Signifor LAR
UcraniaContenido
INSTRUCCIONES PARA USO MÉDICO DEL MEDICAMENTO SIGIFOR LAR (SIGNIFOR® LAR®)
Composición:
Principio activo: pasireotida;
1 frasco con polvo para suspensión inyectable contiene 20 mg, 40 mg o 60 mg de pasireotida (en forma de pasireotida pamoato).
Excipientes:
polvo: poli (D,L-láctido-co-glicólido) (50-60:40-50), poli (D,L-láctido-co-glicólido) (50:50);
disolvente: 1 jeringa precargada con disolvente de 2 ml contiene: carmelosa sódica, manitol (E421), poloxámero 188, agua para preparaciones inyectables.
Forma farmacéutica. Polvo para suspensión inyectable.
Principales propiedades físico-químicas:
polvo: polvo de color de ligeramente amarillento a amarillento;
disolvente: solución transparente incolora a amarillenta o ligeramente marrón.
Grupo farmacoterapéutico. Hormonas hipofisarias, hipotalámicas y sus análogos. Somatostatina y sus análogos. Código ATC H01C B05.
Propiedades farmacológicas.
Farmacodinámica.
Mecanismo de acción. Pasireótido es un ciclexapeptido, análogo inyectable de la somatostatina. Al igual que los péptidos hormonales naturales somatostatina-14 y somatostatina-28 (también conocidos como factor que inhibe la liberación de la hormona del crecimiento [SRIF]) y otros análogos de la somatostatina, el pasireótido ejerce su acción farmacológica mediante la unión a los receptores de somatostatina. Se conocen cinco subtipos de receptores humanos de somatostatina: hsst1, 2, 3, 4 y 5. En condiciones fisiológicas normales, estos subtipos de receptores se expresan en diferentes tejidos. Los análogos de la somatostatina se unen a los receptores hsst con diferente potencia (ver tabla a continuación). El pasireótido se une con alta afinidad a cuatro de los cinco receptores hsst.
Afinidad de unión de la somatostatina (SRIF-14), pasireótido, octreótido y lanreótido a los cinco subtipos humanos de receptores sst (hsst1–5).
| Medicamento |
hsst1 |
hsst2 |
hsst3 |
hsst4 |
hsst5 |
| Somatostatina (SRIF-14) |
0,93 ± 0,12 |
0,15 ± 0,02 |
0,56 ± 0,17 |
1,5 ± 0,4 |
0,29 ± 0,04 |
| Pasireotido |
9,3 ± 0,1 |
1,0 ± 0,1 |
1,5 ± 0,3 |
> 100 |
0,16 ± 0,01 |
| Octreótido |
280 ± 80 |
0,38 ± 0,08 |
7,1 ± 1,4 |
>1000 |
6,3 ± 1,0 |
| Lanreotido |
180 ± 20 |
0,54 ± 0,08 |
14 ± 9 |
230 ± 40 |
17 ± 5 |
Los resultados se presentan como media ± error estándar de la media (SEM) del IC50, expresado en nmol/l.
Efectos farmacodinámicos. Los receptores de somatostatina se expresan en muchos tejidos, especialmente en tumores neuroendocrinos, en cuya presencia se observa una secreción excesiva de hormonas, incluyendo la hormona del crecimiento (GH) en acromegalia y la hormona adrenocorticotropa (ACTH) en la enfermedad de Cushing.
Los estudios in vitro mostraron que las células tumorales corticotropas obtenidas de pacientes con enfermedad de Cushing presentan una marcada expresión de hsst5, mientras que los receptores de otros subtipos o no se expresan o lo hacen de forma mínima. Pasireótido se une y activa cuatro de los cinco hsst, especialmente hsst5, en adenomas corticotropos productores de ACTH, provocando la inhibición de la secreción de ACTH.
Debido a su amplio perfil de unión a los receptores de somatostatina, pasireótido es capaz de estimular los subtipos de receptores hsst2 y hsst5, necesarios para la inhibición de la secreción de GH y del factor de crecimiento similar a la insulina (IGF-1), por lo que puede ser eficaz en el tratamiento de la acromegalia.
Metabolismo de la glucosa. En un estudio aleatorizado, doble ciego, realizado con voluntarios sanos, el desarrollo de hiperglucemia tras la administración subcutánea de pasireótido en dosis de 0,6 mg y 0,9 mg dos veces al día se asoció con una reducción significativa en la secreción de insulina y de incretinas (específicamente, péptido glucagón-símil-1 (GLP-1) y polipéptido insulínico dependiente de glucosa (GIP)). Pasireótido no afectó la sensibilidad a la insulina.
Población pediátrica. La Agencia Europea de Medicamentos ha eximido al medicamento Signifor LAR del requisito de presentar resultados de estudios en niños de todas las subcategorías con acromegalia y gigantismo hipofisario, así como con enfermedad de Cushing dependiente de la hipófisis, hipersecreción hipofisaria de ACTH e hipercortisolismo dependiente de la hipófisis (para información sobre el uso en niños, véase la sección «Posología y forma de administración»).
Farmacocinética.
Pasireótido para administración intramuscular se presenta en forma de microesferas de acción prolongada. Tras una inyección única, la concentración plasmática de pasireótido muestra una liberación inicial «en bolo» el primer día de la inyección, seguida de una disminución de la concentración entre los días 2 y 7. Posteriormente, se observó un aumento gradual hasta alcanzar la concentración máxima aproximadamente al día 21 y una fase de disminución progresiva durante las siguientes semanas, junto con la fase final de degradación de la matriz polimérica de la formulación.
Absorción. La biodisponibilidad relativa de pasireótido por vía intramuscular en comparación con la administración subcutánea es completa. No se han realizado estudios para evaluar la biodisponibilidad absoluta de pasireótido en humanos.
Reparto. En voluntarios sanos, tras la administración intramuscular, pasireótido se distribuye ampliamente con un gran volumen de distribución (Vz/F > 100 litros). La distribución entre células sanguíneas y plasma es independiente de la concentración y muestra que pasireótido se localiza principalmente en el plasma sanguíneo (91 %). La unión a proteínas plasmáticas es moderada (88 %) e independiente de la concentración.
