Primovist
Ukraina
Spis treści
ULOTKA DOLECZNA DO PREPARATU LECZNICZEGO PRIMOVIST (PRIMOVIST®)
Skład:
substancja czynna: sól dwu-sodowa kwasu gadoksetowego (Gd-EOB-DTPA);
1 ml roztworu do wstrzykiwań zawiera 181,43 mg soli dwu-sodowej kwasu gadoksetowego (Gd-EOB-DTPA) (równowartość 0,25 mmol);
substancje pomocnicze: sól trój-sodowa kwasu kaloksetowego (Ca-EOB-DTPA), trometamol, kwas chlorowodorowy rozcieńczony, wodorotlenek sodu, woda do wstrzykiwań.
Postać leku. Roztwór do wstrzykiwań.
Podstawowe właściwości fizykochemiczne: przezroczysty roztwór bez zawiesin.
Grupa farmakoterapeutyczna. Środki kontrastowe paramagnetyczne.
Kod ATC V08CA10.
Właściwości farmakologiczne
Farmakodynamika.
Mechanizm działania
Lek Primovist to paramagnetyczny środek kontrastowy do rezonansu magnetycznego.
Właściwości kontrastujące są uzależnione od gadoksetatu (Gd-EOB-DTPA), jonowego kompleksu składającego się z gadolinu (III) oraz ligandy etoksybenzylo-dietylenotryaminopentakwasu (EOB-DTPA). W czasie skanowania z wykorzystaniem sekwencji ważonych T1 skrócenie czasu relaksacji podłużnej wzbudzonych jąder atomowych, wywołane przez jony gadolinu, zwiększa intensywność sygnału i tym samym poprawia kontrast obrazu niektórych tkanek.
Wpływy farmakodynamiczne
Dwusodowy gadoksetat zmniejsza czas relaksacji nawet przy niskich stężeniach. Przy pH 7, indukcji pola magnetycznego 0,47 T oraz temperaturze 40 °C relaksywność r1, określana na podstawie wpływu na czas relaksacji podłużnej T1 protonów w osoczu, wynosi około 8,7 l/mmol/s, relaksywność (r2), określana na podstawie wpływu na czas relaksacji poprzecznej (T2), wynosi około 8,56 l/mmol/s. Przy indukcji pola magnetycznego 1,5 T oraz temperaturze 37 °C relaksywność w osoczu wynosi: r1 = 6,9 l/mmol/s, r2 = 8,7 l/mmol/s. Relaksywność zależy w niewielkim stopniu od indukcji pola magnetycznego.
EOB-DTPA tworzy stabilny kompleks z paramagnetycznym jonem gadolinu o niezwykle wysokiej stabilności termodynamicznej (log KGdl = 23,46). Gd-EOB-DTPA jest związkem hydrofilowym o wysokiej rozpuszczalności w wodzie oraz współczynniku podziału między n-butanol a buforem przy pH 7,6 wynoszącym około 0,011. Ze względu na lipofilowość fragmentu etoksybenzylowego, dwusodowy gadoksetat wykazuje dwufazowy mechanizm działania: rozkład w przestrzeni pozakomórkowej po wstrzyknięciu bolusowym oraz późniejsze selektywne wychwytanie przez hepatocyty. Relaksywność r1 w tkankach wątroby wynosi 16,6 l/mmol/s (przy 0,47 T), co zwiększa intensywność sygnału tkanek wątroby. Następnie dwusodowy gadoksetat jest wydzielany z żółcią.
Urazy z brakiem lub obniżoną funkcją hepatocytów (torbiel, przerzuty, większość nowotworów hepatocelularnych) nie gromadzą Primovista. Dobrze zróżnicowany nowotwór hepatocelularny może zawierać funkcjonujące hepatocyty i wykazywać pewne kontrastowanie w fazie hepatocelularnej. Dlatego konieczna jest dodatkowa informacja kliniczna w celu ustalenia właściwej diagnozy.
Substancja nie wykazuje wyraźnych właściwości hamujących enzymy w klinicznie istotnych stężeniach.
Wizualizacja
Po wstrzyknięciu bolusowym leku Primovist, dynamiczna wizualizacja w fazie tętniczej, wrotno-żylnej oraz w fazie równowagi pozwala uzyskać różny obraz czasowy kontrastowania różnych typów zmian w wątrobie, stanowiący podstawę do charakterystyki radiologicznej zmian.
Kontrastowanie miąższu wątroby w trakcie fazy hepatocelularnej pozwala określić liczbę zmienionych ognisk w wątrobie, ich rozmieszczenie segmentarne, wizualizację oraz granice, co poprawia wykrywanie zmian. Różnicowanie zmian wątroby według charakteru dynamiki kontrastowania/wypłukiwania środka kontrastowego pozwala uzyskać dodatkowe informacje.
