Ozempic®
Ukraina
Spis treści
- INSTRUKCJA DOTYCZĄCA STOSOWANIA LEKU OZEMPIC®
- Skład:
- Właściwości farmakologiczne
- Właściwości kliniczne
- Szczególne ostrzeżenia i środki ostrożności dotyczące stosowania
- Sposób stosowania i dawki
- Efekty uboczne
- Efekty uboczne
- Skład:
- Właściwości farmakodynamiczne
- Właściwości kliniczne
- Szczególne wskazania dotyczące stosowania
- Sposób stosowania i dawki
- Niepożądane reakcje
- Efekty uboczne
INSTRUKCJA DOTYCZĄCA STOSOWANIA LEKU OZEMPIC®
Skład:
substancja czynna: semaglutyd;
1 ml roztworu zawiera 1,34 mg semaglutydu – analogu ludzkiego peptydu podobnego do glukagonu-1 (GLP-1), wyprodukowanego w Saccharomyces cerevisiae metodą rekombinowanego DNA;
jeden wypełniony wcześniej strzykacz długopisowy zawiera 2 mg semaglutydu w 1,5 ml roztworu;
substancje pomocnicze: fosforan sodu dwuwodny; glikol propylenowy; fenol; kwas solny (do regulacji pH); wodorotlenek sodu (do regulacji pH); woda do wstrzykiwań.
Postać farmaceutyczna. Roztwór do wstrzykiwań.
Główne właściwości fizykochemiczne: przezroczysty, bezbarwny lub prawie bezbarwny roztwór izotoniczny; pH = 7,4.
Grupa farmakoterapeutyczna. Leki stosowane w cukrzycy, analogi peptydu podobnego do glukagonu-1 (GLP-1). Kod ATX A10B J06.
Właściwości farmakologiczne
Farmakodynamika
Mechanizm działania
Semaglutyd jest analogiem GLP-1 z 94 % homologią do GLP-1 człowieka. Działa jako agonista receptorów GLP-1, wiąże się i aktywuje selektywnie receptory GLP-1, które są celem działania naturalnego GLP-1.
GLP-1 jest hormonem fizjologicznym, który wpływa na regulację stężenia glukozy i apetytu oraz na układ sercowo-naczyniowy. Wpływ na stężenie glukozy i apetyt jest pośrednictwem receptorów GLP-1, zlokalizowanych w trzustce i mózgu.
Semaglutyd obniża stężenie glukozy we krwi, działając w sposób zależny od glukozy, stymulując wydzielanie insuliny i hamując wydzielanie glukagonu przy podwyższonym stężeniu glukozy we krwi. Mechanizm obniżania stężenia glukozy we krwi towarzyszy również nieznaczne opóźnienie opróżniania żołądka w wczesnej fazie poposiłkowej. W przypadku hipoglikemii semaglutyd zmniejsza wydzielanie insuliny i nie przeszkadza w wydzielaniu glukagonu.
Stosowanie semaglutyd powoduje zmniejszenie masy ciała i masy tkanki tłuszczowej poprzez zmniejszenie spożycia kalorii oraz obniżenie apetytu. Ponadto semaglutyd zmniejsza popęd do jedzenia żywności o wysokiej zawartości tłuszczu.
Ekspresja receptorów GLP-1 zachodzi również w sercu, układzie naczyniowym, układzie odpornościowym i nerkach.
W trakcie badań klinicznych semaglutyd pozytywnie wpływał na poziom lipidów w osoczu krwi, obniżał ciśnienie tętnicze skurczowe i zmniejszał stan zapalny. W badaniach na zwierzętach semaglutyd hamował rozwój miażdżycy, zapobiegając postępowi blaszek aorty i zmniejszając stan zapalny w blaszkach.
Skutki farmakodynamiczne
Wszystkie badania farmakodynamiczne przeprowadzono po 12 tygodniach leczenia (w tym z etapem nasycenia dawkowania) w stanie równowagi po zastosowaniu semaglutyd w dawce 1 mg raz w tygodniu.
Stężenie glukozy na czczo i stężenie glukozy w fazie poposiłkowej
Semaglutyd prowadzi do zmniejszenia stężenia glukozy na czczo i stężenia glukozy w fazie poposiłkowej. U pacjentów z cukrzycą typu 2 leczenie semaglutydem w dawce 1 mg prowadziło do zmniejszenia stężenia glukozy pod względem zmiany absolutnej od wartości wyjściowej (mmol/l) oraz względnej redukcji w porównaniu do placebo (%) dla parametrów stężenia glukozy na czczo (1,6 mmol/l; 22 %), stężenia glukozy 2 godziny po posiłku (4,1 mmol/l; 37 %), średniego dobowego stężenia glukozy (1,7 mmol/l; 22 %) oraz wahań stężenia glukozy poposiłkowej w trakcie trzech posiłków (0,6–1,1 mmol/l) w porównaniu do placebo. Semaglutyd obniżał stężenie glukozy na czczo już po pierwszym zastosowaniu.
Funkcja komórek β i wydzielanie insuliny
Semaglutyd poprawia funkcję komórek β. W porównaniu do placebo semaglutyd poprawiał pierwszą i drugą fazę odpowiedzi insuliny z trzykrotnym i dwukrotnym wzrostem odpowiednio, a także zwiększał maksymalną aktywność sekrecyjną komórek β u pacjentów z cukrzycą typu 2. Ponadto, w porównaniu do placebo leczenie semaglutydem prowadziło do zwiększenia stężenia insuliny na czczo.
Wydzielanie glukagonu
Semaglutyd zmniejsza stężenie glukagonu na czczo i stężenie glukagonu w fazie poposiłkowej. U pacjentów z cukrzycą typu 2 zastosowanie semaglutyd prowadziło do względnej redukcji stężenia glukagonu w porównaniu do placebo: stężenie glukagonu na czczo (8–21 %), odpowiedź glukagonu w fazie poposiłkowej (14–15 %) oraz średnie dobowe stężenie glukagonu (12 %).