Basándose en datos de estudios in vitro, pasireótido es sustrato del transportador de eflujo proteína P. Según datos de estudios in vitro, pasireótido no es sustrato del transportador de eflujo BCRP (proteína de resistencia al cáncer de mama) ni de los transportadores de influxo OCT1 (transportador de catiónicos orgánicos 1), OATP (polipéptido transportador de aniones orgánicos) 1B1, 1B3 o 2B1. A las dosis terapéuticas, pasireótido tampoco es inhibidor de UGT1A1, OATP1B1 ni 1B3, OAT1 ni OAT3, OCT1 ni OCT2, P-gp, BCRP, MRP2 ni BSEP.
Biotransformación. Pasireótido es altamente estable desde el punto de vista metabólico. Los datos de estudios in vitro indican que pasireótido no es sustrato, inhibidor ni inductor de ninguna de las principales enzimas CYP450. En voluntarios sanos, pasireótido se detecta principalmente sin cambios en el plasma, orina y heces.
Eliminación. Pasireótido se elimina principalmente por aclaramiento hepático (excreción biliar), con una participación mínima de la vía renal. En un estudio ADME (absorción, distribución, metabolismo y excreción) en humanos, se recuperó el 55,9 ± 6,63 % de la dosis radiactiva durante los primeros 10 días tras la administración, incluyendo el 48,3 ± 8,16 % de la radiactividad en heces y el 7,63 ± 2,03 % en orina.
El aclaramiento (CL/F) de pasireótido intramuscular en voluntarios sanos es de media entre 4,5 y 8,5 l/h. Según los resultados del análisis farmacocinético poblacional, el CL/F medido fue aproximadamente de 4,8 a 6,5 l/h en pacientes con enfermedad de Cushing típica y de aproximadamente 5,6 a 8,2 l/h en pacientes con acromegalia típica.
Linealidad y dependencia del tiempo. El estado de equilibrio farmacocinético tras la administración intramuscular de pasireótido se alcanza tras 3 meses. Tras varias dosis mensuales en el rango de 10 mg a 60 mg cada 4 semanas, pasireótido por vía intramuscular muestra un efecto farmacocinético aproximadamente proporcional a la dosis en pacientes.
Categorías especiales de pacientes.
Población pediátrica. No se han realizado estudios en niños.
Pacientes con alteración de la función renal. El aclaramiento renal desempeña un papel mínimo en la eliminación de pasireótido en humanos. En un estudio clínico, una dosis subcutánea única de pasireótido de 900 µg en pacientes con disfunción renal, con alteración leve, moderada o grave de la función renal o enfermedad renal en estadio terminal (ESRD), no tuvo un impacto significativo en la exposición plasmática total de pasireótido. La exposición plasmática de pasireótido no unido (AUCinf,u) aumentó en pacientes con alteración de la función renal (leve: 33 %; moderada: 25 %; grave: 99 %; ESRD: 143 %) en comparación con los pacientes del grupo control.
Pacientes con alteración de la función hepática. No se han realizado estudios clínicos con pasireótido intramuscular en pacientes con alteración de la función hepática. En un estudio clínico con administración subcutánea única de pasireótido en pacientes con alteración de la función hepática, se observaron diferencias estadísticamente significativas en pacientes con alteración hepática moderada y grave (clases B y C según Child-Pugh). En estos pacientes, el AUCinf aumentó un 60 % y un 79 %, la Cmax aumentó un 67 % y un 69 %, y el CL/F disminuyó un 37 % y un 44 %, respectivamente.
Pacientes de edad avanzada (≥ 65 años). La edad no es un parámetro significativo en el análisis farmacocinético poblacional de pacientes.
Características demográficas. El análisis farmacocinético poblacional de la administración intramuscular de pasireótido indica que la raza y el sexo del paciente no influyen en los parámetros farmacocinéticos del medicamento. El peso corporal tuvo un efecto leve en la farmacocinética en un estudio con pacientes previamente no tratados, pero no tuvo efecto en un estudio con pacientes con enfermedad insuficientemente controlada. Las mujeres con acromegalia mostraron una mayor exposición (32 % y 51 %) en comparación con los hombres en estudios con pacientes no tratados previamente y con enfermedad insuficientemente controlada, respectivamente; estas diferencias no fueron clínicamente relevantes según los datos de eficacia y seguridad.
Características clínicas.
Indicaciones.
Tratamiento de pacientes adultos con acromegalia en quienes la intervención quirúrgica no es óptima o ha fracasado y en quienes no se logra un control adecuado con otro análogo de la somatostatina.
Tratamiento de pacientes adultos con enfermedad de Cushing en quienes la intervención quirúrgica no es óptima o ha fracasado.
La dosis de 60 mg se utiliza únicamente para el tratamiento de la acromegalia.
Contraindicaciones.
Hipersensibilidad al principio activo o a cualquiera de los excipientes del medicamento. Alteración grave de la función hepática (Clase C según Child-Pugh).
Interacción con otros medicamentos y otras formas de interacción.
Interacciones farmacocinéticas esperadas que afectan al pasireótido. Se estudió el efecto del inhibidor de la glucoproteína P, verapamilo, sobre la farmacocinética del pasireótido tras administración subcutánea en un estudio de interacción con medicamentos realizado en voluntarios sanos. No se observaron cambios en la farmacocinética (velocidad ni grado de absorción) del pasireótido.
Interacciones farmacocinéticas esperadas que afectan a otros medicamentos. El pasireótido puede reducir la biodisponibilidad relativa de la ciclosporina. Puede ser necesaria la ajuste de la dosis de ciclosporina cuando se administre concomitantemente con pasireótido para mantener concentraciones terapéuticas.
Interacciones farmacodinámicas esperadas. Medicamentos que prolongan el intervalo QT.