Fazę opóźnioną (hepatocelularną) można badać po 20 minutach od wstrzyknięcia, przy czym okres wizualizacji trwa nie krócej niż 120 minut.
Wyniki badań diagnostycznych i skuteczności technicznej w trakcie badań klinicznych wskazują na minimalną poprawę wizualizacji po 20 minutach od wstrzyknięcia w porównaniu z okresem 10 minut po wstrzyknięciu.
Okres wizualizacji skraca się do 60 minut u pacjentów wymagających dializy oraz u pacjentów z podwyższonym poziomem bilirubiny (> 3 mg/dl).
Wykrzepianie leku Primovist przez wątrobę powoduje kontrastowanie układu wyprowadzającego żółć.
Właściwości fizykochemiczne gotowego do stosowania roztworu leku Primovist
| Osmolarność przy 37 °C (mOsm/kg H2O) |
688 |
| Lepkość przy 37 °C (mPa·s) |
1,19 |
| Gęstość przy 37 °C (g/ml) |
1,0881 |
| pH |
7,4 |
Pacjenci pediatryczni
Przeprowadzono badanie obserwacyjne z udziałem 52 pacjentów pediatrycznych (w wieku od 2 miesięcy do 18 lat). Pacjenci byli kierowani na rezonans magnetyczny (RM) wątroby z zastosowaniem środka kontrastującego Primovist w celu oceny podejrzanych lub znanych ogniskowych zmian wątroby. Dodatkowe informacje diagnostyczne uzyskano porównując obrazy rezonansu magnetycznego wątroby z kontrastem i bez kontrastu. Rejestrowano ciężkie działania niepożądane, żadne jednak nie zostało ocenione przez badacza jako powiązane z zastosowaniem środka kontrastującego Primovist. Ze względu na charakter retrospektywny i małą liczbę przypadków w tym badaniu, nie można wyciągnąć wniosków dotyczących skuteczności i bezpieczeństwa stosowania leku u dzieci.
Farmakokinetyka.
Rozkład
Po wewnątrzżylnym podaniu profil „stężenie–czas” dla dinatriowej soli kwasu gadoksetowego charakteryzuje się dwueksponencjalnym spadkiem. Dinatriowa sól kwasu gadoksetowego rozkłada się w przestrzeni pozakomórkowej (objętość rozkładu w stanie równowagi wynosi około 0,21 l/kg). Substancja wykazuje niewielkie wiązanie z białkami (poniżej 10%). Lek przenika przez barierę łożyskową w niewielkich ilościach.
Gadoksetat dinatriowy jest liniowym środkiem kontrastującym zawierającym gadolin (GKZ). Badania wykazały, że po podaniu GKZ gadolin może gromadzić się w organizmie, w szczególności w mózgu oraz innych tkankach i narządach.
Zastosowanie GKZ może powodować zależne od dawki zwiększenie intensywności sygnału T1-ważonego w mózgu, szczególnie w jądrze ząbkowanym, prążku bladym i podwzgórzu. Wzrost intensywności sygnału oraz dane przedkliniczne wskazują, że gadolin może uwalniać się z liniowych środków kontrastujących zawierających gadolin.
Biotransformacja
Gadoksetat dinatriowy nie ulega metabolizmowi.
Wydalanie
Dinatriowa sól kwasu gadoksetowego (Gd-EOB-DTPA) wydala się w równych ilościach przez nerki i wątrobę. Okres półtrwania Gd-EOB-DTPA wynosił około 1 godziny. Farmakokinetyka była liniowa do dawki 0,4 ml/kg (100 µmol/kg). Całkowity klirens (Cltot) wynosił około 250 ml/min, natomiast klirens nerkowy (Clr) – około 120 ml/min.
Charakterystyka u pacjentów z określonych grup
Pacjenci w wieku podeszłym (od 65 roku życia). Wskutek fizjologicznych zmian funkcji nerek z wiekiem klirens gadoksetatu dinatriowego w osoczu obniżał się z 210 ml/min u młodszych pacjentów do 163 ml/min u pacjentów w wieku od 65 lat. Okres półtrwania końcowego oraz rozkład systemowy były wyższe u starszych pacjentów (2,3 godziny i 197 µmol × godz/l w porównaniu do 1,6 godziny i 153 µmol × godz/l). Wydalanie dawki podanej dobowo przez nerki zachodzi w pełnym zakresie w ciągu 24 godzin; nie stwierdzono różnic w wydalaniu gadoksetatu dinatriowego między zdrowymi starszymi pacjentami a młodymi.
