Primovist
Ucrania
Contenido
INSTRUCCIONES PARA USO MÉDICO DEL MEDICAMENTO PRIMOVISt (PRIMOVIST®)
Composición:
Principio activo: sal disódica del ácido gadoxético (Gd-EOB-DTPA);
1 ml de solución inyectable contiene 181,43 mg de sal disódica del ácido gadoxético (Gd-EOB-DTPA) (equivalente a 0,25 mmol);
Sustancias auxiliares: sal trisódica del ácido caloxético (Ca-EOB-DTPA), trometamol, ácido clorhídrico diluido, hidróxido de sodio, agua para preparaciones inyectables.
Forma farmacéutica. Solución inyectable.
Propiedades fisicoquímicas principales: solución transparente, sin partículas visibles.
Grupo farmacoterapéutico. Agentes de contraste paramagnéticos.
Código ATC V08CA10.
Propiedades farmacológicas
Farmacodinámica.
Mecanismo de acción
El medicamento PrimoVist es un agente de contraste paramagnético para la visualización por resonancia magnética.
El efecto de realce del contraste se debe al gadoxetato (Gd-EOB-DTPA), un complejo iónico formado por gadolinio (III) y el ligando ácido etoxibencil-dietilentriaminopentaacético (EOB-DTPA). Durante la adquisición con secuencias ponderadas en T1, la reducción del tiempo de relajación longitudinal de los núcleos atómicos excitados, provocada por los iones de gadolinio, incrementa la intensidad de la señal y, por lo tanto, mejora el contraste de la imagen en ciertos tejidos.
Efectos farmacodinámicos
El gadoxetato disódico reduce el tiempo de relajación incluso a bajas concentraciones. A pH 7, un campo magnético de 0,47 T y una temperatura de 40 °C, la relajividad r1, definida por su influencia sobre el tiempo de relajación longitudinal T1 de los protones en plasma, es aproximadamente de 8,7 L/mmol/s; la relajividad r2, definida por su influencia sobre el tiempo de relajación transversal T2, es aproximadamente de 8,56 L/mmol/s. Con una intensidad de campo magnético de 1,5 T y una temperatura de 37 °C, la relajividad en plasma es: r1 = 6,9 L/mmol/s, r2 = 8,7 L/mmol/s. La relajividad depende ligeramente de la intensidad del campo magnético.
El EOB-DTPA forma un complejo estable con el ion paramagnético gadolinio, con una estabilidad termodinámica extremadamente alta (log KGdL = 23,46). El Gd-EOB-DTPA es un compuesto hidrofílico con alta solubilidad en agua y un coeficiente de reparto entre n-butanol y tampón a pH 7,6 de aproximadamente 0,011. Debido a la lipofilia del fragmento etoxibencil, el gadoxetato disódico presenta un mecanismo de acción bifásico: distribución en el espacio extracelular tras la inyección en bolo seguida de una captación selectiva por los hepatocitos. La relajividad r1 en el tejido hepático es de 16,6 L/mmol/s (a 0,47 T), lo que incrementa la intensidad de la señal en el tejido hepático. Posteriormente, el gadoxetato disódico se excreta por la vía biliar.
Las lesiones con ausencia o disminución de la función de los hepatocitos (quistes, metástasis, la mayoría de los carcinomas hepatocelulares) no acumulan PrimoVist. Los carcinomas hepatocelulares bien diferenciados pueden contener hepatocitos funcionales y mostrar cierto realce en la fase hepatocelular. Por ello, se requiere información clínica adicional para establecer un diagnóstico correcto.
La sustancia no muestra propiedades inhibitorias significativas sobre enzimas a concentraciones clínicamente relevantes.
Visualización
Tras la inyección en bolo del medicamento PrimoVist, la visualización dinámica en las fases arterial, portal y de equilibrio permite obtener un patrón temporal diferencial del realce de distintos tipos de lesiones hepáticas, sirviendo como base para la caracterización radiológica de las mismas.
El realce del parénquima hepático durante la fase hepatocelular permite determinar el número de lesiones hepáticas, su distribución segmentaria, visualización y límites, mejorando así la detección de las lesiones. La diferenciación de las lesiones hepáticas según el patrón dinámico de realce/eliminación del agente de contraste proporciona información adicional.
La fase retardada (hepatocelular) puede evaluarse a partir de los 20 minutos tras la inyección, y el periodo de visualización dura al menos 120 minutos.
