Primovist
Ucraina
Indice
ISTRUZIONI PER L'USO MEDICO DEL MEDICINALE PRIMOVIST (PRIMOVIST®)
Composizione:
Principio attivo: sale disodico dell'acido gadossietico (Gd-EOB-DTPA);
1 ml di soluzione iniettabile contiene 181,43 mg di sale disodico dell'acido gadossietico (Gd-EOB-DTPA) (equivalente a 0,25 mmol);
Eccipienti: sale trisodico dell'acido caossietico (Ca-EOB-DTPA), trometamolo, acido cloridrico diluito, idrossido di sodio, acqua per preparazioni iniettabili.
Forma farmaceutica. Soluzione iniettabile.
Proprietà fisico-chimiche principali: soluzione limpida, priva di inclusioni.
Gruppo farmacoterapeutico. Agenti di contrasto paramagnetici.
Codice ATC V08CA10.
Proprietà farmacologiche
Farmacodinamica.
Mecanismo d'azione
Il medicinale Primovist è un agente di contrasto paramagnetico per la risonanza magnetica.
L'effetto di potenziamento del contrasto è determinato dal gadossetato (Gd-EOB-DTPA), un complesso ionico costituito da gadolinio (III) e dal ligando acido etossibenzil-dietilentriammina-pentacetico (EOB-DTPA). Durante la scansione con sequenze impulsive pesate in T1, l'accorciamento del tempo di rilassamento longitudinale dei nuclei atomici eccitati, indotto dagli ioni di gadolinio, aumenta l'intensità del segnale e quindi migliora il contrasto dell'immagine di alcuni tessuti.
Effetti farmacodinamici
Il gadossietato disodico riduce il tempo di rilassamento anche a basse concentrazioni. A pH 7, campo magnetico di 0,47 T e temperatura di 40 °C, la rilassività r1, definita dall'effetto sul tempo di rilassamento longitudinale T1 dei protoni nel plasma, è di circa 8,7 l/mmol/s; la rilassività r2, definita dall'effetto sul tempo di rilassamento trasversale T2, è di circa 8,56 l/mmol/s. A un campo magnetico di 1,5 T e temperatura di 37 °C, la rilassività nel plasma è: r1 = 6,9 l/mmol/s, r2 = 8,7 l/mmol/s. La rilassività dipende in misura trascurabile dall'intensità del campo magnetico.
L'EOB-DTPA forma un complesso stabile con l'ione paramagnetico gadolinio, caratterizzato da un'elevatissima stabilità termodinamica (log KGdl = 23,46). Il Gd-EOB-DTPA è un composto idrofilo con elevata solubilità in acqua e un coefficiente di ripartizione tra n-butanol e tampone a pH 7,6 di circa 0,011. A causa della lipofilia del frammento etossibenzilico, il gadossietato disodico mostra un meccanismo d'azione bifasico: distribuzione nello spazio extracellulare dopo iniezione bolus, seguita da un successivo rapido e selettivo uptake da parte degli epatociti. La rilassività r1 nei tessuti epatici è di 16,6 l/mmol/s (a 0,47 T), il che aumenta l'intensità del segnale nei tessuti del fegato. Successivamente, il gadossietato disodico viene escreto con la bile.
Le lesioni con assenza o ridotta funzionalità degli epatociti (cisti, metastasi, la maggior parte dei carcinomi epatocellulari) non accumulano Primovist. Il carcinoma epatocellulare ben differenziato può contenere epatociti funzionanti e mostrare un certo grado di contrasto nell'immagine durante la fase epatocitaria. Pertanto, è necessaria un'ulteriore informazione clinica per stabilire una diagnosi corretta.
La sostanza non mostra alcun effetto inibitorio significativo sugli enzimi a concentrazioni clinicamente rilevanti.
Visualizzazione
Dopo iniezione bolus del medicinale Primovist, la visualizzazione dinamica nelle fasi arteriosa, portovenosa ed equilibrio consente di ottenere un diverso andamento temporale del contrasto nei vari tipi di lesioni epatiche, fornendo la base per la caratterizzazione radiologica delle lesioni stesse.
Il contrasto del parenchima epatico durante la fase epatocitaria permette di determinare il numero di aree interessate dal fegato, la loro distribuzione segmentaria, la visualizzazione e i margini, migliorando così l'individuazione delle lesioni. La differenziazione delle lesioni epatiche in base al comportamento dinamico del contrasto/eliminazione dell'agente di contrasto fornisce informazioni aggiuntive.