Wydzielanie insuliny i glukagonu zależne od glukozy
Semaglutyd obniżał wysokie stężenie glukozy we krwi poprzez stymulację wydzielania insuliny i zmniejszenie wydzielania glukagonu, działając w sposób zależny od glukozy. Szybkość wydzielania insuliny po zastosowaniu semaglutyd u pacjentów z cukrzycą typu 2 była porównywalna do tej u osób zdrowych.
W przypadku wywołanej hipoglikemii semaglutyd w porównaniu do placebo nie zmieniał reakcji kontrregulacyjnej podwyższenia stężenia glukagonu i nie nasilał zmniejszenia stężenia peptydu C u pacjentów z cukrzycą typu 2.
Opróżnianie żołądka
Semaglutyd powodował nieznaczne opóźnienie wczesnego poposiłkowego opróżniania żołądka, co prowadziło do zmniejszenia szybkości napływu glukozy do krwi w okresie poposiłkowym.
Apetyt, spożycie kalorii i wybór produktów spożywczych
W porównaniu do placebo semaglutyd prowadził do zmniejszenia spożycia kalorii o 18–35 % podczas trzech kolejnych dowolnych posiłków ad libitum. Wspomagało to wywołane przez semaglutyd hamowanie apetytu zarówno na czczo, jak i w okresie poposiłkowym, poprawę kontroli spożycia pokarmu, osłabienie popędu do przekąsek oraz względnie mniejszy popęd do jedzenia żywności o wysokiej zawartości tłuszczu.
Lipidy na czczo i w fazie poposiłkowej
W porównaniu do placebo semaglutyd zmniejszał stężenia trójglicerydów na czczo i cholesterolu lipoprotein o bardzo niskiej gęstości (VLDL) na czczo o 12 % i 21 % odpowiednio. Reakcja trójglicerydów i reakcja cholesterolu VLDL na posiłek o wysokiej zawartości tłuszczu zmniejszały się o ponad 40 %.
Elektrofizjologia serca (QTc)
Wpływ semaglutyd na proces repolaryzacji serca oceniano w starannie przeprowadzonym badaniu QTc. Semaglutyd nie wydłużał odstępów QTc w dawkach do 1,5 mg w stanie równowagi.
Skuteczność kliniczna i bezpieczeństwo
Zarówno poprawa kontroli glikemicznej, jak i zmniejszenie zachorowań i śmiertelności sercowo-naczyniowej są integralną częścią terapii cukrzycy typu 2.
Skuteczność i bezpieczeństwo stosowania semaglutyd w dawkach 0,5 mg i 1 mg raz w tygodniu oceniano w sześciu randomizowanych, kontrolowanych badaniach klinicznych fazy 3a z udziałem 7215 pacjentów z cukrzycą typu 2 (4107 pacjentów otrzymywało semaglutyd). Głównym celem pięciu badań klinicznych (SUSTAIN 1–5) była ocena skuteczności kontroli glikemicznej, a jednego badania (SUSTAIN 6) – skutków sercowo-naczyniowych leczenia.
Przeprowadzono dodatkowe badanie kliniczne fazy 3b (SUSTAIN 7) z udziałem 1201 pacjentów w celu porównania skuteczności i bezpieczeństwa stosowania semaglutyd w dawkach 0,5 mg i 1 mg raz w tygodniu z przyjmowaniem dulaglutyd w dawkach 0,75 mg i 1,5 mg odpowiednio raz w tygodniu. Badanie fazy 3b (SUSTAIN 9) zostało przeprowadzone w celu zbadania skuteczności i bezpieczeństwa stosowania semaglutyd jako uzupełnienia leczenia inhibitorem współtransportera sodu-glukozy typu 2 (SGLT-2).
Leczenie semaglutyd wykazało trwałe, statystycznie bardziej znaczące i klinicznie istotne zmniejszenie wartości HbA1c i masy ciała w okresie do dwóch lat w porównaniu do placebo i aktywnego leczenia kontrolnego (sitagliptyna, insulinę glarginy, egzekutyd ER i dulaglutyd).
Skuteczność semaglutyd nie zależała od wieku, płci, rasy, pochodzenia etnicznego, początkowego wskaźnika BMI, początkowej masy ciała (kg), czasu trwania cukrzycy i stopnia zaburzenia funkcji nerek pacjenta.
Wartości osiągnęły cele badania we wszystkich randomizowanych grupach pacjentów (analizy oparte na mieszanych modelach dla powtarzanych pomiarów lub wielokrotnym warunkowym obliczaniu).
Ponadto przeprowadzono badanie fazy 3b (SUSTAIN 11) w celu zbadania efektu semaglutyd w porównaniu z efektem insuliny aspart jako uzupełnienia do metforminy i zoptymalizowanej insuliny glarginy (100 J).
Szczegółowe informacje można znaleźć poniżej.
Badanie SUSTAIN 1. Monoterapia
W podwójnie ślepej, kontrolowanej placebo, trwającej 30 tygodni, 388 pacjentów z niewystarczającą kontrolą choroby przy stosowaniu diety i aktywności fizycznej zostało zrandomizowanych do grup przyjmujących raz w tygodniu semaglutyd w dawce 0,5 mg lub 1 mg albo placebo.
Tabela 1
Badanie SUSTAIN 1: wyniki w 30. tygodniu
| Wskaźnik |
Semaglutyd 0,5 mg |
Semaglutyd 1 mg |
Placebo |
| Liczba pacjentów |
128 |
130 |
129 |
| HbA1c (%) |
|||
| Na początku (wartość średnia) |
8,1 |
8,1 |
8,0 |
| Zmiana od początku do 30. tygodnia |
-1,5 |
-1,6 |
0 |
| Różnica w porównaniu z placebo (95 % CI) |
-1,4 [-1,7, -1,1]a |
-1,5 [-1,8, -1,2]a |
- |
| Pacjenci (%), którzy osiągnęli HbA1c < 7 % |
74 |
72 |
25 |
| Glukoza we krwi na czczo (mmol/l) |
|||
| Na początku (wartość średnia) |
9,7 |
9,9 |
9,7 |
| Zmiana od początku do 30. tygodnia |
-2,5 |
-2,3 |
-0,6 |
| Masa ciała (kg) |
|||
| Na początku (wartość średnia) |
89,8 |
96,9 |
89,1 |
| Zmiana od początku do 30. tygodnia |
-3,7 |
-4,5 |
-1,0 |
| Różnica w porównaniu z placebo (95 % CI) |
-2,7 [-3,9, -1,6]a |
-3,6 [-4,7, -2,4]a |
- |
a p < 0,0001 (dwustronny) na korzyść.