Los resultados de eficacia diagnóstica y técnica en estudios clínicos indican una mejora mínima en la visualización a los 20 minutos tras la inyección en comparación con los 10 minutos tras la inyección.
El periodo de visualización se reduce a 60 minutos en pacientes que requieren diálisis y en aquellos con niveles elevados de bilirrubina (> 3 mg/dL).
La excreción hepática del medicamento PrimoVist produce el realce del sistema biliar.
Propiedades físico-químicas de la solución lista para usar del medicamento PrimoVist
| Osmolalidad a 37 °C (mOsm/kg H2O) |
688 |
| Viscosidad a 37 °C (mPa·s) |
1,19 |
| Densidad a 37 °C (g/ml) |
1,0881 |
| pH |
7,4 |
Pacientes pediátricos
Se realizó un estudio observacional con 52 pacientes pediátr游戏副本
Características clínicas.
Indicaciones.
Primovist está indicado para la detección de lesiones focales hepáticas y para obtener información adicional sobre la naturaleza de dichas lesiones mediante imágenes ponderadas en T1 mediante resonancia magnética (RM).
Primovist debe utilizarse únicamente cuando la información diagnóstica sea importante y no pueda obtenerse mediante imágenes por resonancia magnética (RM) sin el uso de un agente de contraste, y cuando sea necesaria una fase de imagen posterior inmediata.
El medicamento debe administrarse para diagnóstico exclusivamente por vía intravenosa.
Contraindicaciones.
Hipersensibilidad al principio activo o a cualquiera de los excipientes del medicamento.
Precauciones especiales de seguridad.
Inspección. El medicamento debe examinarse cuidadosamente antes de su uso. No se debe utilizar Primovist si se observa un cambio significativo en su coloración, la presencia de partículas sólidas o la pérdida de integridad del envase.
Manejo. El medicamento Primovist está listo para su uso. La jeringa precargada debe prepararse para inyección inmediatamente antes de su uso. La tapa de la jeringa debe retirarse únicamente inmediatamente antes de la administración del fármaco.
Eliminación. El medicamento no utilizado o los residuos deben eliminarse de acuerdo con los requisitos de la legislación local.
La etiqueta desprendible de la jeringa precargada debe pegarse en la historia clínica del paciente con el fin de garantizar una documentación clara sobre el uso de agentes de contraste que contienen gadolinio. También debe indicarse la dosis administrada. Si se utiliza una historia clínica electrónica, debe registrarse el nombre del medicamento, el número de lote y la dosis.
Interacción con otros medicamentos y otras formas de interacción.
El transporte del gadoxetato al hígado puede estar mediado por los péptidos transportadores de aniones orgánicos (OATP). No puede descartarse que los inhibidores potentes de OATP puedan provocar una interacción y reducir el efecto de contraste hepático. Sin embargo, no existen datos clínicos que respalden esta teoría.
Estudios de interacción en voluntarios sanos han demostrado que la administración concomitante de eritromicina no afectó la eficacia ni la farmacocinética del medicamento Primovist. No se han realizado estudios clínicos sobre interacciones con otros medicamentos.
Efecto de niveles elevados de bilirrubina o ferritina en pacientes
Los niveles elevados de bilirrubina o ferritina pueden reducir el efecto de contraste hepático provocado por el medicamento Primovist (ver sección «Propiedades farmacológicas»).
Interacción con pruebas diagnósticas
En la determinación del contenido de hierro en suero mediante métodos complejométricos (por ejemplo, el método de formación de complejos con ferrozina), pueden obtenerse valores erróneos durante las 24 horas posteriores al estudio con el medicamento Primovist, debido a la presencia de agente complejante libre en la solución del agente de contraste.
Características de uso.
Se deben observar las normas generales de seguridad durante la realización de la resonancia magnética, especialmente excluyendo marcapasos y dispositivos implantados ferromagnéticos.
Los procedimientos diagnósticos que implican el uso de agentes de contraste deben realizarse bajo supervisión médica por un profesional con experiencia en el uso de agentes de contraste y con conocimientos suficientes sobre el procedimiento.
Después de la inyección, el paciente debe permanecer bajo observación durante al menos 30 minutos, ya que la experiencia con agentes de contraste indica que la mayoría de las reacciones adversas ocurren precisamente en este período.
Alteraciones de la función renal
Antes de la administración del medicamento Primovist, todos los pacientes deben someterse a una evaluación de detección de disfunción renal mediante análisis de laboratorio.