La fase ritardata (epatocitaria) può essere esaminata a partire da 20 minuti dopo l'iniezione, con un periodo di visualizzazione che dura almeno 120 minuti.
I risultati di efficacia diagnostica e tecnica ottenuti negli studi clinici indicano un miglioramento minimo della visualizzazione a 20 minuti dall'iniezione rispetto al periodo di 10 minuti dopo l'iniezione.
Il periodo di visualizzazione si riduce a 60 minuti nei pazienti sottoposti a dialisi e in quelli con livelli elevati di bilirubina (> 3 mg/dl).
L'escrezione epatica del medicinale Primovist determina il contrasto del sistema biliare.
Proprietà fisico-chimiche della soluzione pronta all'uso del medicinale Primovist
| Osmolarità a 37 °C (mOsm/kg H2O) |
688 |
| Viscosità a 37 °C (mPa·s) |
1,19 |
| Densità a 37 °C (g/ml) |
1,0881 |
| pH |
7,4 |
Pazienti pediatrici
Uno studio osservazionale è stato condotto su 52 pazienti pediatrici (di età compresa tra 2 mesi e 18 anni). I pazienti sono stati sottoposti a risonanza magnetica (RM) del fegato con l'uso del medicinale Primovist per valutare lesioni focali epatiche sospette o note. Informazioni diagnostiche aggiuntive sono state ottenute confrontando le immagini di risonanza magnetica del fegato con e senza mezzo di contrasto. Sono state riportate reazioni avverse gravi, ma nessuna è stata valutata dagli investigatori come correlata all'uso del medicinale Primovist. A causa della natura retrospettiva e del ridotto numero di campioni di questo studio, non è possibile trarre conclusioni sull'efficacia e sulla sicurezza dell'uso del medicinale nei bambini.
Farmacocinetica.
Distribuzione
Dopo somministrazione endovenosa, il profilo concentrazione-tempo del sale disodico dell'acido gadoxetico è caratterizzato da una riduzione bifasica. Il sale disodico dell'acido gadoxetico si distribuisce nello spazio extracellulare (volume di distribuzione allo stato stazionario di circa 0,21 l/kg). La sostanza mostra un legame trascurabile con le proteine (inferiore al 10%). Il medicinale attraversa la barriera placentare in quantità trascurabili.
Il gadoxetato disodico è un mezzo di contrasto lineare a base di gadolinio (GBCM). Gli studi hanno dimostrato che dopo l'uso di GBCM il gadolinio viene trattenuto nell'organismo, in particolare nel cervello e in altri tessuti e organi.
L'uso di GBCM può causare un aumento dose-dipendente dell'intensità del segnale ponderato in T1 nel cervello, in particolare nel nucleo dentato, nella substantia nigra e nel talamo. L'aumento dell'intensità del segnale e i dati preclinici indicano che il gadolinio viene rilasciato dai mezzi di contrasto lineari a base di gadolinio.
Biotrasformazione
Il gadoxetato disodico non subisce metabolismo.
Eliminazione
Il sale disodico dell'acido gadoxetico (Gd-EOB-DTPA) viene eliminato in quantità equimolari attraverso i reni e il fegato. L'emivita di eliminazione del Gd-EOB-DTPA è di circa 1 ora. La farmacocinetica è risultata lineare fino alla dose di 0,4 ml/kg (100 µmol/kg). La clearance totale (Cltot) è di circa 250 ml/min, mentre la clearance renale (Clr) è di circa 120 ml/min.
Caratteristiche nei pazienti di gruppi specifici
Pazienti anziani (65 anni e oltre). A causa dei cambiamenti fisiologici della funzione renale con l'età, la clearance plasmatica del gadoxetato disodico diminuisce da 210 ml/min nei pazienti più giovani a 163 ml/min nei pazienti di età pari o superiore a 65 anni. Il periodo finale di emivita e l'esposizione sistemica risultano più elevati nei pazienti anziani (2,3 ore e 197 µmol × h/l rispetto a 1,6 ore e 153 µmol × h/l). L'eliminazione urinaria della dose somministrata avviene completamente entro 24 ore; non sono state osservate differenze significative nell'eliminazione del gadoxetato disodico tra pazienti anziani sani e pazienti più giovani.