Niewydolność nerek i/lub wątroby. U pacjentów z łagodnym i umiarkowanym zaburzeniem funkcji wątroby w porównaniu do zdrowych ochotników obserwowano lekkie do umiarkowanego zwiększenie stężeń w osoczu, wydłużenie okresu półtrwania i zmniejszenie wydalania z moczem, jak również zmniejszenie wydalania przez wątrobę. Nie zaobserwowano jednak klinicznie istotnych różnic w kontrastowaniu sygnału wątroby. U pacjentów z ciężkim zaburzeniem funkcji wątroby, szczególnie u tych z nieprawidłowo wysokimi (> 3 mg/dl) stężeniami bilirubiny, AUC wzrastała do 259 µmol × godz/l w porównaniu do 160 µmol × godz/l w grupie kontrolnej. Okres półtrwania wydłużał się do 2,6 godziny w porównaniu do 1,8 godziny w grupie kontrolnej. Wydalanie przez układ hepatobilarny znacząco zmniejszało się do 5,7% podanej dawki, a także obserwowano zmniejszenie kontrastowania sygnału wątroby.
U pacjentów z niewydolnością nerek w stadium końcowym AUC wzrastała 6-krotnie do 903 µmol × godz/l, a końcowy okres półtrwania wydłużał się do 20 godzin. Hemodializa zwiększa klirens gadoksetatu dinatriowego (patrz sekcja „Szczególne środki ostrożności stosowania”). Ogólnie po 3-godzinnym zabiegu dializy około 30% dawki gadoksetatu dinatriowego wydalało się w ciągu 1 godziny po wstrzyknięciu. Oprócz wpływu hemodializy na klirens, znaczna frakcja podanej dawki gadoksetatu dinatriowego wydala się u tych pacjentów z żółcią, o czym świadczy wykrycie średnio około 50% w kale w ciągu 4 dni (zakres od 24,6 do 74,0%; n = 6 pacjentów).
Dane przedkliniczne dotyczące bezpieczeństwa
W standardowych badaniach toksyczności ostrej i subchronicznej, genotoksyczności oraz potencjału uczulającego nie stwierdzono specyficznego ryzyka dla organizmu ludzkiego.
Bezpieczeństwo sercowo-naczyniowe. U psów obserwowano niewielkie i przemijające wydłużenie interwału QT w badaniu przy najwyższej dawce 0,5 mmol/kg, co odpowiada 20-krotnej dawce dla człowieka. W wysokich stężeniach Gd-EOB-DTPA blokuje kanały potasowe serca (gen hERG) i wydłuża czas trwania potencjału czynnościowego w izolowanych mięśniach brodawkowatych świnki morskiej. Wskazuje to na możliwość wydłużenia interwału QT przy przedawkowaniu środka kontrastującego Primovist. W badaniach farmakologicznej bezpieczeństwa nie zaobserwowano wpływu na inne układy narządów.
Toxycologia reprodukcyjna i laktacja. W badaniach embriotoksyczności u królików po wielokrotnym podaniu 0,2 mmol/kg Gd-EOB-DTPA, co odpowiada 25,9-krotnej (w przeliczeniu na powierzchnię ciała) lub 80-krotnej (w przeliczeniu na masę ciała) zalecanej dawce dla człowieka, zaobserwowano zwiększenie liczby porzucenia ciąży po implantacji oraz częstotliwość poronień. U szczurów mniej niż 0,5% podanej wewnętrznie dawki (0,1 mmol/kg) radioznakowanego gadoksetatu wydzielano z mlekiem matki, a absorpcja po podaniu doustnym była bardzo niska (0,4%).
Dane dotyczące młodych zwierząt. Wyniki badań toksyczności jednorazowych i wielokrotnych dawek u noworodków i młodych szczurów jakościowo nie różniły się od tych obserwowanych u dorosłych szczurów, jednak młodsze zwierzęta są bardziej wrażliwe.
Lokalna tolerancja. Reakcje lokalnej nietolerancji obserwowano wyłącznie po wstrzyknięciu do mięśnia Gd-EOB-DTPA.
Kancerogenność. Badania kancerogenności nie były prowadzone.
Dane kliniczne.
Wskazania.
Primovist jest wskazany do wykrywania ogniskowych zmian w wątrobie oraz uzyskiwania dodatkowych informacji dotyczących charakteru tych zmian w obrazowaniu metodą rezonansu magnetycznego (MRI) z ważeniem T1.
Primovist należy stosować tylko wtedy, gdy informacja diagnostyczna jest istotna i nie może być uzyskana za pomocą rezonansu magnetycznego (MRI) bez środka kontrastującego oraz gdy konieczne jest uzyskanie opóźnionych faz obrazowania.
Lekarstwo stosuje się do diagnostyki wyłącznie drogą dożylną.
Przeciwwskazania.
Podwyższona wrażliwość na substancję czynną lub którykolwiek inny składnik lekarstwa.
Szczególne środki ostrożności.