En pacientes con insuficiencia renal aguda o crónica grave (velocidad de filtración glomerular < 30 ml/min/1,73 m²) se han observado casos de fibrosis sistémica nefrogénica (FSN) asociados al uso de agentes de contraste que contienen gadolinio. Los pacientes sometidos a trasplante hepático tienen un riesgo particular, ya que la frecuencia de insuficiencia renal aguda en este grupo es alta. Debido al riesgo de FSN, el uso del medicamento Primovist debe evitarse en pacientes con insuficiencia renal grave y en pacientes durante el período perioperatorio del trasplante hepático, excepto cuando la información diagnóstica es esencial y no puede obtenerse mediante resonancia magnética sin contraste.
La hemodiálisis realizada poco después de la administración del medicamento Primovist puede ser útil para eliminar el agente del organismo. No existen datos sobre el uso de hemodiálisis para prevenir o tratar la FSN en pacientes que previamente no estaban en diálisis.
Pacientes de edad avanzada
Dado que el aclaramiento renal del gadoxetato puede estar alterado en pacientes de edad avanzada, es especialmente importante evaluar la posible disfunción renal en pacientes de 65 años o más.
Pacientes con enfermedades cardiovasculares
Debe tenerse precaución al administrar el medicamento Primovist a pacientes con enfermedades cardiovasculares graves debido a la limitación de datos disponibles.
Primovist no debe administrarse a pacientes con hipokalemia no controlada.
Debe tenerse especial precaución al administrar el medicamento Primovist a pacientes:
- con síndrome congénito de QT prolongado presente o en antecedentes;
- con antecedentes de arritmias tras la administración de medicamentos que prolongan la repolarización cardíaca;
- que estén tomando medicamentos que prolongan la repolarización cardíaca, por ejemplo, antiarrítmicos de clase III (amiodarona, sotalol).
La administración del medicamento Primovist puede provocar un alargamiento transitorio del intervalo QT en algunos pacientes (ver sección «Propiedades farmacológicas»).
Hipersensibilidad
Pueden producirse reacciones alérgicas, incluyendo anafilaxia, tras la administración de agentes de contraste con gadolinio para RM. La mayoría de estas reacciones adversas ocurren dentro de los 30 minutos posteriores a la administración del agente de contraste. Sin embargo, como con otros agentes de contraste de esta clase, también pueden ocurrir reacciones retardadas que aparecen desde varias horas hasta varios días después de la inyección. Siempre deben estar disponibles los medicamentos adecuados para tratar reacciones de hipersensibilidad, así como equipos de soporte vital.
El riesgo de reacciones de hipersensibilidad es mayor en presencia de las siguientes condiciones o enfermedades:
- reacción previa a la administración de agentes de contraste;
- asma bronquial en antecedentes;
- antecedentes de reacciones alérgicas.
La decisión de administrar el medicamento Primovist a pacientes con predisposición a alergias debe tomarse tras una evaluación cuidadosa de la relación riesgo-beneficio.
Las reacciones de hipersensibilidad pueden ser más graves en pacientes que toman betabloqueantes, especialmente si tienen asma bronquial. Debe tenerse en cuenta que los pacientes que toman betabloqueantes pueden ser refractarios al tratamiento estándar de reacciones de hipersensibilidad con betamiméticos.
Si se produce una reacción de hipersensibilidad, se debe interrumpir inmediatamente la administración del agente de contraste.
Intolerancia local
La administración intramuscular puede provocar reacciones de intolerancia local, incluyendo necrosis focal; por lo tanto, se recomienda evitar esta vía de administración (ver sección «Propiedades farmacológicas»).
Acumulación en el organismo
Tras la administración de gadoxetato disódico, el gadolinio puede retenerse en el cerebro y otros tejidos del organismo (huesos, hígado, riñones, piel) y provocar un aumento dependiente de la dosis en la intensidad de la señal ponderada en T1 en el cerebro, especialmente en el núcleo dentado, la sustancia pálida y el tálamo. La relevancia clínica de esta acumulación es desconocida. En pacientes que requieran escáneres repetidos, se debe considerar cuidadosamente el beneficio diagnóstico del uso de gadoxetato disódico frente al posible depósito de gadolinio en el cerebro y otros tejidos.
Sustancias auxiliares
Este medicamento contiene 11,7 mg de sodio por 1 ml, lo que equivale al 0,585 % de la ingesta diaria máxima recomendada por la OMS de 2 g de sodio para adultos (4,1 % (82 mg) basado en la dosis administrada a una persona de 70 kg). La dosis es de 0,1 ml/kg de peso corporal.