El pasireótido debe administrarse con precaución a pacientes que toman simultáneamente medicamentos que prolongan el intervalo QT, tales como antiarrítmicos de clase Ia (por ejemplo, quinidina, procainamida, disopiramida), antiarrítmicos de clase III (por ejemplo, amiodarona, dronedarona, sotalol, dofetilida, ibutilida), ciertos antibacterianos (eritromicina intravenosa, inyecciones de pentamidina, claritromicina, moxifloxacino), ciertos antipsicóticos (por ejemplo, clorpromacina, tiotixeno, flufenacina, pimozida, haloperidol, tiaprido, amisulprida, sertindol, metadona), ciertos antihistamínicos (por ejemplo, terfenadina, astemizol, mizolastina), medicamentos antimaláricos (por ejemplo, cloroquina, halofantrina, lumefantrina), y ciertos antifúngicos (ketoconazol, excepto en champú).
Medicamentos que causan bradicardia. Se recomienda el monitoreo clínico del ritmo cardíaco, especialmente al inicio del tratamiento, en pacientes que reciben pasireótido junto con medicamentos que causan bradicardia, tales como betabloqueantes (por ejemplo, metoprolol, carteolol, propranolol, sotalol), inhibidores de la colinesterasa (por ejemplo, rivastigmina, fisostigmina), ciertos bloqueadores de canales de calcio (por ejemplo, verapamilo, diltiazem, bepridilo) y ciertos antiarrítmicos.
Insulina y medicamentos antidiabéticos. Puede ser necesaria la corrección de la dosis (disminución o aumento) de insulina y medicamentos antidiabéticos (por ejemplo, metformina, liraglutida, vildagliptina, nateglinida) cuando se administren concomitantemente con pasireótido.
Características de uso.
Alteraciones del metabolismo de la glucosa. Se han observado con frecuencia cambios en los niveles de glucosa en sangre en voluntarios sanos y pacientes tras el tratamiento con pasireótido. Se han notificado hiperglucemia e, más raramente, hipoglucemia en pacientes que participaron en estudios clínicos con pasireótido (véase la sección «Reacciones adversas»).
Los pacientes que desarrollaron hiperglucemia generalmente respondieron a la terapia antidiabética. La reducción de la dosis o la interrupción del tratamiento con pasireótido debido a hiperglucemia fueron eventos infrecuentes durante los estudios clínicos con pasireótido.
El desarrollo de hiperglucemia está relacionado con la disminución de la secreción de insulina y de hormonas incretinas (específicamente, el péptido glucagón-simil 1 [GLP-1] y el polipéptido insulinotrópico dependiente de glucosa [GIP]).
Se debe evaluar el estado glucémico (glucosa en plasma en ayunas/hemoglobina A1c [GPA/HA1c]) antes de iniciar el tratamiento con pasireótido. El monitoreo de GPA/HbA1c durante el tratamiento debe realizarse según las recomendaciones disponibles. El autocontrol del nivel de glucosa en sangre y/o la evaluación de GPA debe realizarse semanalmente durante los primeros 3 meses y posteriormente de forma periódica, según la práctica clínica habitual, así como durante las primeras 4-6 semanas tras cualquier aumento de dosis. Además, se debe realizar monitoreo de GPA a las 4 semanas y de hemoglobina A1c a los 3 meses tras la finalización del tratamiento.
Si un paciente desarrolla hiperglucemia durante el tratamiento con el medicamento Signifor LAR, se recomienda iniciar o ajustar la terapia antidiabética según las recomendaciones disponibles para controlar la hiperglucemia. Si la hiperglucemia no controlada persiste a pesar del tratamiento farmacológico adecuado, se debe considerar reducir la dosis de Signifor LAR o interrumpir el tratamiento (véase la sección «Interacción con otros medicamentos y otras formas de interacción»).
Se han notificado casos poscomercialización de cetoacidosis con el uso del medicamento Signifor LAR en pacientes con y sin antecedentes de diabetes mellitus. Los pacientes que presenten signos y síntomas compatibles con acidosis metabólica grave deben ser evaluados para descartar el desarrollo de cetoacidosis, independientemente de si tienen o no antecedentes de diabetes mellitus.
En pacientes con mal control glucémico (definido como un valor de HbA1c > 8 % a pesar de la terapia antidiabética), se debe intensificar el control de la diabetes y el monitoreo antes y durante el tratamiento con pasireótido.
Pruebas hepáticas. En pacientes que recibieron pasireótido, generalmente se observó un ligero aumento transitorio en los niveles de aminotransferasas. También se han notificado casos raros de aumento simultáneo de ALT (alanina aminotransferasa) por encima de 3 × LSN (límite superior normal) y bilirrubina por encima de 2 × LSN (véase la sección «Reacciones adversas»).
El monitoreo de la función hepática debe realizarse según indicaciones clínicas. Se recomienda controlar la función hepática antes de iniciar la administración intramuscular de pasireótido y a las 2-3 semanas de tratamiento, y posteriormente mensualmente durante los primeros 3 meses tras el inicio del tratamiento.
Los pacientes con aumento de los niveles de transaminasas requieren un monitoreo frecuente de la función hepática hasta que los valores regresen a los niveles previos al tratamiento. El tratamiento con pasireótido debe interrumpirse si el paciente desarrolla ictericia u otros signos de disfunción hepática clínicamente significativa, si hay un aumento persistente de AST (aspartato aminotransferasa) o ALT por encima de 5 × LSN, o si ALT o AST superan 3 × LSN simultáneamente con un aumento de bilirrubina superior a 2 × LSN. Tras la interrupción del tratamiento con pasireótido, se debe continuar el control del paciente hasta la desaparición de los síntomas. No se debe reiniciar el tratamiento si se sospecha que las alteraciones de la función hepática están relacionadas con pasireótido.
Eventos relacionados con el sistema cardiovascular. Se han notificado casos de bradicardia durante el uso de pasireótido. Se recomienda un monitoreo cuidadoso en pacientes con enfermedades cardíacas y/o factores de riesgo de bradicardia, como bradicardia clínicamente significativa o infarto de miocardio reciente, bloqueos cardíacos de alto grado, insuficiencia cardíaca congestiva (clase III o IV según la clasificación de la New York Heart Association - NYHA), angina inestable, taquicardia ventricular sostenida o fibrilación ventricular. Puede ser necesario ajustar la dosis de medicamentos, por ejemplo betabloqueantes, bloqueadores de canales de calcio o fármacos destinados al control del equilibrio electrolítico.