Insufficienza renale e/o epatica. Nei pazienti con compromissione epatica lieve o moderata, rispetto ai volontari sani, si è osservato un aumento da lieve a moderato delle concentrazioni plasmatiche, del periodo di emivita e dell'eliminazione urinaria, nonché una riduzione dell'eliminazione epatica. Tuttavia, non è stata osservata alcuna differenza clinicamente rilevante nel contrasto del segnale epatico. Nei pazienti con grave compromissione epatica, in particolare in quelli con livelli anomali elevati (> 3 mg/dl) di bilirubina, l'AUC aumentava a 259 µmol × h/l rispetto a 160 µmol × h/l nel gruppo di controllo. Il periodo di emivita risultava aumentato a 2,6 ore rispetto a 1,8 ore nel gruppo di controllo. L'eliminazione attraverso il sistema epatobiliare risultava notevolmente ridotta al 5,7% della dose somministrata, con conseguente riduzione del contrasto del segnale epatico.
Nei pazienti con insufficienza renale allo stadio terminale, l'AUC aumentava di 6 volte fino a 903 µmol × h/l e il periodo finale di emivita si prolungava fino a 20 ore. L'emodialisi aumenta la clearance del gadoxetato disodico (vedere sezione «Indicazioni particolari per l'uso»). In generale, durante una procedura di dialisi della durata di 3 ore, circa il 30% della dose di gadoxetato disodico viene eliminato entro 1 ora dall'iniezione. Oltre all'effetto dell'emodialisi sulla clearance, una frazione significativa della dose somministrata di gadoxetato disodico viene eliminata attraverso la bile in questi pazienti, come indicato dal ritrovamento mediamente di circa il 50% nelle feci entro 4 giorni (intervallo dal 24,6 al 74,0%; n = 6 pazienti).
Dati preclinici di sicurezza
Negli studi standard di tossicità acuta e subcronica, genotossicità e potenziale sensibilizzante non è stato identificato alcun rischio specifico per l'organismo umano.
Sicurezza cardiovascolare. Un lieve e transitorio prolungamento dell'intervallo QT è stato osservato nei cani durante uno studio alla dose più alta di 0,5 mmol/kg, corrispondente a 20 volte la dose umana. A concentrazioni elevate, il Gd-EOB-DTPA blocca i canali del potassio cardiaci specifici (gene hERG) e prolunga la durata del potenziale d'azione nei muscoli papillari isolati dei criceti dorati. Ciò indica la possibilità di prolungamento dell'intervallo QT in caso di sovradosaggio del medicinale Primovist. Negli studi di sicurezza farmacologica non sono stati osservati effetti su altri sistemi organici.
Tossicologia riproduttiva e allattamento. Negli studi di embriotossicità nei conigli, dopo somministrazione ripetuta di 0,2 mmol/kg di Gd-EOB-DTPA, pari a 25,9 volte (in base alla superficie corporea) o 80 volte (in base al peso corporeo) la dose raccomandata per l'uomo, è stato osservato un aumento delle perdite post-impianto e della frequenza di aborti. Nei ratti, meno dello 0,5% della dose endovenosa (0,1 mmol/kg) di gadoxetato marcato radioattivamente è stato escreto nel latte materno, con un'assorbimento dopo somministrazione orale molto basso (0,4%).
<Dati sugli animali giovani. I risultati dello studio sulla tossicità di dosi singole e multiple in ratti neonati e giovani non differivano qualitativamente da quelli osservati nei ratti adulti, ma gli animali giovani risultano più sensibili.
Tollerabilità locale. Reazioni di intolleranza locale sono state osservate solo dopo somministrazione intramuscolare di Gd-EOB-DTPA.
Carcinogenicità. Non sono stati condotti studi sulla carcinogenicità.
Caratteristiche cliniche.
Indicazioni.
Primovist è indicato per il rilevamento di lesioni focali epatiche e per ottenere informazioni aggiuntive riguardo alla natura di tali lesioni mediante imaging a risonanza magnetica pesata in T1 (MRI).
Primovist deve essere utilizzato solo quando l'informazione diagnostica è importante e non può essere ottenuta mediante imaging a risonanza magnetica (MRI) senza l'uso di un mezzo di contrasto, e quando è necessaria una fase di imaging ritardata.
Il medicinale deve essere utilizzato per via diagnostica esclusivamente per via endovenosa.
Controindicazioni.