Sprawdzenie. Lekarstwo należy dokładnie sprawdzić przed zastosowaniem. Primovist nie wolno stosować w przypadku istotnej zmiany zabarwienia, pojawienia się cząstek stałych lub naruszenia integralności opakowania.
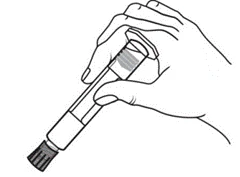
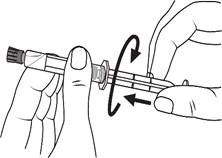
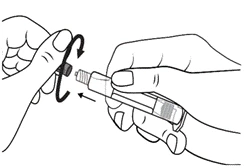
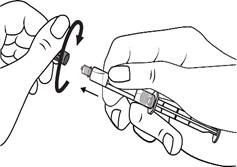
Postępowanie. Lekarstwo Primovist jest gotowe do użycia. Wstępnie napełniony strzykawkę należy przygotować do iniekcji bezpośrednio przed zastosowaniem. Nakrywkę ze strzykawki należy zdejmować bezpośrednio przed wstrzygnięciem leku.
Unieszkodliwienie. Nieużytego lekarstwa lub odpadów należy pozbyć się zgodnie z obowiązującymi przepisami lokalnymi.
Nalepkę z wstępnie napełnionej strzykawki należy wkleić do karty medycznej pacjenta w celu zapewnienia przejrzystej dokumentacji dotyczącej stosowania środków kontrastujących zawierających gadolin. Należy również podać podaną dawkę. W przypadku stosowania elektronicznej karty pacjenta, należy wprowadzić nazwę lekarstwa, numer serii oraz dawkę.
Interakcje z innymi lekarstwami i inne rodzaje interakcji.
Transport gadoxetatu do wątroby może być pośredniczony przez organiczne anionowe białka transportowe (OATP). Nie można wykluczyć, że silne inhibitory OATP mogą powodować interakcje i zmniejszać efekt kontrastowania wątroby. Brakuje jednak danych klinicznych potwierdzających tę teorię.
Badania interakcji przeprowadzone u zdrowych ochotników wykazały, że jednoczesne stosowanie erytromycyny nie wpływało na skuteczność i farmakokinetykę lekarstwa Primovist. Nie przeprowadzono badań klinicznych interakcji z innymi lekarstwami.
Wpływ podwyższonego poziomu bilirubiny lub ferrytyny u pacjentów
Podwyższone poziomy bilirubiny lub ferrytyny mogą zmniejszyć efekt kontrastowania wątroby wywołany lekarstwem Primovist (patrz sekcja „Właściwości farmakologiczne”).
Interakcja z testami diagnostycznymi
Podczas oznaczania zawartości żelaza w surowicy krwi metodami kompleksometrycznymi (np. metodą kompleksowania z ferrocyminem) w okresie do 24 godzin po badaniu z zastosowaniem lekarstwa Primovist mogą wystąpić wyniki fałszywie dodatnie z powodu obecności wolnej substancji wiążącej się z kompleksami w roztworze środka kontrastującego.
Szczególne zagadnienia dotyczące stosowania.
Należy przestrzegać ogólnych zasad bezpieczeństwa podczas wykonywania obrazowania metodą rezonansu magnetycznego, w szczególności wykluczyć obecność stymulatora serca oraz implantów ferromagnetycznych.
Procedury diagnostyczne wykorzystujące środki kontrastujące powinny być wykonywane pod nadzorem lekarza posiadającego doświadczenie w stosowaniu środków kontrastujących oraz wystarczającą wiedzę na temat przeprowadzania procedury.
Po wstrzyknięciu pacjent powinien pozostawać pod obserwacją przez co najmniej 30 minut, ponieważ doświadczenie z zastosowania środków kontrastujących wskazuje, że większość działań niepożądanych pojawia się właśnie w tym okresie.
Uszkodzenie funkcji nerek
Przed podaniem leku Primovist należy wykonać badania przesiewowe u wszystkich pacjentów w celu wykrycia zaburzeń czynności nerek na podstawie danych laboratoryjnych.
U pacjentów z ostrym lub przewlekłym, ciężkim uszkodzeniem nerek (przepływ kłębuszkowy < 30 ml/min/1,73 m²) obserwowano przypadki nefrogennego fibrozy systemowego (NFS) związanych ze stosowaniem środków kontrastujących zawierających gadolin. Szczególnie narażeni są pacjenci poddawani przeszczepieniu wątroby, ponieważ częstość ostrego uszkodzenia nerek w tej grupie jest wysoka. Z uwagi na ryzyko rozwoju NFS należy unikać stosowania leku Primovist u pacjentów z ciężkim uszkodzeniem nerek oraz w okresie okołoperacyjnym przeszczepienia wątroby, z wyjątkiem przypadków, gdy informacja diagnostyczna jest absolutnie konieczna i nie może być uzyskana podczas rezonansu magnetycznego bez kontrastowania.