En dos estudios con voluntarios sanos se demostró que pasireótido prolongaba el intervalo QT en el ECG. El significado clínico de esta prolongación no se conoce. En estudios clínicos de fase III en pacientes con acromegalia no se observaron diferencias clínicamente significativas respecto a la prolongación del intervalo QT entre la administración intramuscular de pasireótido y los análogos de somatostatina utilizados como comparador activo. Todos los eventos relacionados con la prolongación del intervalo QT fueron transitorios y desaparecieron sin intervención terapéutica.
No se han observado episodios de taquicardia tipo torsade de pointes en ningún estudio clínico con pasireótido.
Pasireótido debe usarse con precaución y considerando la relación beneficio-riesgo en pacientes con factores de riesgo de prolongación del intervalo QT, tales como:
- Síndrome congénito de prolongación del intervalo QT;
- Enfermedades cardíacas no controladas o significativas, incluyendo infarto de miocardio reciente, insuficiencia cardíaca congestiva, angina inestable o bradicardia clínicamente significativa;
- Administración concomitante de medicamentos antiarrítmicos u otros fármacos con capacidad conocida de prolongar el intervalo QT;
- Hipokalemia y/o hipomagnesemia.
Antes de iniciar el tratamiento con el medicamento Signifor LAR, se recomienda realizar un ECG basal. Es deseable realizar monitoreo del efecto sobre el intervalo QTc a los 21 días tras el inicio del tratamiento y posteriormente según indicaciones clínicas. La hipokalemia y/o hipomagnesemia deben corregirse antes de iniciar el tratamiento con Signifor LAR y debe realizarse monitoreo periódico adecuado durante el tratamiento.
Hipocortisolismo. La supresión de la secreción de ACTH (hormona adrenocorticotrópica) puede provocar hipocortisolismo en pacientes que reciben Signifor LAR. Por lo tanto, es necesario realizar monitoreo e instruir a los pacientes sobre los signos y síntomas asociados con hipocortisolismo (por ejemplo, debilidad, fatiga, anorexia, náuseas, vómitos, hipotensión arterial, hiperaldosteronismo, hiponatremia, hipoglucemia). En caso de hipocortisolismo confirmado, puede ser necesaria una terapia sustitutiva temporal con esteroides exógenos (glucocorticoides) y/o la reducción de la dosis o una pausa en el tratamiento con Signifor LAR. Una disminución rápida de los niveles de cortisol puede estar asociada con una disminución en el recuento de leucocitos.
Vesícula biliar y eventos relacionados. La litiasis biliar (enfermedad por cálculos biliares) es una reacción adversa establecida asociada con el uso de análogos de somatostatina y se ha notificado frecuentemente en estudios clínicos con pasireótido. Se han notificado casos poscomercialización de colangitis con el uso del medicamento Signifor LAR, la mayoría de los cuales se informaron como complicaciones de la enfermedad por cálculos biliares. Por lo tanto, se recomienda realizar ecografía de la vesícula biliar antes y con intervalos de 6 y 12 meses durante el tratamiento con Signifor LAR. La presencia de cálculos biliares en pacientes que toman Signifor LAR generalmente tiene un curso asintomático; la presencia de cálculos biliares con manifestaciones clínicas debe tratarse según los estándares de la práctica clínica.
Hormonas hipofisarias. Dado que el efecto farmacológico de pasireótido imita al de la somatostatina, no puede descartarse la posibilidad de inhibición de hormonas hipofisarias, además de GH y/o IGF-1, en pacientes con acromegalia, y de ACTH/cortisol en pacientes con enfermedad de Cushing. Por lo tanto, es necesario realizar monitoreo de la función hipofisaria (por ejemplo, niveles de TSH/hormona tiroidea libre T4, hormona del crecimiento) antes y periódicamente durante el tratamiento con Signifor LAR, según los estándares clínicos.
Efecto sobre la función reproductiva en mujeres. El efecto terapéutico de reducción de los niveles de hormona del crecimiento y normalización de la concentración del factor de crecimiento similar a la insulina 1 (IGF-1) en mujeres con acromegalia, y la reducción o normalización de los niveles de cortisol en suero en mujeres con enfermedad de Cushing, puede potencialmente restablecer la función reproductiva. A las pacientes en edad fértil se les debe recomendar el uso de métodos anticonceptivos adecuados, si es necesario, durante el tratamiento con Signifor LAR.
Alteraciones de la coagulación sanguínea. Los pacientes con valores significativamente elevados del tiempo de protrombina (TP) y del tiempo de tromboplastina parcial (TTPa), o pacientes que reciben derivados de cumarina o anticoagulantes basados en heparina, fueron excluidos de los estudios clínicos con pasireótido, ya que no se ha establecido la seguridad de la combinación con estos anticoagulantes. Si no puede evitarse la administración concomitante de derivados de cumarina o anticoagulantes basados en heparina durante la administración intramuscular de Signifor LAR, se debe realizar un control regular de los parámetros de coagulación (TP y TTPa) y ajustar la dosis del anticoagulante según sea necesario.
Alteraciones de la función renal. Debido al aumento de la exposición al medicamento Signifor LAR no unido, debe usarse con precaución en pacientes con insuficiencia renal grave o enfermedad renal en fase terminal.
Contenido de sodio. Este medicamento contiene menos de 1 mmol de sodio (23 mg) por dosis recomendada, es decir, prácticamente carece de sodio.
Uso durante el embarazo o la lactancia.
Embarazo.
Los datos sobre el uso de pasireótido en mujeres embarazadas son limitados. Los estudios en animales con administración subcutánea de pasireótido mostraron toxicidad reproductiva. Pasireótido no se recomienda para su uso en mujeres embarazadas ni en mujeres en edad fértil que no utilicen métodos anticonceptivos.
Lactancia.
No se sabe si pasireótido se excreta en la leche materna. Los datos disponibles de estudios con administración subcutánea de pasireótido en ratas mostraron que pasireótido atraviesa la leche. Durante el tratamiento con el medicamento Signifor LAR, se debe interrumpir la lactancia.
Fertilidad.
Los estudios con administración subcutánea de pasireótido en ratas mostraron efectos sobre parámetros reproductivos en hembras. La relevancia clínica de estos efectos en humanos es desconocida.