Uso durante el embarazo o la lactancia.
Embarazo. Los datos sobre el uso de agentes de contraste basados en gadolinio en mujeres embarazadas son limitados. El gadolinio puede atravesar la placenta. No se conoce si el efecto del gadolinio se asocia con consecuencias adversas para el feto. En estudios en animales, la administración repetida de dosis altas del fármaco mostró toxicidad reproductiva (ver sección «Propiedades farmacológicas»). El medicamento Primovist no se recomienda durante el embarazo a menos que haya indicaciones absolutas.
Lactancia. Las sustancias de contraste que contienen gadolinio pasan a la leche materna en cantidades muy pequeñas (ver sección «Propiedades farmacológicas»). Tras la administración de dosis clínicas, no se espera un efecto sobre el lactante debido a la cantidad insignificante del principio activo excretado en la leche materna y a la escasa absorción desde el tracto gastrointestinal. La decisión sobre continuar o suspender la lactancia durante 24 horas tras la administración del medicamento Primovist debe tomarse conjuntamente por el médico y la mujer lactante.
Fertilidad. Los resultados de los estudios en animales no mostraron alteración de la fertilidad.
Capacidad para afectar la velocidad de reacción al conducir vehículos o manejar máquinas.
El medicamento Primovist no afecta la velocidad de reacción al conducir vehículos o manejar máquinas.
Vía de administración y dosis.
Vía de administración
La solución acuosa lista para usar del medicamento Primovist se administra sin diluir mediante inyección bolus a una velocidad de aproximadamente 2 ml/s. Tras la inyección del agente de contraste, la cánula intravenosa debe lavarse con solución de cloruro de sodio al 0,9 %.
Para información sobre la visualización, véase la sección «Propiedades farmacológicas».
Dosificación
Con fines diagnósticos, debe utilizarse la dosis más baja posible que proporcione un contraste adecuado. La dosis debe calcularse en función del peso corporal del paciente y no debe superar la dosis recomendada por kilogramo de peso corporal indicada en esta sección.
La dosis recomendada del medicamento Primovist es la siguiente:
Adultos: 0,1 ml del medicamento Primovist por 1 kg de peso corporal.
Administración de dosis repetidas
No existe información clínica sobre la administración de dosis repetidas del medicamento Primovist.
Grupos de pacientes específicos
Insuficiencia renal
Debe evitarse la administración del medicamento Primovist en pacientes con insuficiencia renal grave (velocidad de filtración glomerular < 30 ml/min/1,73 m²) y en pacientes en período peroperatorio de trasplante hepático, salvo cuando la información diagnóstica sea extremadamente necesaria y no pueda obtenerse mediante resonancia magnética sin realce con contraste (véase la sección «Precauciones de uso»). Si no puede evitarse el uso de Primovist, la dosis no debe exceder de 0,025 mmol/kg de peso corporal. No debe administrarse más de una dosis en un mismo escaneo. Debido a la falta de información sobre la administración repetida, no debe repetirse la inyección de Primovist, excepto cuando el intervalo entre inyecciones sea de al menos 7 días.
Pacientes con insuficiencia hepática
No es necesaria la corrección de la dosis.
Pacientes de edad avanzada (a partir de 65 años)
No se ha detectado necesidad de corrección de la dosis en pacientes de edad avanzada. No obstante, debe tenerse especial precaución al administrar el medicamento a pacientes ancianos (véase la sección «Precauciones de uso»).
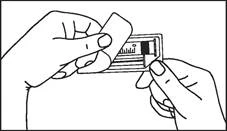
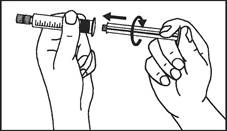
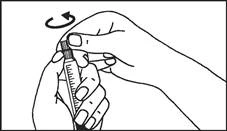
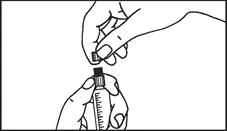
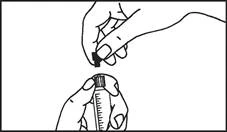
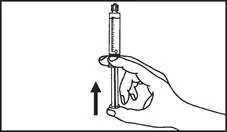
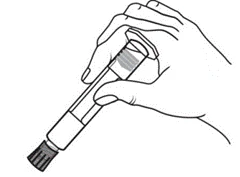
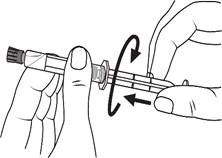
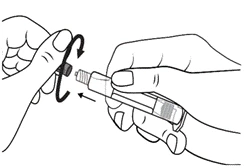
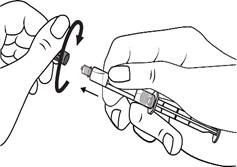
Instrucciones para el uso de la jeringa
Las jeringas precargadas deben prepararse para la inyección inmediatamente antes de su administración. La tapa de la jeringa debe retirarse únicamente inmediatamente antes de la inyección. Cualquier medicamento no utilizado o residuos deben eliminarse de acuerdo con los requisitos de la legislación local.