Hemodializa przeprowadzona w krótkim czasie po podaniu leku Primovist może pomóc w jego usunięciu z organizmu. Brak danych dotyczących stosowania hemodializy w celu zapobiegania lub leczenia NFS u pacjentów, którzy wcześniej nie byli poddawani dializie.
Pacjenci w wieku podeszłym
Ponieważ klirens nerkowy gadoksetatu u pacjentów w wieku podeszłym może być zaburzony, szczególnie ważne jest wykonanie badań w celu wykrycia zaburzeń czynności nerek u pacjentów w wieku 65 lat i starszych.
Pacjenci z chorobami układu sercowo-naczyniowego
Leku Primovist należy stosować z ostrożnością u pacjentów z ciężkimi chorobami układu sercowo-naczyniowego ze względu na ograniczoną dostępność danych.
Leku Primovist nie należy stosować pacjentom z niekontrolowaną hipokaliemią.
Leku Primovist należy stosować z dużą ostrożnością u pacjentów:
- z wrodzonym zespołem wydłużonego interwału QT obecnie lub w wywiadzie;
- z arytmiami w wywiadzie po stosowaniu leków wydłużających repolaryzację serca;
- stosujących leki wydłużające repolaryzację serca, np. leki przeciwarytmiczne klasy III (amiodaron, sotalol).
Stosowanie leku Primovist może powodować przemijające wydłużenie interwału QT u niektórych pacjentów (patrz sekcja „Właściwości farmakologiczne”).
Nadwrażliwość
Reakcje alergiczne, w tym wstrząs, mogą występować po podaniu środków kontrastujących zawierających gadolin do MRI. Większość takich działań niepożądanych pojawia się w ciągu 30 minut po podaniu środka kontrastującego. Jednakże, podobnie jak przy stosowaniu innych środków kontrastujących tej klasy, możliwe są opóźnione reakcje pojawiające się od kilku godzin do kilku dni po wstrzyknięciu. Zawsze należy mieć dostęp do odpowiednich leków stosowanych w leczeniu reakcji nadwrażliwościowych oraz do środków ratunkowych.
Ryzyko rozwoju reakcji nadwrażliwościowych jest większe przy występowaniu poniższych stanów i chorób:
- reakcja na wcześniejsze podanie środków kontrastujących;
- astma oskrzelowa w wywiadzie;
- reakcje alergiczne w wywiadzie.
Decyzję o stosowaniu leku Primovist u pacjentów z tendencją do alergii należy podejmować po szczególnej ocenie stosunku ryzyka do korzyści.
Reakcje nadwrażliwościowe mogą być bardziej nasilone u pacjentów stosujących beta-blokery, szczególnie przy obecności astmy oskrzelowej. Należy wziąć pod uwagę, że pacjenci stosujący beta-blokery mogą być oporni na standardowe leczenie reakcji nadwrażliwościowych beta-agonistami.
W przypadku wystąpienia reakcji nadwrażliwościowej należy natychmiast przerwać podawanie środka kontrastującego.
Nieodporność miejscowa
Podanie do mięśni może powodować reakcje miejscowej nieodporności, w tym martwicę ogniskową, dlatego zaleca się unikanie tej drogi podania (patrz sekcja „Właściwości farmakologiczne”).
Akumulacja w organizmie
Po podaniu gadoksetanu dinatrynowego gadolin może gromadzić się w mózgu i innych tkankach organizmu (kości, wątroba, nerki, skóra) i powodować zależne od dawki zwiększenie intensywności sygnału ważonego T1 w mózgu, szczególnie w jądrze ząbkowanym, bladym płacie i podwzgórzu. Znaczenie kliniczne jest nieznane. U pacjentów, którzy będą wymagać ponownego skanowania, należy rozważyć korzyści z diagnostyki przy użyciu gadoksetanu dinatrynowego oraz potencjalne gromadzenie się gadolinu w mózgu i innych tkankach.
Substancje pomocnicze
Ten lek zawiera 11,7 mg sodu na 1 ml, co odpowiada 0,585 % zalecanego przez WHO maksymalnego dziennego spożycia 2 g sodu dla dorosłego (4,1 % (82 mg) na podstawie dawki przepisanej osobie o masie ciała 70 kg). Dawka wynosi 0,1 ml/kg masy ciała.
Stosowanie w okresie ciąży lub karmienia piersią.