Capacidad para afectar la velocidad de reacción al conducir vehículos o manejar maquinaria.
Signifor LAR puede tener un efecto insignificante sobre la capacidad de conducir vehículos o manejar maquinaria. Se debe aconsejar a los pacientes tener precaución al conducir vehículos o trabajar con maquinaria si experimentan fatiga, mareo o cefalea durante el tratamiento con Signifor LAR.
Vía de administración y dosis.
Dosificación.
Acromegalia.
La dosis recomendada inicial para el tratamiento de la acromegalia es de 40 mg de pasireotida cada 4 semanas.
La dosis puede aumentarse hasta un máximo de 60 mg en pacientes cuyos niveles de la hormona del crecimiento (GH) y/o del factor de crecimiento similar a la insulina 1 (IGF-1) no estén suficientemente controlados tras 3 meses de tratamiento con la dosis de 40 mg.
Puede ser necesario un descenso temporal de la dosis para controlar posibles reacciones adversas o una respuesta excesiva al tratamiento (IGF-1 < límite inferior de la normalidad). La dosis puede reducirse temporal o permanentemente.
Enfermedad de Cushing.
La dosis inicial recomendada para el tratamiento de la enfermedad de Cushing es de 10 mg de pasireotida administrados como inyección intramuscular profunda cada 4 semanas.
Tras el primer mes de tratamiento y periódicamente después, debe evaluarse a los pacientes para determinar el beneficio clínico. El ajuste posológico debe realizarse cada 2-4 meses según la respuesta terapéutica y la tolerabilidad. La dosis máxima de Signifor LAR en la enfermedad de Cushing es de 40 mg cada 4 semanas. Si no se observa beneficio clínico, debe considerarse la posibilidad de interrumpir el tratamiento con el medicamento.
El tratamiento de posibles reacciones adversas o un efecto terapéutico excesivo (niveles de cortisol < límite inferior de la normalidad) puede requerir una reducción de la dosis, la suspensión temporal o la interrupción definitiva del tratamiento con Signifor LAR.
Transición de la administración subcutánea a la intramuscular en la enfermedad de Cushing.
No existen datos de estudios clínicos sobre la transición de la administración subcutánea a la intramuscular de pasireotida. Si dicha transición es necesaria, la dosis inicial recomendada para el tratamiento de la enfermedad de Cushing es de 10 mg de pasireotida como inyección intramuscular profunda cada 4 semanas. Debe vigilarse al paciente para evaluar la respuesta terapéutica y la tolerabilidad; también puede ser necesaria una ajuste adicional de la dosis.
Omisión de dosis.
Si se omite una dosis de Signifor LAR, la inyección omitida debe administrarse tan pronto como sea posible. La siguiente dosis debe administrarse a los 4 semanas tras la inyección realizada para restablecer el esquema posológico de una vez cada 4 semanas.
Pacientes con características especiales.
Pacientes de edad avanzada (≥ 65 años). Los datos sobre el uso de Signifor LAR en pacientes de 65 años o más son limitados, pero no hay evidencia que sugiera la necesidad de ajuste posológico en estos pacientes.
Alteraciones renales. No se requiere ajuste posológico en pacientes con alteración de la función renal.
Alteraciones hepáticas. No se requiere ajuste posológico en pacientes con alteración hepática leve (Clase A según Child-Pugh).
La dosis inicial recomendada en acromegalia para pacientes con alteración hepática moderada (Clase B según Child-Pugh) es de 20 mg cada 4 semanas. La dosis máxima recomendada en estos pacientes es de 40 mg cada 4 semanas.
Enfermedad de Cushing: la dosis inicial recomendada en la enfermedad de Cushing en pacientes con alteración hepática moderada (Clase B según la escala de Child-Pugh) es de 10 mg cada 4 semanas, y la dosis máxima recomendada en estos pacientes es de 20 mg cada 4 semanas.
Signifor LAR está contraindicado en pacientes con alteración hepática grave (Clase C según Child-Pugh).
Vía de administración.
Signifor LAR debe administrarse mediante inyección intramuscular profunda por un profesional sanitario debidamente capacitado. La suspensión del medicamento debe prepararse inmediatamente antes de la administración.
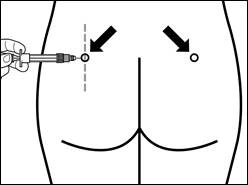
Los sitios para las inyecciones intramusculares repetidas deben alternarse: músculo glúteo izquierdo y derecho.
Componentes del kit de inyección:
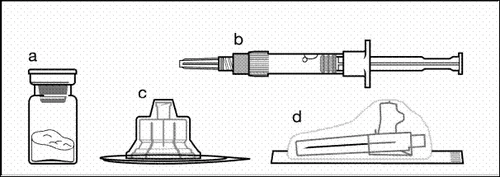
a un frasco con polvo;
b una jeringa precargada con disolvente;
c un adaptador para frasco para la preparación de la suspensión;
d una aguja de seguridad para inyección (20G × 1,5ʺ).
Para la correcta preparación de la suspensión del medicamento Signifor LAR, deben seguirse cuidadosamente las instrucciones que se indican a continuación.