Ipersensibilità al principio attivo o a qualsiasi altro componente del medicinale.
Misure precauzionali particolari.
Controllo. Il medicinale deve essere attentamente ispezionato prima dell'uso. Primovist non deve essere utilizzato in caso di marcato cambiamento del colore, presenza di particelle solide o compromissione dell'integrità dell'imballaggio.
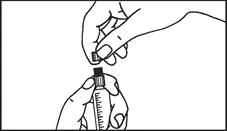
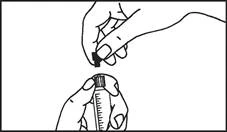
Maneggiamento. Il medicinale Primovist è pronto all'uso. La siringa pre-riempita deve essere preparata per l'iniezione immediatamente prima dell'uso. Il tappo della siringa deve essere rimosso solo immediatamente prima dell'iniezione del prodotto.
Smaltimento. Il medicinale non utilizzato o i rifiuti derivati dal suo utilizzo devono essere smaltiti in conformità con i requisiti della normativa locale.
L'etichetta rimovibile della siringa pre-riempita deve essere incollata sulla cartella clinica del paziente al fine di garantire una documentazione chiara dell'uso di agenti di contrasto contenenti gadolinio. Deve essere indicata anche la dose somministrata. Se si utilizza una cartella clinica elettronica, devono essere registrati il nome del medicinale, il numero di lotto e la dose.
Interazioni con altri medicinali e altre forme di interazione.
Il trasporto del gadossietato nel fegato può essere mediato da trasportatori peptidici organici anionici (OATP). Non si può escludere che potenti inibitori degli OATP possano causare interazioni e ridurre l'effetto di contrasto epatico. Tuttavia, non esistono dati clinici a sostegno di questa ipotesi.
Studi sull'interazione in volontari sani hanno dimostrato che la somministrazione contemporanea di eritromicina non ha influenzato l'efficacia e la farmacocinetica del medicinale Primovist. Non sono stati condotti studi clinici sull'interazione con altri medicinali.
Effetto dei livelli elevati di bilirubina o ferritina nei pazienti
Livelli elevati di bilirubina o ferritina possono ridurre l'effetto di contrasto epatico indotto dal medicinale Primovist (vedere sezione «Proprietà farmacologiche»).
Interazione con test diagnostici
Nei test per la determinazione del ferro sierico mediante metodi complessometrici (ad esempio il metodo con ferrocina), nei 24 ore successive all'esame con l'uso del medicinale Primovist, possono essere ottenuti valori falsati a causa della presenza della sostanza complessante libera nella soluzione del mezzo di contrasto.
Caratteristiche di impiego.
Devono essere rispettate le norme generali di sicurezza per l'esecuzione della risonanza magnetica, in particolare escludendo l'uso di pacemaker e impianti ferromagnetici.
Le procedure diagnostiche che prevedono l'uso di agenti di contrasto devono essere effettuate sotto la supervisione di un medico esperto nell'utilizzo di tali agenti e con sufficiente conoscenza della procedura.
Dopo l'iniezione, il paziente deve essere tenuto sotto osservazione per almeno 30 minuti, poiché l'esperienza con gli agenti di contrasto dimostra che la maggior parte delle reazioni avverse si manifesta proprio in questo periodo.
Disfunzione renale
Prima della somministrazione del medicinale Primovist, tutti i pazienti devono essere sottoposti a screening per la disfunzione renale mediante esami di laboratorio.
In pazienti con insufficienza renale acuta o cronica grave (velocità di filtrazione glomerulare < 30 ml/min/1,73 m²) sono stati osservati casi di fibrosi sistemica nefrogena (FSN) associata all'uso di agenti di contrasto contenenti gadolinio. Un rischio particolare riguarda i pazienti sottoposti a trapianto epatico, nei quali la frequenza di insufficienza renale acuta è elevata. A causa del rischio di sviluppare FSN, l'uso di Primovist deve essere evitato in pazienti con insufficienza renale grave e in pazienti nel periodo perioperatorio del trapianto epatico, salvo nei casi in cui l'informazione diagnostica sia essenziale e non possa essere ottenuta mediante risonanza magnetica senza mezzo di contrasto.
L'emodialisi effettuata a breve distanza di tempo dalla somministrazione di Primovist può essere utile per l'eliminazione del farmaco dall'organismo. Tuttavia, non esistono dati sull'uso dell'emodialisi per la prevenzione o il trattamento della FSN in pazienti non precedentemente in dialisi.