Ciąża. Dane dotyczące stosowania środków kontrastujących opartych na gadolinie u kobiet w ciąży są ograniczone. Gadolin może przenikać przez łożysko. Nie wiadomo, czy wpływ gadolinu wiąże się z niekorzystnymi skutkami dla płodu. W badaniach na zwierzętach po podawaniu wielokrotnych wysokich dawek lek wykazywał toksyczność reprodukcyjną (patrz sekcja „Właściwości farmakologiczne”). Leku Primovist nie zaleca się stosować w okresie ciąży, chyba że istnieją absolutne wskazania.
Karmienie piersią. Substancje kontrastujące zawierające gadolin przechodzą do mleka matki w bardzo niewielkich ilościach (patrz sekcja „Właściwości farmakologiczne”). Po zastosowaniu dawek klinicznych nie należy oczekiwać wpływu na niemowlę ze względu na niewielką ilość substancji czynnej wydzielanej z mlekiem matki oraz słabe wchłanianie z przewodu pokarmowego. Decyzję o kontynuowaniu karmienia piersią lub jego przerwaniu na 24 godziny po podaniu leku Primovist powinny podejmować lekarz i kobieta karmiąca piersią.
Plodność. Wyniki badań na zwierzętach nie wykazały zaburzeń płodności.
Wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn.
Leku Primovist nie wpływa na szybkość reakcji podczas prowadzenia pojazdów lub obsługi maszyn.
Sposób i dawki stosowania.
Sposób stosowania
Gotowy do użycia wodny roztwór leku Primovist podaje się w formie nierozcieńczonej w sposób bolusowy z szybkością około 2 ml/s. Po wstrzyknięciu środka kontrastującego kaniulę dożylną należy przemyć 0,9% roztworem chlorku sodu.
Informacje dotyczące wizualizacji znajdują się w punkcie „Właściwości farmakologiczne”.
Dawkowanie
W celach diagnostycznych należy stosować najniższą dawkę zapewniającą wystarczającą kontrastowość. Dawka powinna być obliczana z uwzględnieniem masy ciała pacjenta i nie powinna przekraczać zalecanej dawki na kilogram masy ciała, podanej w niniejszym punkcie.
Zalecana dawka leku Primovist to:
dorośli: 0,1 ml leku Primovist na 1 kg masy ciała.
Powtarzane dawkowanie
Brak danych klinicznych dotyczących stosowania powtórnego dawkowania leku Primovist.
Osobne grupy pacjentów
Niewydolność nerek
Należy unikać stosowania leku Primovist u pacjentów z ciężką niewydolnością nerek (przepływ kłębuszkowy < 30 ml/min/1,73 m2) oraz u pacjentów w okresie okołotransplantacyjnym przeszczepu wątroby, z wyjątkiem przypadków, gdy informacja diagnostyczna jest absolutnie konieczna i nie może być uzyskana podczas przeprowadzania rezonansu magnetycznego bez kontrastowania (zob. punkt „Szczególne ostrzeżenia i środki ostrożności podczas stosowania”). Jeśli nie można uniknąć stosowania leku Primovist, dawka nie powinna przekraczać 0,025 mmol/kg masy ciała. Podczas jednego skanowania nie należy stosować więcej niż jednej dawki. Ze względu na niewystarczającą ilość danych dotyczących ponownego stosowania, wstrzyknięcia leku Primovist nie należy powtarzać, z wyjątkiem przypadków, gdy okres między wstrzyknięciami wynosi co najmniej 7 dni.
Pacjenci z niewydolnością wątroby
Nie ma potrzeby dostosowywania dawki.
Pacjenci w wieku podeszłym (65 lat i więcej)
Nie stwierdzono potrzeby dostosowywania dawek u pacjentów w wieku podeszłym. Podczas stosowania u pacjentów w wieku podeszłym należy zachować szczególną ostrożność (zob. punkt „Szczególne ostrzeżenia i środki ostrożności podczas stosowania”).
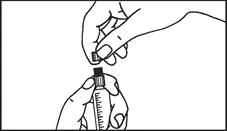
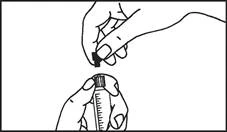
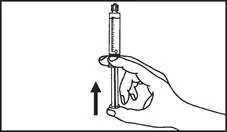
Instrukcja dotycząca stosowania strzykawki
Strzykawki wstępnie napełnione należy przygotowywać do wstrzyknięcia bezpośrednio przed jego wykonaniem. Czapkę strzykawki należy zdejmować dopiero bezpośrednio przed podaniem leku. Nieużywany lek lub odpady należy utylizować zgodnie z lokalnymi przepisami prawa.
Nalepkę zdejmowaną ze strzykawki wstępnie napełnionej należy wkleić do karty medycznej pacjenta w celu zapewnienia czytelnego dokumentowania stosowania środków kontrastujących zawierających gadolin. Należy również podać podaną dawkę. Jeśli stosowana jest elektronicza karta pacjenta, należy wprowadzić nazwę leku, numer serii i dawkę.