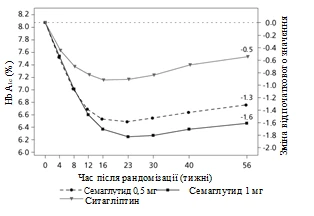
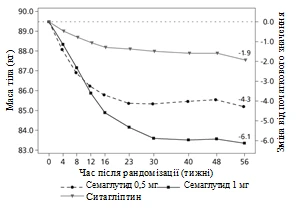
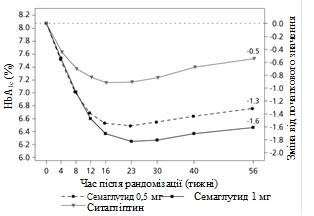
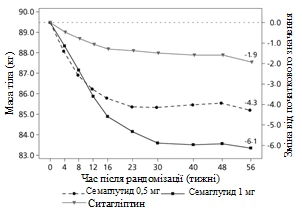
Badanie kliniczne SUSTAIN 2. Semaglutyd w porównaniu z sytagliptyną, oba w kombinacji z jednym-dwuoma doustnymi lekami przeciwcukrzycowymi (metforminą i/lub tiazolidonynami)
W podwójnie zaślepionym badaniu z aktywnym kontrolem trwającym 56 tygodni 1231 pacjentów zostało przydzielonych randomizacyjnie do grupy przyjęcia albo semaglutydu 0,5 mg raz w tygodniu albo 1 mg raz w tygodniu, albo sytagliptyny 100 mg raz dziennie; wszystkie w kombinacji z metforminą (94 %) i/lub tiazolidonynami (6 %).
Tabela 2
Badanie SUSTAIN 2: wyniki w 56. tygodniu
| Wskaźnik |
Semaglutyd 0,5 mg |
Semaglutyd 1 mg |
Sytagliptyna 100 mg |
| Liczba pacjentów |
409 |
409 |
407 |
| HbA1c (%) |
|||
| Na początku (wartość średnia) |
8,0 |
8,0 |
8,2 |
| Zmiana od początku do 56. tygodnia |
-1,3 |
-1,6 |
-0,5 |
| Różnica w porównaniu z sytagliptyną (95 % CI) |
-0,8 [-0,9, -0,6]a |
-1,1 [-1,2, -0,9]a |
- |
| Pacjenci (%), którzy osiągnęli HbA1c < 7 % |
69 |
78 |
36 |
| Glukoza we krwi na czczo (mmol/l) |
|||
| Na początku (wartość średnia) |
9,3 |
9,3 |
9,6 |
| Zmiana od początku do 56. tygodnia |
-2,1 |
-2,6 |
-1,1 |
| Masa ciała (kg) |
|||
| Na początku (wartość średnia) |
89,9 |
89,2 |
89,3 |
| Zmiana od początku do 56. tygodnia |
-4,3 |
-6,1 |
-1,9 |
| Różnica w porównaniu z sytagliptyną (95 % CI) |
-2,3 [-3,1, -1,6]a |
-4,2 [-4,9, -3,5]a |
- |
a p < 0,0001 (dwustronny) na korzyść.
**
**
Rys. 1. Średnia zmiana HbA1c (%) oraz masy ciała (kg) od początku do 56. tygodnia
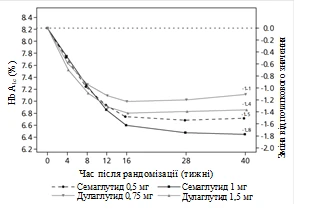
Badanie kliniczne SUSTAIN 7. Semaglutyd w porównaniu z dulaglutidem, oba w połączeniu z metforminą
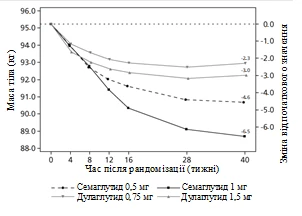
Uczestników otwartego badania klinicznego trwającego 40 tygodni (1201 pacjentów), którzy przyjmowali metforminę, przydzielono losowo w stosunku 1:1:1:1 do grup otrzymujących raz w tygodniu semaglutyd w dawce 0,5 mg lub dulaglutid w dawce 0,75 mg, lub semaglutyd w dawce 1 mg, lub dulaglutid w dawce 1,5 mg.
W badaniu porównywano wyniki stosowania semaglutydu 0,5 mg z dulaglutidem 0,75 mg oraz semaglutydu 1 mg z dulaglutidem 1,5 mg.
Najczęstszymi reakcjami niepożądanymi były zaburzenia ze strony przewodu pokarmowego, które występowały u takiej samej frakcji pacjentów otrzymujących semaglutyd 0,5 mg (129 pacjentów [43 %]), semaglutyd 1 mg (133 [44 %]) oraz dulaglutid 1,5 mg (143 [48 %]); zaburzenia ze strony przewodu pokarmowego występowały rzadziej u pacjentów leczonych dulaglutidem 0,75 mg (100 [33 %]).
W 40. tygodniu zwiększenie częstości tętna pod wpływem semaglutydu (0,5 mg i 1 mg) oraz dulaglutidu (0,75 mg i 1,5 mg) wynosiło odpowiednio 2,4 i 4,0 oraz 1,6 i 2,1 uderzenia na minutę.