Pazienti anziani
Poiché la clearance renale del gadossietato in pazienti anziani può essere compromessa, è particolarmente importante effettuare un'indagine sulla presenza di disfunzione renale nei pazienti di età pari o superiore a 65 anni.
Pazienti con patologie cardiovascolari
Primovist deve essere utilizzato con cautela in pazienti con gravi patologie cardiovascolari a causa della limitatezza dei dati disponibili.
Primovist non deve essere somministrato a pazienti con ipokaliemia non controllata.
Primovist deve essere utilizzato con particolare cautela in pazienti:
- con sindrome congenita di allungamento dell'intervallo QT attuale o anamnestica;
- con anamnesi di aritmie durante il trattamento con farmaci che prolungano la repolarizzazione cardiaca;
- che assumono farmaci che prolungano la repolarizzazione cardiaca, come antiaritmici di classe III (amiodarone, sotalolo).
L'uso di Primovist può causare un prolungamento transitorio dell'intervallo QT in alcuni pazienti (vedere sezione «Proprietà farmacologiche»).
Ipersensibilità
Reazioni allergiche, compreso lo shock anafilattico, possono verificarsi dopo la somministrazione di agenti di contrasto per risonanza magnetica contenenti gadolinio. La maggior parte di queste reazioni avverse si manifesta entro 30 minuti dalla somministrazione del mezzo di contrasto. Tuttavia, come con altri agenti di contrasto di questa classe, possono verificarsi reazioni ritardate, che si manifestano da alcune ore a diversi giorni dopo l'iniezione. Devono sempre essere immediatamente disponibili i farmaci appropriati per il trattamento delle reazioni di ipersensibilità e le attrezzature per il soccorso d'emergenza.
Il rischio di sviluppare reazioni di ipersensibilità è maggiore in presenza delle seguenti condizioni o patologie:
- reazioni a precedenti somministrazioni di agenti di contrasto;
- asma bronchiale in anamnesi;
- reazioni allergiche in anamnesi.
La decisione di utilizzare Primovist in pazienti con predisposizione allergica deve essere presa solo dopo un'attenta valutazione del rapporto rischio/beneficio.
Le reazioni di ipersensibilità possono essere più gravi in pazienti in trattamento con beta-bloccanti, specialmente in caso di asma bronchiale. Si deve considerare che i pazienti in terapia con beta-bloccanti possono essere refrattari al trattamento standard delle reazioni di ipersensibilità con beta-agonisti.
In caso di comparsa di una reazione di ipersensibilità, la somministrazione del mezzo di contrasto deve essere immediatamente interrotta.
Intolleranza locale
La somministrazione intramuscolare può causare reazioni di intolleranza locale, compreso il necrosi focale; pertanto si raccomanda di evitare questa via di somministrazione (vedere sezione «Proprietà farmacologiche»).
Accumulo nell'organismo
Dopo la somministrazione di gadossietato disodico, il gadolinio può accumularsi nel cervello e in altri tessuti corporei (ossa, fegato, reni, pelle), causando un aumento dose-dipendente del segnale ponderato in T1 nel cervello, in particolare nel nucleo dentato, nel pallido e nel talamo. L'importanza clinica di questo fenomeno è sconosciuta. Nei pazienti che potrebbero necessitare di scansioni ripetute, si deve valutare attentamente il beneficio diagnostico derivante dall'uso del gadossietato disodico rispetto al potenziale accumulo di gadolinio nel cervello e in altri tessuti.
Eccipienti
Questo medicinale contiene 11,7 mg di sodio per 1 ml, pari allo 0,585% della dose giornaliera massima raccomandata dall'OMS di 2 g di sodio per l'adulto (4,1% (82 mg) sulla base della dose prescritta per un individuo di 70 kg di peso). La dose è di 0,1 ml/kg di peso corporeo.
Uso durante la gravidanza o l’allattamento.
Gravidanza. I dati sull'uso di agenti di contrasto a base di gadolinio in donne in gravidanza sono limitati. Il gadolinio può attraversare la placenta. Non è noto se l'esposizione al gadolinio sia associata a esiti avversi per il feto. Negli studi sugli animali, con somministrazioni ripetute di alte dosi, il farmaco ha mostrato tossicità riproduttiva (vedere sezione «Proprietà farmacologiche»). Primovist non è raccomandato durante la gravidanza se non in caso di assoluta necessità.