La etiqueta desprendible de la jeringa precargada debe pegarse en la historia clínica del paciente con el fin de garantizar una documentación clara del uso de agentes de contraste que contienen gadolinio. También debe registrarse la dosis administrada. Si se utiliza una historia clínica electrónica, debe incluirse el nombre del medicamento, el número de lote y la dosis.
Jeringa de vidrio
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
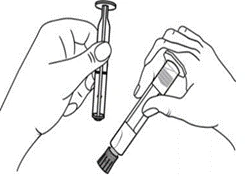
Jeringa de plástico
| Administración manual
|
Administración mediante bomba de infusión
|
|
|
|
|
|
|
|
|
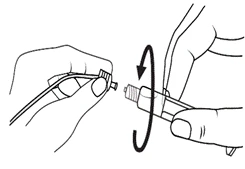
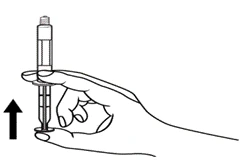
Niños.
La seguridad y eficacia del medicamento Primovist en pacientes menores de 18 años no han sido establecidas, por lo tanto, no se utiliza en esta categoría de pacientes. Los datos disponibles actualmente se presentan en la sección «Propiedades farmacológicas».
Sobredosificación.
No se han notificado casos de sobredosificación; no existen características descritas de los síntomas por sobredosificación.
Dosis únicas del medicamento Primovist de 0,4 ml/kg (0,1 mmol/kg) de peso corporal fueron bien toleradas. En un número limitado de pacientes, se estudió una dosis de 0,2 ml/kg (0,5 mmol/kg) de peso corporal durante ensayos clínicos; la frecuencia de reacciones adversas fue mayor, pero no se observaron nuevos efectos adversos en estos pacientes.
En caso de sobredosificación accidental, se recomienda la vigilancia del paciente, incluyendo el monitoreo del sistema cardiovascular, ya que es posible la inducción del alargamiento del intervalo QT (ver sección «Propiedades farmacológicas»).
El medicamento Primovist se elimina mediante diálisis. Sin embargo, no existen datos sobre la posibilidad de utilizar hemodiálisis para prevenir la fibrosis sistémica nefrogénica.
Reacciones adversas.
El perfil de seguridad del medicamento Primovist se basa en los resultados de estudios clínicos realizados con más de 1900 pacientes y en datos de vigilancia poscomercialización.
Las reacciones adversas más frecuentes (≥ 0,5 %) en pacientes que recibieron el medicamento Primovist fueron: náuseas, cefalea, sensación de calor, aumento de la presión arterial, dolor de espalda y mareo.
La reacción adversa más grave observada en pacientes que recibieron el medicamento Primovist fue el shock anafiláctico. Las reacciones anafilactoides retardadas (ocurridas desde varias horas hasta varios días después de la administración) se observaron raramente. La mayoría de los efectos adversos fueron de carácter transitorio y de intensidad leve o moderada.
Las reacciones adversas observadas con el uso del medicamento Primovist se indican en la tabla siguiente. Se han clasificado por órganos y sistemas afectados (Diccionario Médico para la Actividad Regulatoria, MedDRA, versión 12.1). Se utilizaron los términos MedDRA correspondientes para describir reacciones específicas, sus síntomas y estados clínicamente similares.
Las reacciones adversas que se enumeran a continuación, registradas durante estudios clínicos, se han clasificado según su frecuencia de aparición: frecuentes (de ≥ 1/100 a < 1/10), poco frecuentes (de ≥ 1/1000 a < 1/100) y raras (de ≥ 1/10000 a < 1/1000). La frecuencia de las reacciones adversas detectadas únicamente durante el período de vigilancia poscomercialización se indica como «frecuencia desconocida». Dentro de cada grupo, las reacciones adversas se enumeran en orden decreciente de gravedad.