Tabela 3
Badanie SUSTAIN 7: wyniki w 40. tygodniu
| Wskaźnik |
Semaglutyd 0,5 mg |
Semaglutyd 1 mg |
Dulaglutyd 0,75 mg |
Dulaglutyd 1,5 mg |
| Liczba pacjentów |
301 |
300 |
299 |
299 |
| HbA1c (%) |
||||
| Na początku (wartość średnia) |
8,3 |
8,2 |
8,2 |
8,2 |
| Zmiana od początku do 40. tygodnia |
-1,5 |
-1,8 |
-1,1 |
-1,4 |
| Różnica w porównaniu z dulaglutydem (95 % CI) |
-0,4b [-0,6, -0,2)a |
-0,4c [-0,6, -0,3]a |
- |
- |
| Pacjenci (%), którzy osiągnęli HbA1c < 7 % |
68 |
79 |
52 |
67 |
| Glukoza we krwi na czczo (mmol/l) |
||||
| Na początku (wartość średnia) |
9,8 |
9,8 |
9,7 |
9,6 |
| Zmiana od początku do 40. tygodnia |
-2,2 |
-2,8 |
-1,9 |
-2,2 |
| Masa ciała (kg) |
||||
| Na początku (wartość średnia) |
96,4 |
95,5 |
95,6 |
93,4 |
| Zmiana od początku do 40. tygodnia |
-4,6 |
-6,5 |
-2,3 |
-3,0 |
| Różnica w porównaniu z dulaglutydem (95 % CI) |
-2,3b [-3,0, -1,5)a |
-3,6c [-4,3, -2,8]a |
- |
- |
a p < 0,0001 (dwustronny) na korzyść.
b Semaglutyd 0,5 mg w porównaniu z dulaglutydem 0,75 mg.
c Semaglutyd 1 mg w porównaniu z dulaglutydem 1,5 mg.
**
**
Rys. 2. Średnia zmiana HbA1c (%) oraz masy ciała (kg) od początku do 40. tygodnia
Badanie kliniczne SUSTAIN 3. Semaglutyd w porównaniu z eksenatydem ER, oba w połączeniu z metforminą lub metforminą i sulfonylomocznikiem
W otwartym badaniu trwającym 56 tygodni 813 pacjentów przyjmujących wyłącznie metforminę (49 %), metforminę ze związkami sulfonylomocznymi (45 %) lub inne leki (6 %) zostało zrandomizowanych do grupy przyjmującej semaglutyd 1 mg lub eksenatyd ER 2 mg raz w tygodniu.
Tabela 4
Badanie SUSTAIN 3: wyniki w 56. tygodniu
| Wskaźnik |
Semaglutyd 1 mg |
Eksyenatyd ER 2 mg |
| Liczba pacjentów |
404 |
405 |
| HbA1c (%) |
||
| Na początku (wartość średnia) |
8,4 |
8,3 |
| Zmiana od początku do 56. tygodnia |
-1,5 |
-0,9 |
| Różnica w porównaniu z eksyenatydem (95 % CI) |
-0,6 [-0,8, -0,4]a |
- |
| Pacjenci (%), którzy osiągnęli HbA1c < 7 % |
67 |
40 |
| Glukoza we krwi na czczo (mmol/l) |
||
| Na początku (wartość średnia) |
10,6 |
10,4 |
| Zmiana od początku do 56. tygodnia |
-2,8 |
-2,0 |
| Masa ciała (kg) |
||
| Na początku (wartość średnia) |
96,2 |
95,4 |
| Zmiana od początku do 56. tygodnia |
-5,6 |
-1,9 |
| Różnica w porównaniu z eksyenatydem (95 % CI) |
-3,8 [-4,6, -3,0]a |
- |
a p < 0,0001 (dwustronny) na korzyść.
Badanie kliniczne SUSTAIN 4. Semaglutyd w porównaniu do insuliny glarginy, obu stosowanych w połączeniu z jednym-dwoma doustnymi lekami przeciwcukrzycowymi (metformyną lub metformyną i sulfonemoleem)
W otwartym badaniu porównawczym trwającym 30 tygodni 1089 pacjentów zostało losowo przydzielonych do grupy otrzymującej semaglutyd w dawce 0,5 mg raz w tygodniu, semaglutyd w dawce 1 mg raz w tygodniu lub insulinę glarginę raz dziennie, jednocześnie stosując metformynę (48 %) lub metformynę i sulfonemole (51 %).
Tabela 5
Badanie SUSTAIN 4: wyniki w 30. tygodniu
| Wskaźnik |
Semaglutyd 0,5 mg |
Semaglutyd 1 mg |
Insulin glargina |
| Liczba pacjentów |
362 |
360 |
360 |
| HbA1c (%) |
|||
| Na początku (wartość średnia) |
8,1 |
8,2 |
8,1 |
| Zmiana od początku do 30. tygodnia |
-1,2 |
-1,6 |
-0,8 |
| Różnica w porównaniu do insuliny glarginy (95 % CI) |
-0,4 [-0,5, -0,2]a |
-0,8 [-1,0, -0,7]a |
- |
| Pacjenci (%), którzy osiągnęli HbA1c < 7 % |
57 |
73 |
38 |
| Glukoza we krwi na czczo (mmol/l) |
|||
| Na początku (wartość średnia) |
9,6 |
9,9 |
9,7 |
| Zmiana od początku do 30. tygodnia |
-2,0 |
-2,7 |
-2,1 |
| Masa ciała (kg) |
|||
| Na początku (wartość średnia) |
93,7 |
94,0 |
92,6 |
| Zmiana od początku do 30. tygodnia |
-3,5 |
-5,2 |
+1,2 |
| Różnica w porównaniu do insuliny glarginy (95 % CI) |
-4,6 [-5,3, -4,0]a |
-6,34 [-7,0, -5,7]a |
- |
a p < 0,0001 (dwustronny) na korzyść.
Badanie kliniczne SUSTAIN 5. Semaglutyd w porównaniu z placebo, oba stosowane w połączeniu z insuliną bazalną
W podwójnie ślepej, kontrolowanej placebo studium trwającym 30 tygodni, 397 pacjentów z niewystarczającym kontrolowaniem leczenia insuliną bazalną bez stosowania lub ze stosowaniem metforminy, zostało losowo przydzielonych do grup otrzymujących semaglutyd 0,5 mg raz w tygodniu, semaglutyd 1 mg raz w tygodniu lub placebo.