| Paso 1 Saque Signifor LAR del refrigerador. Atención El juego de inyección debe alcanzar la temperatura ambiente. Deje el juego de inyección a temperatura ambiente durante al menos 30 minutos (pero no más de 24 horas). Nota: si el juego de inyección no se utiliza dentro de las 24 horas, puede volver a guardarse en el refrigerador. |
|
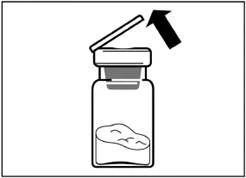
| Paso 2 Retire la tapa de plástico del frasco y limpie el tapón de goma del frasco con una toallita de alcohol. |
|
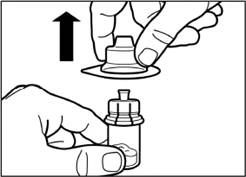
| Retire la película del envoltorio que contiene el adaptador para frasco, pero no saque el adaptador de este envoltorio. Sujetando el adaptador para frasco por el envoltorio, colóquelo sobre la parte superior del frasco e introdúzcalo completamente hacia abajo hasta que encaje (se oirá un clic característico). |
|
| Retire verticalmente hacia arriba el recipiente de plástico del adaptador para frasco. |
|
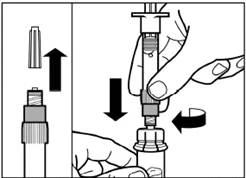
| Paso 3 Retire la tapa de la jeringa precargada con el disolvente y enrosque la jeringa en el adaptador para frasco. |
|
| Presione lentamente el émbolo hacia abajo hasta la posición final para transferir todo el disolvente al frasco. |
|
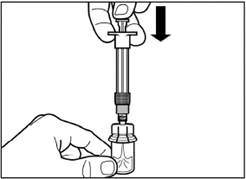
| Paso 4 Atención: manteniendo el émbolo presionado, mueva cuidadosamente el frasco en plano horizontal durante al menos 30 segundos hasta que se forme una suspensión homogénea. Si el polvo no se hubiera suspendido completamente, mezcle nuevamente con cuidado el contenido moviendo el frasco en plano horizontal durante 30 segundos. |
|
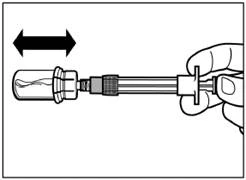
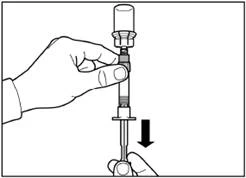
| Paso 5 Gire el frasco con la jeringa acoplada boca abajo y lentamente tire del émbolo hacia abajo para transferir el contenido del frasco a la jeringa. |
|
| Desenrosque la jeringa del adaptador del frasco. |
|
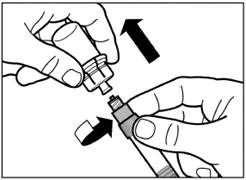
| Paso 6 Coloque la aguja de seguridad para inyección en la jeringa. |
|
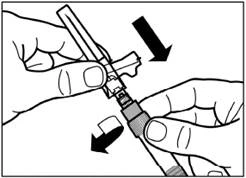
| Retire el protector de la aguja tirando de él hacia arriba a lo largo de la línea de la aguja. Para evitar la sedimentación, puede mantener la homogeneidad de la suspensión agitando suavemente la jeringa. Dele ligeros golpes suaves a la jeringa para que las burbujas de aire visibles suban hacia arriba, luego elimínelas presionando suavemente el émbolo. La suspensión ya está lista para su uso inmediato. |
|
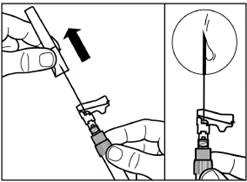
| Paso 7 Signifor LAR solo puede administrarse por vía intramuscular profunda. Prepare el lugar de inyección limpiándolo con una torunda de alcohol. Introduzca completamente la aguja en el músculo glúteo derecho o izquierdo, en un ángulo de 90° respecto a la superficie de la piel. Tire ligeramente del émbolo hacia atrás para asegurarse de que la aguja no ha entrado en un vaso sanguíneo (si la aguja ha entrado en un vaso sanguíneo, inyéctela en otro lugar). Presione lentamente el émbolo hasta que la jeringa quede vacía. Retire la aguja del lugar de inyección y active el mecanismo de seguridad (como se muestra en la figura inferior). |
|
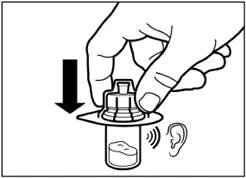
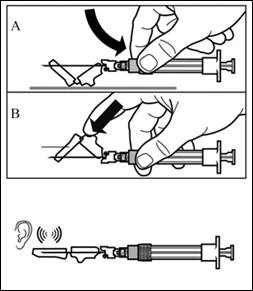
| Paso 8 Active el mecanismo de protección de la aguja mediante uno de los métodos indicados a continuación:
Un clic audible confirma la activación correcta del mecanismo de protección. Deseche inmediatamente el vial y la jeringa con la aguja en un contenedor para objetos punzantes. |
|
Niños.
No se han estudiado la seguridad y la eficacia del medicamento Signifor LAR en niños y adolescentes (de 0 a 18 años de edad). No existen datos disponibles.
Sobredosis.
En caso de sobredosis, se recomienda iniciar un tratamiento de soporte adecuado, determinado por el estado clínico del paciente, que debe continuar hasta la desaparición de los síntomas.
Reacciones adversas.
Resumen del perfil de seguridad. El perfil de seguridad tras la administración intramuscular de pasireótido es coherente con el observado con los medicamentos de la clase de análogos de la somatostatina, excepto por una mayor frecuencia e intensidad de hiperglucemia observadas tras la administración intramuscular de pasireótido. El perfil de seguridad tras la administración intramuscular de pasireótido es en gran parte similar entre los diferentes indicaciones: acromegalia y enfermedad de Cushing.
Acromegalia. En acromegalia, la evaluación de la seguridad se realizó en 491 pacientes que recibieron pasireótido (419 pacientes recibieron pasireótido por vía intramuscular y 72 recibieron pasireótido por vía subcutánea) durante estudios de Fase I, II y III.
Las reacciones adversas más frecuentes (frecuencia ≥ 1/10), según datos reunidos de seguridad de los estudios de Fase III C2305 y C2402, fueron (en orden descendente): diarrea (más frecuente en el estudio C2305), litiasis biliar, hiperglucemia (más frecuente en el estudio C2402) y diabetes mellitus. Las reacciones adversas de Grado 3 y 4 según los Criterios Comunes de Toxicidad (CCT) fueron principalmente relacionadas con la hiperglucemia.