Tabla. Reacciones adversas registradas durante estudios clínicos o durante el período de vigilancia poscomercialización en pacientes tratados con el medicamento Primovist
| Sistema de órganos |
Frecuentes |
Poco frecuentes |
Aislados |
Frecuencia desconocida |
| Trastornos del sistema inmunitario |
reacciones de hipersensibilidad/anafilactoides (por ejemplo, shock*, hipotensión, edema faringolaríngeo, urticaria, edema facial, rinitis, conjuntivitis, dolor abdominal, hipoestesia, estornudos, tos, palidez) |
|||
| Trastornos del sistema nervioso |
dolor de cabeza |
vértigo, mareo, disgeusia, parestesia, parosmia |
temblor, acatisia |
inquietud |
| Trastornos cardíacos |
bloqueo de rama de His, palpitaciones |
taquicardia |
||
| Trastornos vasculares |
aumento de la presión arterial, sofocos |
|||
| Trastornos respiratorios, del tórax y del mediastino |
trastornos respiratorios (disnea*, distress respiratorio) |
|||
| Trastornos gastrointestinales |
náuseas |
vómitos, sequedad de boca |
molestias en la cavidad oral, hipersecreción salival |
|
| Trastornos de la piel y del tejido subcutáneo |
erupción cutánea, prurito** |
erupción máculo-papulosa, hiperhidrosis |
||
| Trastornos del sistema músculo-esquelético y del tejido conjuntivo |
dolor de espalda |
|||
| Trastornos generales y en el lugar de inyección |
dolor en el pecho, reacciones en el lugar de inyección (de diferentes tipos)***, sensación de calor, fiebre, fatiga, sensaciones atípicas |
malestar, sensación de malestar general |
* Se han notificado estados amenazantes para la vida y/o resultados letales. Esta información se obtuvo durante el período poscomercialización.
** Prurito (prurito generalizado, prurito ocular).
*** Las reacciones en el lugar de inyección (diversos tipos) se definen mediante los siguientes términos: hematoma en el lugar de inyección, sensación de ardor en el lugar de inyección, sensación de frío en el lugar de inyección, irritación en el lugar de inyección, dolor en el lugar de inyección.
Descripción de reacciones adversas individuales
Durante los estudios clínicos se notificaron alteraciones en los parámetros de laboratorio, tales como: aumento del hierro en suero, aumento de la bilirrubina, aumento de las transaminasas hepáticas, disminución de la hemoglobina, aumento de la amilasa, leucocituria, hiperglucemia, aumento de la relación albúmina/creatinina en orina, hiponatremia, aumento de los fosfatos inorgánicos, disminución de las proteínas en suero, leucocitosis, hipopotasemia, aumento de la lactato deshidrogenasa. Durante los estudios clínicos se realizaron electrocardiogramas (ECG) de forma rutinaria, y en algunos pacientes se observó un alargamiento transitorio del intervalo QT sin eventos adversos asociados.
Se han notificado casos de fibrosis sistémica nefrogénica (FSN) con el uso de otros agentes de contraste que contienen gadolinio (ver sección «Precauciones de uso»).
Notificación de reacciones adversas sospechosas
La notificación de reacciones adversas tras la autorización del medicamento es importante. Permite continuar con el seguimiento de la relación beneficio/riesgo del medicamento. Los profesionales médicos y farmacéuticos, así como los pacientes o sus representantes legales, deben notificar todos los casos de reacciones adversas sospechosas y de falta de eficacia del medicamento a través del sistema automatizado de información de farmacovigilancia en el siguiente enlace: https://aisf.dec.gov.ua.
Período de validez.
5 años (jeringa de vidrio);
3 años (jeringa de plástico).
Condiciones de conservación.
No requiere condiciones especiales de almacenamiento.
Incompatibilidades.
Dado que no se han realizado estudios de compatibilidad, este medicamento no debe mezclarse con otros medicamentos.
Envase.
10 ml en jeringa de vidrio; 1 jeringa en caja plástica transparente cerrada con papel, incluida en una caja de cartón;
10 ml en jeringa de plástico; 1 jeringa colocada en un soporte de cartón y empaquetada en una caja de cartón.
Categoría de dispensación.
Medicamento sujeto a prescripción médica.
Fabricante.
Bayer AG.
Dirección del fabricante y lugar de actividad.
Müllerstrasse 178, 13353, Berlín, Alemania.