Tabela 6
Badanie SUSTAIN 5: wyniki w 30. tygodniu
| Wskaźnik |
Semaglutyd 0,5 mg |
Semaglutyd 1 mg |
Placebo |
| Liczba pacjentów |
132 |
131 |
133 |
| HbA1c (%) |
|||
| Na początku (wartość średnia) |
8,4 |
8,3 |
8,4 |
| Zmiana od początku do 30. tygodnia |
-1,4 |
-1,8 |
-0,1 |
| Różnica w porównaniu z placebo (95 % CI) |
-1,4 [-1,6, -1,1]a |
-1,8 [-2,0, -1,5]a |
- |
| Pacjenci (%), którzy osiągnęli HbA1c < 7 % |
61 |
79 |
11 |
| Glukoza we krwi na czczo (mmol/l) |
|||
| Na początku (wartość średnia) |
8,9 |
8,5 |
8,6 |
| Zmiana od początku do 30. tygodnia |
-1,6 |
-2,4 |
-0,5 |
| Masa ciała (kg) |
|||
| Na początku (wartość średnia) |
92,7 |
92,5 |
89,9 |
| Zmiana od początku do 30. tygodnia |
-3,7 |
-6,4 |
-1,4 |
| Różnica w porównaniu z placebo (95 % CI) |
-2,3 [-3,3, -1,3]a |
-5,1 [-6,1, -4,0]a |
- |
a p < 0,0001 (dwustronny) na korzyść.
Badanie kliniczne SUSTAIN 9. Semaglutyd w porównaniu z placebo, oba w połączeniu z inhibitorem SGLT-2 ± metformyną lub sulfonylomocznem
W podwójnie ślepej, kontrolowanej placebo studii trwającej 30 tygodni, 302 pacjentów z niewystarczającą kontrolą leczenia inhibitorem SGLT-2 bez stosowania lub ze stosowaniem metformyny lub sulfonylomoczniny zostało losowo przydzielonych do grupy przyjmującej semaglutyd 1,0 mg raz w tygodniu lub do grupy placebo.
Tabela 7
Badanie SUSTAIN 9: wyniki na 30. tydzień
| Wskaźnik |
Semaglutyd 1 mg |
Placebo |
| Liczba pacjentów |
151 |
151 |
| HbA1c (%) |
||
| Na początku (wartość średnia) |
8,0 |
8,1 |
| Zmiana od początku do 30. tygodnia |
-1,5 |
-0,1 |
| Różnica w porównaniu do placebo (95 % CI) |
-1,4 [-1,6, -1,2]a |
- |
| Pacjenci (%), którzy osiągnęli HbA1c < 7 % |
78,7 |
18,7 |
| Glukoza we krwi na czczo (mmol/l) |
||
| Na początku (wartość średnia) |
9,1 |
8,9 |
| Zmiana od początku do 30. tygodnia |
-2,2 |
0,0 |
| Masa ciała (kg) |
||
| Na początku (wartość średnia) |
89,6 |
93,8 |
| Zmiana od początku do 30. tygodnia |
-4,7 |
-0,9 |
| Różnica w porównaniu do placebo (95 % CI) |
-3,8 [-4,7, -2,9]a |
- |
a p < 0,0001 (dwustronny) dla korzyści, skorygowany pod względem wielokrotności na podstawie hierarchicznego testowania wartości HbA1c i masy ciała.
Badanie kliniczne SUSTAIN 11 – semaglutyd w porównaniu z insuliną aspart jako dodatek do insuliny glarginy + metforminy
W 52-tygodniowym otwartym badaniu 1748 pacjentów z niedostatecznie kontrolowanym cukrzycą typu 2 po 12-tygodniowym okresie wprowadzenia insuliny glarginy i metforminy podzielono losowo w stosunku 1:1 na grupę otrzymującą semaglutyd raz w tygodniu (0,5 mg lub 1,0 mg) lub insuliny aspart trzy razy dziennie. W populacji włączonych średnia długość trwania cukrzycy wynosiła 13,4 roku, a średni poziom HbA1c 8,6% z docelowym HbA1c w zakresie 6,5–7,5%.
Leczenie semaglutydem doprowadziło do obniżenia HbA1c w 52. tygodniu (–1,5% dla semaglutydu w porównaniu z –1,2% dla insuliny aspart).
Liczba epizodów hipoglikemii ciężkiej w obu grupach leczenia była niska (4 epizody przy stosowaniu semaglutydu w porównaniu z 7 epizodami przy stosowaniu insuliny aspart).
Średnia początkowa masa ciała zmniejszyła się przy stosowaniu semaglutydu (–4,1 kg) i wzrosła przy stosowaniu insuliny aspart (+2,8 kg), przewidywana różnica zależna od leczenia wyniosła –6,99 kg (95% CI od –7,41 do –6,57) w 52. tygodniu.
Stosowanie łączone z monoterapią sulfonylomocznikami
W badaniu klinicznym SUSTAIN 6 (patrz sekcja „Choroby układu sercowo-naczyniowego”) 123 pacjentów początkowo stosowało monoterapię sulfonylomocznikami. Początkowy poziom HbA1c wynosił 8,2%, 8,4% i 8,4% odpowiednio przy stosowaniu semaglutydu 0,5 mg, semaglutydu 1 mg i placebo. Po 30 tygodniach poziom HbA1c zmniejszył się o 1,6%, 1,5% odpowiednio przy stosowaniu semaglutydu 0,5 mg i semaglutydu 1 mg oraz wzrósł o 0,1% przy stosowaniu placebo.
Stosowanie łączone z insuliną premiks ± jeden-dwa doustne leki obniżające poziom glukozy we krwi (POPG)
W badaniu SUSTAIN 6 (patrz sekcja „Choroby układu sercowo-naczyniowego”) 867 pacjentów stosowało insulinę premiks (na początku razem z POPG lub bez nich). Początkowy poziom HbA1c wynosił 8,8%, 8,9% i 8,9% odpowiednio przy stosowaniu semaglutydu 0,5 mg, semaglutydu 1 mg i placebo. Po 30 tygodniach poziom HbA1c zmniejszył się o 1,3%, 1,8% i 0,4% odpowiednio przy stosowaniu semaglutydu 0,5 mg, semaglutydu 1 mg i placebo.