Allattamento. Le sostanze di contrasto contenenti gadolinio sono escrete nel latte materno in quantità molto ridotte (vedere sezione «Proprietà farmacologiche»). Con dosi cliniche, non ci si aspetta un effetto sui neonati a causa della bassa quantità di principio attivo escretato nel latte e del suo scarso assorbimento dal tratto gastrointestinale. La decisione di continuare o interrompere l’allattamento per 24 ore dopo la somministrazione di Primovist deve essere presa congiuntamente dal medico e dalla madre che allatta.
Fertilità. I risultati degli studi sugli animali non hanno evidenziato alterazioni della fertilità.
Capacità di guidare veicoli o utilizzare macchinari.
Primovist non influenza la capacità di guidare veicoli o utilizzare macchinari.
Modalità e dosaggio di somministrazione.
Modalità di somministrazione
La soluzione pronta all’uso del medicinale Primovist viene somministrata in forma non diluita mediante iniezione bolus ad una velocità di circa 2 ml/sec. Dopo l’iniezione del mezzo di contrasto, la cannula endovenosa deve essere risciacquata con soluzione fisiologica allo 0,9%.
Per informazioni riguardo alla visualizzazione, vedere la sezione «Proprietà farmacologiche».
Dosaggio
Ai fini diagnostici, deve essere utilizzata la dose più bassa possibile che garantisca un contrasto sufficiente. La dose deve essere calcolata in base al peso corporeo del paziente e non deve superare la dose raccomandata per chilogrammo di peso corporeo indicata in questa sezione.
La dose raccomandata del medicinale Primovist è la seguente:
adulti: 0,1 ml di medicinale Primovist per 1 kg di peso corporeo.
Ripetizione della dose
Non sono disponibili informazioni cliniche sull’utilizzo di dosi ripetute del medicinale Primovist.
Gruppi di pazienti particolari
Insufficienza renale
L’uso del medicinale Primovist deve essere evitato nei pazienti con grave insufficienza renale (velocità di filtrazione glomerulare < 30 ml/min/1,73 m²) e nei pazienti nel periodo perioperatorio del trapianto epatico, salvo nei casi in cui l’informazione diagnostica sia estremamente necessaria e non possa essere ottenuta mediante risonanza magnetica senza mezzo di contrasto (vedere la sezione «Avvertenze particolari e precauzioni di impiego»). Se l’uso del medicinale Primovist non può essere evitato, la dose non deve superare 0,025 mmol/kg di peso corporeo. Non deve essere somministrata più di una dose durante una singola scansione. Poiché le informazioni riguardo alla somministrazione ripetuta sono insufficienti, l’iniezione del medicinale Primovist non deve essere ripetuta, salvo nei casi in cui l’intervallo tra le iniezioni sia di almeno 7 giorni.
Pazienti con insufficienza epatica
Non è necessaria alcuna correzione della dose.
Pazienti anziani (età ≥ 65 anni)
Non è necessaria alcuna correzione della dose nei pazienti anziani. Tuttavia, si raccomanda particolare cautela nell’uso nei pazienti anziani (vedere la sezione «Avvertenze particolari e precauzioni di impiego»).
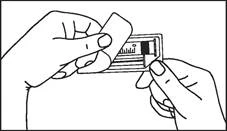
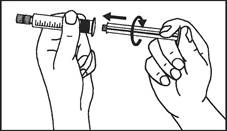
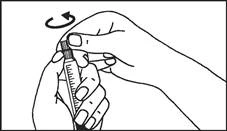
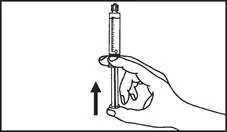
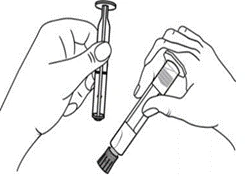
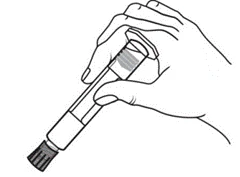
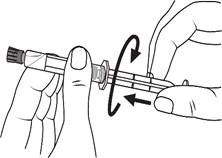
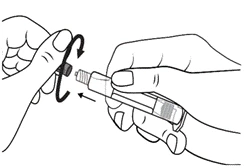
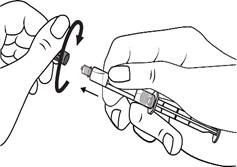
Istruzioni per l’uso della siringa
Le siringhe preriempite devono essere preparate per l’iniezione immediatamente prima della somministrazione. Il tappo della siringa deve essere rimosso solo immediatamente prima dell’iniezione del prodotto. Il medicinale non utilizzato o i rifiuti devono essere smaltiti in conformità con i requisiti della legislazione locale.