Strzykawka szklana
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
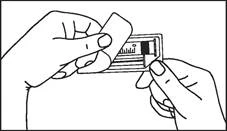
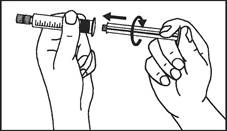
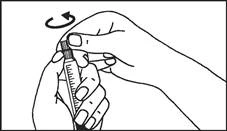
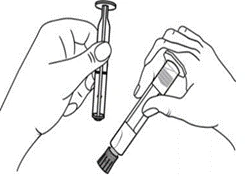
Plastikowy strzykawka
| Ręczne podawanie
|
Podawanie za pomocą dożylnej pompy wlewu
|
|
|
|
|
|
|
|
|
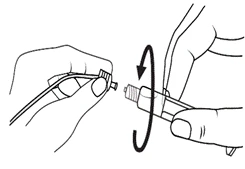
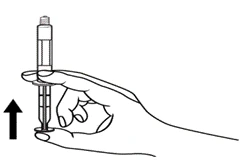
- Dzieci *
Bezpieczeństwo i skuteczność stosowania leku Primovist u pacjentów w wieku do 18 roku życia nie zostały ustalone, dlatego lek ten nie jest stosowany u tej grupy wiekowej. Dane dostępne do chwili obecnej przedstawiono w sekcji „Właściwości farmakologiczne”.
*** Przedawkowanie ***
Nie odnotowano przypadków przedawkowania, brak charakterystyki objawów przedawkowania.
Jednorazowe dawki leku Primovist w dawce 0,4 ml/kg (0,1 mmol/kg) masy ciała były dobrze tolerowane. W ograniczonej liczbie pacjentów badano dawkę 0,2 ml/kg (0,5 mmol/kg) masy ciała w trakcie badań klinicznych; częstotliwość reakcji niepożądanych była wyższa, jednak nie zaobserwowano nowych zjawisk niepożądanych u tych pacjentów.
W przypadku przypadkowego przedawkowania zaleca się obserwację stanu pacjenta, w tym monitorowanie układu sercowo-naczyniowego, ze względu na możliwość wydłużenia interwału QT (patrz sekcja „Właściwości farmakologiczne”).
Lek Primovist jest usuwany z organizmu za pomocą dializy. Brakuje jednak danych dotyczących możliwości stosowania hemodializy w celu zapobiegania włóknieniu systemowemu nefrogennemu.
Efekty uboczne
Profil bezpieczeństwa leku Primovist oparto na wynikach badań klinicznych przeprowadzonych u ponad 1900 pacjentów oraz na danych z okresu postmarketingowego.
Najczęstszymi reakcjami ubocznymi (≥ 0,5 %) u pacjentów otrzymujących lek Primovist były: nudności, ból głowy, uczucie ciepła, podwyższenie ciśnienia tętniczego, ból pleców oraz zawroty głowy.
Najpoważniejszą zarejestrowaną reakcją uboczną u pacjentów otrzymujących lek Primovist był wstrząs anafilaktyczny. Rzadko obserwowano opóźnione reakcje anafilaktyczne (w okresie od kilku godzin do kilku dni po podaniu). Większość efektów ubocznych miała charakter przejściowy i była łagodna lub umiarkowana.
Poniżej wymieniono reakcje uboczne obserwowane podczas stosowania leku Primovist, sklasyfikowane według klas narządów i układów (Medyczny Słownik Terminologii Regulatoryjnej (MedDRA), wersja 12.1). Do opisu poszczególnych reakcji, ich objawów oraz stanów podobnych pod względem objawów zastosowano odpowiednie terminy MedDRA.
Poniżej wymienione reakcje uboczne zarejestrowane w trakcie badań klinicznych zostały sklasyfikowane według częstości występowania: często (od ≥ 1/100 do < 1/10), rzadko (od ≥ 1/1000 do < 1/100), pojedynczo (od ≥ 1/10000 do < 1/1000). Częstość reakcji ubocznych wykrytych wyłącznie w okresie obserwacji postmarketingowej oznaczono jako „częstość nieznana”. W każdej grupie reakcje uboczne wymieniono w kolejności malejącej według nasilenia.