Choroby układu sercowo-naczyniowego
W podwójnie ślepej, klinicznej próbie trwającej 104 tygodnie (SUSTAIN 6) 3297 pacjentów z cukrzycą typu 2 i wysokim ryzykiem sercowo-naczyniowym zostało losowo przydzielonych do grupy otrzymującej semaglutyd 0,5 mg lub semaglutyd 1 mg raz w tygodniu lub placebo jako dodatek do standardowego leczenia przez następne 2 lata. Ogółem 98% pacjentów ukończyło badanie, a na końcu dane dotyczące funkcji życiowych były dostępne dla 99,6% z nich.
Rozkład populacji w badaniu według wieku był następujący: 1598 pacjentów (48,5%) w wieku ≥ 65 lat, 321 (9,7%) w wieku ≥ 75 lat i 20 (0,6%) w wieku ≥ 85 lat. U 2358 pacjentów funkcja nerek była normalna lub z lekkim zaburzeniem, u 832 pacjentów zaburzenie funkcji nerek było umiarkowane, a u 107 pacjentów ciężkie lub zaawansowane do stadium końcowego niewydolności nerek. W populacji było 61% mężczyzn, średni wiek wynosił 65 lat, a średni wskaźnik BMI – 33 kg/m². Średnia długość trwania cukrzycy wynosiła 13,9 roku.
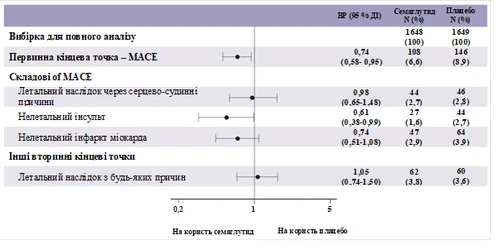
Pierwotnym punktem końcowym był czas od randomizacji do pierwszego wystąpienia poważnego niekorzystnego zdarzenia sercowo-naczyniowego (MACE): zgon z przyczyn sercowo-naczyniowych, niezgonny zawał serca lub niezgonny udar.
Całkowita liczba zdarzeń w składniku pierwotnego punktu końcowego MACE wyniosła 254, w tym 108 (6,6%) przy stosowaniu semaglutydu i 146 (8,9%) przy placebo. Zobacz rysunek 4, przedstawiający wyniki dla pierwotnego i wtórnych punktów końcowych sercowo-naczyniowych. Leczenie semaglutydem doprowadziło do zmniejszenia o 26% ryzyka wystąpienia złożonego pierwotnego punktu końcowego obejmującego zgon z przyczyn sercowo-naczyniowych, niezgonny zawał serca i niezgonny udar. Całkowita liczba zgonów z przyczyn sercowo-naczyniowych, niezgonnych zawałów serca i niezgonnych udarów wyniosła odpowiednio 90, 111 i 71, w tym 44 (2,7%), 47 (2,9%) i 27 (1,6%) odpowiednio przy stosowaniu semaglutydu (rysunek 4). Zmniejszenie ryzyka w pierwotnym złożonym punkcie końcowym wynikało głównie ze zmniejszenia częstości występowania niezgonnych udarów (39%) i niezgonnych zawałów serca (26%) (rysunek 3).
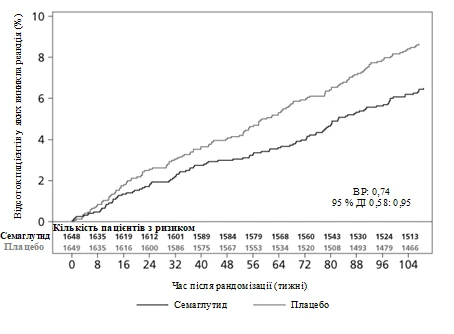
Rys. 3. Krzywa Kaplana-Meiera przedstawiająca czas do pierwszego wystąpienia złożonego zdarzenia: zgon z przyczyn sercowo-naczyniowych, niezgonny zawał serca i niezgonny udar (badanie kliniczne SUSTAIN 6).
**
**
Rys. 4. Diagram lasu: analiza czasu do pierwszego wystąpienia złożonego zdarzenia, jego składowe oraz przypadki zgonów z dowolnej przyczyny (badanie SUSTAIN 6)
Zarejestrowano 158 przypadków nowo zdiagnozowanej lub nasilonej nefropatii. HR [95% CI] czasu do wystąpienia nefropatii, zdefiniowanej jako trwała makroalbuminuria (pierwsze wystąpienie trwałej makroalbuminurii, trwałe podwojenie stężenia kreatyniny w surowicy, potrzeba ciągłej terapii zastępczej nerek lub zgon z przyczyn nerek), wyniósł 0,64 [0,46; 0,88].
Masa ciała
Po roku leczenia utrata masy ciała na poziomie ≥ 5% i ≥ 10% występowała u większej liczby pacjentów przy stosowaniu semaglutydu 0,5 mg (46% i 13%) i 1 mg (52–62% i 21–24%) niż przy stosowaniu aktywnego środka porównawczego sitagliptyny (18% i 3%) i eksenatydu ER (17% i 4%).
W badaniu porównawczym z dulaglutidem trwającym 40 tygodni utrata masy ciała na poziomie ≥ 5% i ≥ 10% została osiągnięta u większej liczby pacjentów przy stosowaniu semaglutydu 0,5 mg (44% i 14%) niż dulaglutidu 0,75 mg (23% i 3%), a także przy stosowaniu semaglutydu 1 mg (do 63% i 27%) niż dulaglutidu 1,5 mg (30% i 8%).
W trakcie badania klinicznego SUSTAIN 6 obserwowano istotne i trwałe zmniejszenie masy ciała od początku leczenia do 104. tygodnia przy stosowaniu semaglutydu 0,5 mg i 1 mg w porównaniu z placebo 0,5 mg i 1 mg jako dodatek do standardowego leczenia (–3,6 kg i –4,9 kg w porównaniu z –0,7 kg i –0,5 kg odpowiednio).