Enfermedad de Cushing. En la enfermedad de Cushing, la evaluación de seguridad de la formulación para administración intramuscular se basó en datos recopilados de 150 pacientes que recibieron pasireótido durante el estudio de Fase III G2304 (duración media de exposición: 57 semanas). Los pacientes fueron aleatorizados en una proporción 1:1 para recibir dosis iniciales de 10 mg o 30 mg de pasireótido, con posibilidad de aumentar la dosis hasta la dosis máxima de 40 mg cada 28 días. Las reacciones adversas más frecuentes (frecuencia ≥ 1/10) durante el estudio de Fase III G2304 fueron hiperglucemia, diarrea, litiasis biliar y diabetes mellitus. La frecuencia y gravedad de las reacciones adversas aumentaron con el incremento de la dosis inicial a 30 mg, aunque no fue uniforme para todas las reacciones adversas.
Las reacciones adversas incluyen eventos notificados durante los estudios fundamentales con la formulación intramuscular en pacientes con acromegalia y enfermedad de Cushing. Las reacciones adversas se presentan clasificadas por sistemas orgánicos según el Diccionario Médico para la Actividad Regulatoria (MedDRA). Dentro de cada sistema orgánico, las reacciones adversas se ordenan por frecuencia. Dentro de cada grupo de frecuencia, las reacciones adversas se presentan en orden descendente de gravedad. La frecuencia se define como: muy frecuente (≥ 1/10); frecuente (≥ 1/100 a < 1/10); poco frecuente (≥ 1/1000 a < 1/100); desconocida (no puede evaluarse debido a la limitación de los datos disponibles).
Reacciones adversas (según términos preferentes) con la administración intramuscular de pasireótido
Trastornos de la sangre y del sistema linfático: frecuente – anemia.
Trastornos del sistema endocrino: frecuente – insuficiencia suprarrenal*.
Trastornos del metabolismo y de la nutrición: muy frecuente – hiperglucemia, diabetes mellitus; frecuente – diabetes mellitus tipo II, alteración de la tolerancia a la glucosa, disminución del apetito; desconocida – cetoacidosis diabética.
Trastornos del sistema nervioso: frecuente – cefalea, vértigo.
Trastornos cardíacos: frecuente – bradicardia sinusal**, prolongación del intervalo QT.
Trastornos gastrointestinales: muy frecuente – diarrea, náuseas, dolor abdominal***; frecuente – distensión abdominal, vómitos; desconocida – esteatorrea, heces acolúricas.
Trastornos hepáticos y de la vía biliar: muy frecuente – litiasis biliar; frecuente – colecistitis***, colestasis.
Trastornos de la piel y del tejido subcutáneo: frecuente – alopecia, prurito.
Trastornos generales y condiciones en el sitio de administración: muy frecuente – astenia***; frecuente – reacciones en el sitio de inyección***.
Investigaciones: frecuente – aumento de hemoglobina glucosilada, aumento de alanina aminotransferasa, aumento de aspartato aminotransferasa, aumento de gamma-glutamil transferasa, aumento de glucosa en sangre, aumento simultáneo de glucosa en sangre y creatinfosfocinasa, aumento de lipasa; poco frecuente – aumento de amilasa, prolongación del tiempo de protrombina.
* La insuficiencia suprarrenal incluye insuficiencia adrenal y disminución de los niveles séricos de cortisol.
** La bradicardia sinusal incluye bradicardia y bradicardia sinusal.
*** El dolor abdominal incluye dolor abdominal y dolor en la parte superior del abdomen. Las reacciones en el sitio de inyección incluyen dolor en el sitio de inyección, nódulo en el sitio de inyección, molestia en el sitio de inyección, aparición de equimosis en el sitio de inyección, prurito en el sitio de inyección, reacciones en el sitio de inyección, hipersensibilidad en el sitio de inyección y edema en el sitio de inyección. La colecistitis incluye colecistitis aguda y crónica. La astenia incluye astenia y fatiga.
Descripción de reacciones adversas individuales.
Alteraciones del metabolismo de la glucosa.
Acromegalia. En pacientes con acromegalia, el aumento de los niveles de glucosa en ayunas fue la desviación de laboratorio más frecuente de Grado 3/4 durante los dos estudios de Fase III. En el estudio C2305, los aumentos de glucosa en ayunas de Grado 3 se observaron en el 9,7 % y 0,6 %, y de Grado 4 en el 0,6 % y 0 % de los pacientes con acromegalia que recibieron pasireótido intramuscular y octreótido intramuscular, respectivamente. En el estudio C2402, se notificó aumento de glucosa en ayunas de Grado 3 en el 14,3 % y 17,7 % de los pacientes con acromegalia que recibieron pasireótido intramuscular a dosis de 40 mg y 60 mg, respectivamente, y en el 0 % de los pacientes del grupo control activo. Dos casos graves relacionados con hiperglucemia (cetoacidosis diabética y coma hiperosmolar diabético) se registraron tras el aumento de la dosis de pasireótido a 60 mg en pacientes previamente no tratados: uno en un paciente con hiperglucemia no tratada y HbA1c > 8 % antes del inicio del tratamiento con pasireótido, y otro en un paciente con hiperglucemia no tratada y glucemia plasmática en ayunas de 359 mg/dl. En ambos estudios, los niveles de FPG y HbA1c aumentaron durante los primeros tres meses de administración intramuscular de pasireótido. En pacientes previamente no tratados (estudio C2305), el aumento absoluto medio de FPG y HbA1c fue principalmente similar en todos los pacientes que recibieron pasireótido intramuscular, independientemente de los valores basales.
El grado y frecuencia de hiperglucemia observados en los dos estudios principales con pacientes con acromegalia fueron más altos en el grupo de administración intramuscular de Signifor LAR que en el grupo control activo (octreótido intramuscular o lanreótido mediante inyección subcutánea profunda). En un análisis combinado de los dos estudios principales, la frecuencia total de reacciones adversas desfavorables relacionadas con hiperglucemia fue del 58,6 % (todos los grados) y del 9,9 % (Criterios Comunes de Toxicidad Grados 3 y 4) en el grupo de administración intramuscular de Signifor LAR, frente al 18,0 % (todos los grados) y 1,1 % (CCT Grados 3 y 4) en el grupo control activo. En un estudio pivotal con pacientes en los que no se lograba un control adecuado con otro análogo de somatostatina, la proporción de pacientes previamente no tratados con agentes antidiabéticos que requirieron terapia antidiabética durante el estudio fue del 17,5 % y 16,1 % en los grupos de Signifor LAR 40 mg y 60 mg, respectivamente, frente al 1,5 % en el grupo control activo; en un estudio pivotal con pacientes no previamente tratados, la proporción de pacientes que requirieron terapia antidiabética durante el estudio fue del 36 % en el grupo de Signifor LAR frente al 4,4 % en el grupo control activo.