Tabela. Reakcje uboczne zarejestrowane w trakcie badań klinicznych lub w okresie obserwacji postmarketingowej u pacjentów otrzymujących lek Primovist
| Układ narządów |
Częste |
Nieczeście |
Pojedyncze |
Częstotliwość nieznana |
| Zaburzenia ze strony układu odpornościowego |
reakcje nadwrażliwości / anafilaktyczne (np. szok*, hipotensja, obrzęk gardła i krtani, pokrzywka, obrzęk twarzy, kichanie, kaszel, bladość, ból brzucha, hipestezja, kichanie, kaszel, zapalenie spojówek, zapalenie błony śluzowej nosa) |
|||
| Zaburzenia ze strony układu nerwowego |
ból głowy |
obrzydzenie, zawroty głowy, dysgezja, parestezja, parosmia |
drżenie, akatyzja |
niespokój |
| Zaburzenia ze strony serca |
blokada pęczka Hisa, kołatanie serca |
tachykardia |
||
| Zaburzenia ze strony naczyń |
wzrost ciśnienia tętniczego, napływy gorąca |
|||
| Zaburzenia ze strony dróg oddechowych, klatki piersiowej i jamy śródpiersia |
zaburzenia ze strony dróg oddechowych (duszność*, napięcie oddechowe) |
|||
| Zaburzenia żołądkowo-jelitowe |
nudności |
wymioty, suchość w ustach |
dyskomfort w jamie ustnej, nadmierna sekrecja śliny |
|
| Zaburzenia ze strony skóry i tkanki podskórnej |
wysypka, swędzenie** |
wysypka makularna i grudkowa, nadpotliwość |
||
| Zaburzenia ze strony układu mięśniowo-szkieletowego i tkanki łącznej |
ból pleców |
|||
| Zaburzenia ogólne i stan w miejscu wstrzyknięcia |
ból w klatce piersiowej, reakcje w miejscu wstrzyknięcia (różne rodzaje)***, uczucie ciepła, gorączka, zmęczenie, nietypowe uczucia |
dyskomfort, uczucie niedobytu |
* Zgłaszano stan grożące life oraz skutki śmiertelne. Te doniesienia pochodzą z okresu po rejestracji.
** Świąd (świąd ogólny, świąd oczu).
*** Reakcje w miejscu wstrzyknięcia (różne rodzaje) określane są następującymi terminami: krwawienie w miejscu wstrzyknięcia, pieczenie w miejscu wstrzyknięcia, uczucie zimna w miejscu wstrzyknięcia, podrażnienie w miejscu wstrzyknięcia, ból w miejscu wstrzyknięcia.
Opis poszczególnych działań niepożądanych
W trakcie badań klinicznych zgłaszano zmiany parametrów laboratoryjnych, takie jak: podwyższenie stężenia żelaza w surowicy, podwyższenie stężenia bilirubiny, podwyższenie stężenia transaminaz wątrobowych, obniżenie stężenia hemoglobiny, podwyższenie stężenia amylazy, leukocyturia, hipertriglicerydemia, zwiększenie stosunku albuminy do kreatyniny w moczu, hiponatremia, podwyższenie stężenia nieorganicznego fosforanu, obniżenie stężenia białka w surowicy, leukocytoza, hipokaliemia, podwyższenie stężenia dehydrogenazy mleczanowej. W trakcie badań klinicznych regularnie przeprowadzano EKG, u niektórych pacjentów obserwowano przejściowe wydłużenie odcinka QT bez towarzyszących działań niepożądanych.
Zgłaszano przypadki nefrogennego fibrozy systemowej (NFS) po stosowaniu innych kontrastowych środków zawierających gadolinę (patrz sekcja „Szczególne ostrzeżenia i środki ostrożności stosowania”).
Zgłaszanie podejrzewanych działań niepożądanych
Zgłaszanie działań niepożądanych po rejestracji leku ma istotne znaczenie. Umożliwia to monitorowanie stosunku korzyści do ryzyka w trakcie stosowania tego leku. Osoby pracujące w zawodach medycznych i farmaceutycznych, jak również pacjenci lub ich ustawowo uprawnione reprezentanty, powinny zgłaszać wszystkie przypadki podejrzewanych działań niepożądanych oraz braku skuteczności leku za pośrednictwem zautomatyzowanego systemu informacyjnego nadzoru farmakologicznego pod adresem: https://aisf.dec.gov.ua.
Okres ważności.
5 lat (szczotka szklana);
3 lata (szczotka plastikowa).
Warunki przechowywania.
Nie wymaga specjalnych warunków przechowywania.
Niezgodność.
Ze względu na brak przeprowadzonych badań dotyczących zgodności, tego leku nie należy mieszać z innymi lekami.
Opakowanie.
10 ml w szklanej strzykawce; 1 strzykawka w przezroczystym plastikowym pojemniku, zamkniętym papierem, umieszczonym w tekturowej puszce;
10 ml w plastikowej strzykawce; 1 strzykawka umieszczona w tekturowym uchwycie i włożona do tekturowej puszki.
Kategoria wydawania.
Na receptę.
Producent.
Bayer AG.
Miejsce położenia producenta oraz adres miejsca prowadzenia działalności.
Müllerstraße 178, 13353 Berlin, Niemcy.