Ciśnienie tętnicze
Obserwowano istotne zmniejszenie średniego ciśnienia tętniczego skurczowego przy stosowaniu semaglutydu 0,5 mg (3,5–5,1 mm Hg) i 1 mg (5,4–7,3 mm Hg) w połączeniu z doustnymi lekami przeciwcukrzycowymi lub z insuliną bazalną. Nie zaobserwowano istotnej różnicy w wartości ciśnienia tętniczego rozkurczowego między leczeniem semaglutydem a lekami porównawczymi.
Dzieci
Europejska Agencja Leków odroczyła obowiązek przedłożenia wyników badań stosowania leku Ozempic® w jednej lub kilku podgrupach pacjentów pediatrycznych w leczeniu cukrzycy typu 2 (patrz sekcja „Sposób stosowania i dawki”).
Farmakokinetyka
W porównaniu z naturalnym GLP-1 semaglutyd charakteryzuje się wydłużonym okresem półtrwania trwającym 1 tydzień, co umożliwia jego podawanie podskórnie raz w tygodniu. Głównym mechanizmem takiego przedłużonego działania jest wiązanie z albuminą, co prowadzi do zmniejszenia klirensu nerkowego i ochrony przed degradacją metaboliczną. Ponadto semaglutyd jest stabilny wobec degradacji przez enzym DPP-4.
Absorpcja
Maksymalne stężenie osiągane jest w ciągu 1–3 dni po podaniu leku. Stężenie stacjonarne osiągane jest po 4–5 tygodniach po stosowaniu leku według schematu raz w tygodniu. U pacjentów z cukrzycą typu 2 średnie stężenie stacjonarne po podaniu podskórnej dawki 0,5 mg i 1 mg semaglutydu wynosiło odpowiednio około 16 nmol/l i 30 nmol/l. Przy dawkach 0,5 mg i 1 mg ekspozycja na semaglutyd wzrastała w sposób zależny od dawki. Podobna ekspozycja była osiągana po podaniu podskórnej dawki semaglutydu w okolicy przedniej ściany brzucha, uda lub ramienia. Bezpośrednia biodostępność semaglutydu po jego podaniu podskórnym wynosiła 89%.
Rozkład
Średni objętość rozkładu semaglutydu po podaniu podskórnemu u pacjentów z cukrzycą typu 2 wynosił około 12,5 l. Semaglutyd w znacznym stopniu wiązał się z albuminą osocza krwi (>99%).
Metabolizm
Przed wydaleniem semaglutyd jest aktywnie metabolizowany poprzez proteolityczne rozszczepienie peptydowego szkieletu białka i kolejne beta-oksydowanie bocznego łańcucha kwasu tłuszczowego. Przypuszcza się, że w metabolizmie semaglutydu uczestniczy enzym neutralnej endopeptydazy (NEP).
Wydalanie
W badaniu z podaniem podskórnym pojedynczej dawki semaglutydu znakowanego izotopem radioaktywnym stwierdzono, że substancja związana z semaglutydem wydalana jest głównie z moczem i kałem; około 2/3 substancji związanej z semaglutydem wydalało się z moczem i około 1/3 – z kałem. Około 3% podanej dawki wydalało się z moczem w niezmienionej postaci semaglutydu. U pacjentów z cukrzycą typu 2 klirens semaglutydu wynosił około 0,05 l/h. Przy okresie półtrwania trwającym około 1 tydzień semaglutyd pozostaje w krążeniu przez około 5 tygodni po podaniu ostatniej dawki.
Osobliwe grupy pacjentów
Pacjenci w podeszłym wieku
Na podstawie danych uzyskanych w trakcie badań fazy 3a z udziałem pacjentów w wieku 20–86 lat wiek nie wpływa na farmakokinetykę semaglutydu.
Płeć, rasa i pochodzenie etniczne
Płeć, rasa (kaukaska, czarna lub mongoloidalna) i pochodzenie etniczne (hiszpańskie lub latynoamerykańskie, niehiszpańskie lub nielatynoamerykańskie) nie wpływały na farmakokinetykę semaglutydu.
Masa ciała
Masa ciała wpływała na ekspozycję na semaglutyd. Wyższa masa ciała prowadzi do niższej ekspozycji; różnica masy ciała poszczególnych pacjentów na poziomie 20% prowadzi do różnicy w ekspozycji o około 16%. Dawki semaglutydu 0,5 mg i 1 mg zapewniają wystarczającą ekspozycję systemową przy masie ciała w zakresie 40–198 kg.
Zaburzenia funkcji nerek
Zaburzenia funkcji nerek nie miały klinicznie istotnego wpływu na farmakokinetykę semaglutydu. Zostało to wykazane po podaniu pojedynczej dawki 0,5 mg semaglutydu u pacjentów z zaburzeniami funkcji nerek różnego stopnia nasilenia (łagodnym, umiarkowanym, ciężkim i u pacjentów na dializie) w porównaniu z pacjentami z normalną funkcją nerek. Potwierdzono to również danymi z klinicznych badań fazy 3a u pacjentów z cukrzycą typu 2 i zaburzeniami funkcji nerek, choć doświadczenie w stosowaniu u pacjentów z końcowym stadium choroby nerek było ograniczone.
Zaburzenia funkcji wątroby
Zaburzenia funkcji wątroby nie miały żadnego wpływu na ekspozycję na semaglutyd. W badaniach z podaniem pojedynczej dawki 0,5 mg semaglutydu oceniano farmakokinetykę semaglutydu u pacjentów z różnym stopniem zaburzeń funkcji wątroby (łagodnym, umiarkowanym, ciężkim) w porównaniu z pacjentami z normalną funkcją wątroby.
Dzieci
Badania stosowania semaglutydu u pacjentów pediatrycznych nie przeprowadzono.
Dane przedkliniczne dotyczące bezpieczeństwa
Dane przedkliniczne oparte na badaniach farmakologicznej bezpieczeństwa, toksyczności powtarzanych dawek i genotoksyczności nie wykazały żadnego ryzyka dla człowieka.
