation. In study C2305 involving acromegaly patients, the proportion of patients with new-onset notable QT/QTc intervals was comparable between the intramuscular pasireotide and intramuscular octreotide groups prior to the crossover phase, with several notable abnormal values. QTcF > 480 ms was observed in 3 patients versus 2 patients in the intramuscular pasireotide and intramuscular octreotide groups, respectively, and QTcF prolongation > 60 ms from baseline was reported in 2 patients versus 1 patient in the respective groups. In study C2402, the only notable deviation was a QTcF > 480 ms in 1 patient in the intramuscular pasireotide 40 mg group. During study G2304 involving Cushing’s disease patients, QTcF > 480 ms was observed in 2 patients. QTcF > 500 ms was not observed in any of the main studies.
Hepatic enzymes. Transient elevations in hepatic enzymes have been reported with somatostatin analogs, and were also observed in patients receiving pasireotide in clinical trials. Elevations were mainly asymptomatic, mild in degree, and reversible with continued treatment. A few cases of concurrent elevations in ALT > 3 × ULN and bilirubin > 2 × ULN were observed with subcutaneous administration, but not in patients receiving intramuscular pasireotide. All cases of concurrent elevations occurred within 10 days of treatment initiation. Patients recovered without clinical sequelae, and liver function test results returned to baseline after discontinuation of treatment.
Monitoring of hepatic enzyme levels before and during treatment with Signifor LAR is recommended according to clinical standards.
Pancreatic enzymes. Asymptomatic elevations in lipase and amylase levels were observed in patients receiving pasireotide in clinical trials. Elevations were predominantly mild in severity and reversible with continued treatment. Pancreatitis is a potential adverse reaction associated with somatostatin analogs, particularly due to the link between cholelithiasis and acute pancreatitis.
Incompatibility.
Due to lack of compatibility studies, this medicinal product should not be mixed with other medicinal products.
Shelf life. 3 years.
Storage conditions. Store in the original packaging at 2–8 °C. Do not freeze. Keep out of the reach of children.
Packaging. Powder in a 6 mL amber glass vial, stoppered with a grey rubber stopper and sealed with a grey (for 20 mg dose), red (for 40 mg dose), or orange (for 60 mg dose) flip-off aluminum cap, supplied with: solvent in a 3 mL pre-filled syringe made of colorless glass with 2 grey rubber stoppers, finger flange, plunger, and cap; one needle and one adapter, all contained in a cardboard box.
Prescription status. Prescription only.
Manufacturer. Recordati Rare Diseases.
Manufacturer’s location and address of its place of business.
Eco River Park, 30 Rue des Peupliers, Nanterre, 92000, France;
Immeuble Le Wilson, 70 Avenue du Général de Gaulle, Puteaux, 92800, France.