L’etichetta rimovibile della siringa preriempita deve essere incollata sulla cartella clinica del paziente al fine di garantire una documentazione chiara dell’uso di agenti di contrasto contenenti gadolinio. Deve essere inoltre indicata la dose somministrata. Se si utilizza una cartella clinica elettronica, devono essere registrati il nome del medicinale, il numero di lotto e la dose somministrata.
Siringa in vetro
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Siringa di plastica
| Administratione manuale
|
Administratione con iniettore automatico
|
|
|
|
|
|
|
|
|
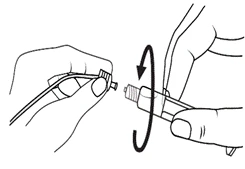
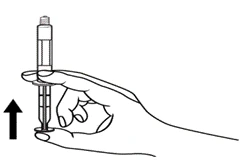
Neonati.
La sicurezza ed efficacia dell'uso del medicinale Primovist nei pazienti di età inferiore a 18 anni non sono state stabilite, pertanto non viene utilizzato in questa categoria di pazienti. I dati attualmente disponibili sono riportati nella sezione «Proprietà farmacologiche».
Sovradosaggio.
Non sono stati riportati casi di sovradosaggio e non sono note le caratteristiche dei sintomi da sovradosaggio.
Singole dosi di medicinale Primovist pari a 0,4 ml/kg (0,1 mmol/kg) di peso corporeo sono state ben tollerate. In un numero limitato di pazienti, è stata studiata una dose pari a 0,2 ml/kg (0,5 mmol/kg) di peso corporeo nell’ambito di studi clinici; la frequenza delle reazioni avverse è risultata maggiore, ma non sono stati osservati nuovi effetti indesiderati in questi pazienti.
In caso di sovradosaggio accidentale, si raccomanda il monitoraggio del paziente, compreso il monitoraggio cardiovascolare, poiché potrebbe verificarsi un’allungamento dell’intervallo QT (vedere la sezione «Proprietà farmacologiche»).
Il medicinale Primovist viene eliminato mediante dialisi. Tuttavia, non sono disponibili dati sull’eventuale utilizzo dell’emodialisi per la prevenzione della fibrosi sistemica nefrogenica.
Effetti indesiderati
Il profilo di sicurezza del medicinale Primovist si basa sui risultati di studi clinici condotti su oltre 1900 pazienti e sui dati provenienti dalla sorveglianza post-commercializzazione.
Gli effetti indesiderati più comuni (≥ 0,5 %) nei pazienti trattati con Primovist sono stati: nausea, cefalea, sensazione di calore, aumento della pressione arteriosa, dolore alla schiena e capogiri.
L’effetto indesiderato più grave osservato nei pazienti trattati con Primovist è stato lo shock anafilattico. Reazioni anafilattoidi ritardate (da alcune ore a diversi giorni dopo la somministrazione) sono state osservate raramente. La maggior parte degli effetti indesiderati aveva carattere transitorio ed era di intensità lieve o moderata.
Gli effetti indesiderati osservati con l’uso del medicinale Primovist sono riportati nella tabella seguente. Essi sono classificati per sistemi e organi (Medical Dictionary for Regulatory Activities (MedDRA), versione 12.1). I corrispondenti termini MedDRA sono stati utilizzati per descrivere specifiche reazioni, i loro sintomi e condizioni correlate.
Gli effetti indesiderati elencati di seguito, osservati durante gli studi clinici, sono classificati in base alla frequenza di insorgenza: comuni (da ≥ 1/100 a < 1/10), non comuni (da ≥ 1/1000 a < 1/100), rari (da ≥ 1/10000 a < 1/1000). La frequenza degli effetti indesiderati rilevati esclusivamente durante il periodo di sorveglianza post-commercializzazione è indicata come «frequenza non nota». All’interno di ciascuna categoria, gli effetti indesiderati sono elencati in ordine decrescente di gravità.