Enfermedad de Cushing. En pacientes con enfermedad de Cushing, el aumento del nivel de glucosa plasmática en ayunas (GPA) fue la desviación de laboratorio más frecuente de Grado 3 según CCT (14,7 % de los pacientes) durante el estudio de Fase III G2304; no se observaron casos de desviación de Grado 4. El aumento medio de HbA1c fue menor en pacientes con niveles normales de glucemia al inicio del estudio en comparación con pacientes con prediabetes o diabetes. Los niveles medios de GPA aumentaron frecuentemente durante el primer mes de tratamiento, y luego disminuyeron y se estabilizaron en meses posteriores. El aumento de los niveles de GPA y HbA1c fue dependiente de la dosis, y los valores generalmente disminuyeron tras la interrupción de la administración intramuscular de pasireótido, aunque permanecieron por encima de los valores basales. La frecuencia total de reacciones adversas relacionadas con hiperglucemia fue del 75,3 % (todos los grados) y del 22,7 % (Grado 3 según CCT). Reacciones adversas como hiperglucemia y diabetes mellitus llevaron a la interrupción del estudio en 3 (2,0 %) y 4 pacientes (2,7 %), respectivamente.
El aumento de los niveles de glucosa y HbA1c en plasma en ayunas observado tras la administración intramuscular de pasireótido fue reversible tras la suspensión del medicamento.
Se recomienda el monitoreo del nivel de glucosa en sangre en pacientes que reciben Signifor LAR.
Trastornos gastrointestinales. Durante el tratamiento con Signifor LAR, se notificaron frecuentemente trastornos gastrointestinales. Estos eventos fueron generalmente de baja gravedad, no requirieron intervención y desaparecieron con la continuación del tratamiento. En pacientes con acromegalia, los trastornos gastrointestinales fueron menos frecuentes en aquellos con control insuficiente previo en comparación con pacientes no previamente tratados.
Reacciones en el sitio de inyección. Durante los estudios de Fase III, las reacciones en el sitio de inyección (por ejemplo, dolor en el sitio de inyección, molestia en el sitio de inyección) fueron principalmente de Grado 1 o 2. La frecuencia de estos eventos fue más alta durante los primeros 3 meses de tratamiento. Durante los estudios con pacientes con acromegalia, las reacciones en el sitio de inyección fueron similares con la administración intramuscular de pasireótido y octreótido, y fueron menos frecuentes en pacientes con control insuficiente previo en comparación con pacientes no previamente tratados.
Prolongación del intervalo QT. En el estudio C2305 con pacientes con acromegalia, la proporción de pacientes con nuevos intervalos QT/QTc significativos fue comparable entre los grupos que recibieron pasireótido intramuscular y octreótido intramuscular antes de la fase cruzada, con algunos valores anómalos notables. Se observaron valores de QTcF > 480 ms en 3 pacientes frente a 2 pacientes en los grupos de administración intramuscular de pasireótido y octreótido, respectivamente, y prolongación de QTcF > 60 ms respecto al valor basal se notificó en 2 pacientes frente a 1 paciente en los grupos correspondientes. En el estudio C2402, la única desviación notable fue un valor de QTcF > 480 ms en 1 paciente en el grupo de administración intramuscular de pasireótido 40 mg. Durante el estudio G2304 con pacientes con enfermedad de Cushing, se observó un valor de QTcF > 480 ms en 2 pacientes. No se observaron valores de QTcF > 500 ms en ninguno de los estudios principales.
Enzimas hepáticas. Se han notificado aumentos transitorios de enzimas hepáticas con análogos de somatostatina, lo cual también se observó en pacientes que recibieron pasireótido durante estudios clínicos. El aumento fue principalmente asintomático, de bajo grado y reversible con la continuación del tratamiento. Se observaron varios casos de aumento simultáneo de ALT > 3 × LSN y bilirrubina > 2 × LSN con la administración subcutánea, pero no en pacientes que recibieron pasireótido intramuscular. Todos los casos de aumento simultáneo ocurrieron dentro de los 10 días posteriores al inicio del tratamiento. Los pacientes se recuperaron sin consecuencias clínicas y los resultados de las pruebas de función hepática regresaron a los valores basales tras la interrupción del tratamiento.
Se recomienda el monitoreo de enzimas hepáticas antes y durante el tratamiento con Signifor LAR según estándares clínicos.
Enzimas pancreáticas. En pacientes que recibieron pasireótido durante estudios clínicos, se observó un aumento asintomático de lipasa y amilasa. El aumento fue principalmente de bajo grado y reversible con la continuación del tratamiento. La pancreatitis es una reacción adversa potencialmente relacionada con el uso de análogos de somatostatina, debido a la asociación entre litiasis biliar y pancreatitis aguda.
Incompatibilidad.
Debido a la falta de estudios de compatibilidad, este medicamento no debe mezclarse con otros medicamentos.
Período de validez. 3 años.
Condiciones de almacenamiento. Conservar en el envase original a una temperatura de 2–8 ºC. No congelar. Mantener fuera del alcance de los niños.
Envase. Polvo en un frasco de vidrio marrón de 6 ml, tapado con un tapón de goma gris, sellado con una tapa de aluminio de sistema flip-off de color gris (para dosis de 20 mg), rojo (para dosis de 40 mg) o naranja (para dosis de 60 mg), acompañado de: un solvente en jeringa precargada de 3 ml de vidrio incoloro con dos tapones de goma grises, tope para dedos, émbolo y tapón; una aguja y un adaptador, todo en caja de cartón.
Categoría de dispensación. Bajo receta médica.
Fabricante. Recordati Rare Diseases.
Ubicación del fabricante y dirección del lugar de actividad.
Eco River Park, 30 Rue des Peupliers, Nanterre, 92000, Francia;
Immeuble Le Wilson, 70 Avenue du Général de Gaulle, Puteaux, 92800, Francia.