Tabella. Effetti indesiderati osservati durante studi clinici o nel periodo di sorveglianza post-commercializzazione in pazienti trattati con il medicinale Primovist
| Sistema degli organi |
Comuni |
Non comuni |
Isolati |
Frequenza sconosciuta |
| Disturbi del sistema immunitario |
reazioni di ipersensibilità/anafilattoidi (ad esempio shock*, ipotensione, edema faringolaringeo, orticaria, edema del volto, rinite, congiuntivite, dolore addominale, ipoestesia, starnuti, tosse, pallore) |
|||
| Disturbi del sistema nervoso |
cefalea |
vertigini, capogiri, disgeusia, parastesia, parosmia |
tremore, acatisia |
agitazione |
| Disturbi cardiaci |
blocco di branca di His, palpitazioni |
tachicardia |
||
| Disturbi vascolari |
aumento della pressione arteriosa, vampate |
|||
| Disturbi del sistema respiratorio, torace e mediastino |
disturbi del sistema respiratorio (dispnea*, distress respiratorio) |
|||
| Disturbi gastrointestinali |
nausea |
vomito, secchezza della bocca |
disagio orale, ipersalivazione |
|
| Disturbi della cute e del tessuto sottocutaneo |
eruzione cutanea, prurito** |
eruzione maculo-papulosa, iperidrosi |
||
| Disturbi del sistema muscoloscheletrico e del tessuto connettivo |
dolore alla schiena |
|||
| Disturbi generali e condizioni nel sito di iniezione |
dolore al petto, reazioni nel sito di iniezione (diverse tipologie)***, sensazione di calore, febbre, affaticamento, sensazioni atipiche |
disagio, malessere |
* Sono stati segnalati stati potenzialmente letali e/o esiti fatali. Queste segnalazioni provengono dal periodo post-commercializzazione.
** Prurito (prurito generalizzato, prurito oculare).
*** Le reazioni nel sito di iniezione (di vario tipo) sono definite dai seguenti termini: ematoma nel sito di iniezione, sensazione di bruciore nel sito di iniezione, sensazione di freddo nel sito di iniezione, irritazione nel sito di iniezione, dolore nel sito di iniezione.
Descrizione di reazioni avverse specifiche
Durante gli studi clinici sono state segnalate alterazioni degli esami di laboratorio, quali: aumento del ferro sierico, aumento della bilirubina, aumento delle transaminasi epatiche, diminuzione dell'emoglobina, aumento dell'amilasi, leucocituria, iperglicemia, aumento del rapporto albumina/creatinina nelle urine, iponatriemia, aumento dei fosfati inorganici, diminuzione delle proteine sieriche, leucocitosi, ipokaliemia, aumento della lattato deidrogenasi. Negli studi clinici è stata eseguita regolarmente l'ECG e in alcuni pazienti è stato osservato un prolungamento transitorio dell'intervallo QT, senza alcuna reazione avversa correlata.
Sono stati segnalati casi di fibrosi sistemica nefrogena (FSN) con l'uso di altri mezzi di contrasto contenenti gadolinio (vedere paragrafo «Informazioni importanti di sicurezza»).
Segnalazione delle reazioni avverse sospette
La segnalazione delle reazioni avverse sospette dopo l'autorizzazione del medicinale è importante. Permette un monitoraggio continuo del rapporto beneficio/rischio del medicinale. I professionisti sanitari e farmaceutici, nonché i pazienti o i loro rappresentanti legali, devono segnalare tutti i casi sospetti di reazioni avverse e l'assenza di efficacia del medicinale attraverso il sistema informativo automatizzato di farmacovigilanza al seguente indirizzo: https://aisf.dec.gov.ua.
Durata della validità.
5 anni (siringa in vetro);
3 anni (siringa in plastica).
Condizioni di conservazione.
Non richiede condizioni particolari di conservazione.
Incompatibilità.
Poiché non sono stati effettuati studi di compatibilità, questo medicinale non deve essere miscelato con altri medicinali.
Confezionamento.
10 ml in siringa di vetro; 1 siringa in un contenitore di plastica trasparente chiuso con carta, inserito in una scatola di cartone;
10 ml in siringa di plastica; 1 siringa inserita in un supporto di cartone e confezionata in una scatola di cartone.
Categoria di prescrizione.
Sotto prescrizione medica.
Produttore.
Bayer AG.
Indirizzo del produttore e sede operativa.
Müllerstrasse 178, 13353 Berlino, Germania.