Nieletalne guzy pochodzące z komórek C tarczycy, obserwowane u gryzoniów, należą do efektów charakterystycznych dla klasy agonistów receptorów GLP-1. W 2-letnim badaniu kancerogennym na szczurach i myszach semaglutyd powodował powstawanie guzów komórek C tarczycy przy klinicznie istotnych poziomach ekspozycji. Nie zaobserwowano żadnych innych guzów, których powstawanie mogłoby być związane z leczeniem. Guzy u gryzoniów są spowodowane niegenotoksycznym, specyficznym, zależnym od receptora GLP-1 mechanizmem pośredniczonym przez receptor, na który częściowo są wrażliwe gryzonie. Znaczenie tego mechanizmu dla człowieka jest wystarczająco niskie, ale nie może być całkowicie wykluczone.
W badaniach niepłodności u szczurów semaglutyd nie wpływał na skuteczność kojarzenia ani na płodność u samców. U samic szczurów obserwowano wydłużenie cyklu estralnego i niewielkie zmniejszenie liczby ciał żółtych (owulacji) przy dawkach towarzyszących utracie masy ciała u samic.
W badaniach rozwoju embrionów i płodów na szczurach semaglutyd wywierał działanie embrionotoksyczne przy ekspozycji niższej niż klinicznie istotne poziomy. Semaglutyd powodował znaczne zmniejszenie masy ciała u samic i zmniejszenie wskaźników przeżycia i wzrostu embrionów. U płodów obserwowano duże wady rozwojowe szkieletu i wady wewnętrzne, w tym zmiany w kościach długich, żebrach, kręgosłupie, kościach ogona, naczyniach krwionośnych i komorach mózgu. Ocena mechanistyczna wykazała, że efekt embrionotoksyczny obejmuje pośredniczone przez receptory GLP-1 zaburzenie dostarczania składników odżywczych do embrionu przez worek żółtkowy u szczurów. Ze względu na różnice w anatomicznej budowie i funkcjach worka żółtkowego u różnych gatunków szczurów oraz brak ekspresji receptorów GLP-1 w worku żółtkowym u małp człekokształtnych, mechanizm ten uważa się za mało prawdopodobny u człowieka. Jednak możliwość bezpośredniego wpływu semaglutydu na płód nie może być wykluczona.
W badaniach toksycznego wpływu na rozwój u królików i makaków jawajskich przy klinicznie istotnych poziomach ekspozycji obserwowano zwiększenie częstości poronień i pewne zwiększenie częstości wad płodu. Takie wyniki pokrywały się ze znaczną utratą masy ciała u samic sięgającą 16%. Nie wiadomo, czy takie efekty są związane ze zmniejszeniem spożycia pokarmu przez samice spowodowanym bezpośrednim działaniem GLP-1.
Postnatalny wzrost i rozwój oceniano u makaków jawajskich. Młode były nieco mniejsze przy urodzeniu, ale ich masa ciała znormalizowała się w okresie karmienia piersią.
U młodych szczurów semaglutyd powodował opóźnienie dojrzewania płciowego zarówno u samców, jak i u samic. Takie opóźnienie nie wpływało ani na płodność i zdolność rozrodczą obu płci, ani na zdolność samic do utrzymania ciąży.
Właściwości kliniczne
Wskazania
Lek Ozempic® stosować w przypadku niedostatecznie kontrolowanej cukrzycy typu 2 jako uzupełnienie diety i aktywności fizycznej:
- jako monoterapię, gdy metformina jest uznawana za niewskazaną z powodu nietolerancji lub przeciwwskazań;
- jako uzupełnienie innych leków stosowanych w leczeniu cukrzycy.
Informacje dotyczące wyników badań stosowania kombinacji leków, wpływu na kontrolę glikemii i zdarzenia sercowo-naczyniowe, jak również dane dotyczące populacji badanych pacjentów, zawarte są w rozdziałach „Szczególne wskazania dotyczące stosowania”, „Współdziałanie z innymi lekami i inne formy oddziaływania” oraz „Farmakodynamika”.
Przeciwwskazania
Podwyższona wrażliwość na substancję czynną lub na którykolwiek z pozostałych składników leku (patrz rozdział „Skład”).
Współdziałanie z innymi lekami i inne formy oddziaływania
Szmaglutyd opóźnia opróżnianie żołądka i może wpływać na szybkość wchłaniania leków doustnych stosowanych jednocześnie. Szmaglutyd należy stosować z ostrożnością u pacjentów przyjmujących leki doustne, które wymagają szybkiego wchłaniania w przewodzie pokarmowym.
Paracetamol
Na podstawie oceny farmakokinetyki paracetamolu podczas standaryzowanego testu z posiłkiem, szmaglutyd opóźnia opróżnianie żołądka. Po jednoczesnym podaniu paracetamolu i szmaglutydu w dawce 1 mg, wartości AUC0-60min i Cmax paracetamolu były niższe odpowiednio o 23% i 27%. Ogólna ekspozycja na paracetamol (AUC0-5h) nie uległa zmianie. Przy jednoczesnym stosowaniu paracetamolu i szmaglutydu nie jest wymagana korekta dawki.
Leki przeciwhamujące (antykoncepcja doustna)
Nie oczekuje się, że szmaglutyd zmniejszy skuteczność doustnych środków antykoncepcyjnych. W przypadku jednoczesnego stosowania szmaglutydu z kombinowanym doustnym środkiem antykoncepcyjnym (etynilostradiol 0,03 mg / lewonorgestrel 0,15 mg), szmaglutyd nie wpływał klinicznie istotnie na ogólną ekspozycję na etynilostradiol i lewonorgestrel. Ekspozycja na etynilostradiol nie była zaburzona; ekspozycja na lewonorgestrel zwiększyła się o 20% w stanie stacjonarnym. Wartości Cmax dla żadnej z tych substancji nie uległy zmianie.
Atorwastatyna
Szmaglutyd nie zmienił ogólnej ekspozycji na atorwastatynę po podaniu jednorazowej dawki atorwastatyny (40 mg). Wartość Cmax atorwastatyny zmniejszyła się o 38%. Uznano to za klinicznie nieistotne.
Digoksyna
Szmaglutyd nie zmienił ogólnej ekspozycji ani wartości Cmax digoksyny po podaniu jednorazowej dawki digoksyny (0,5 mg).
Metformina
Szmaglutyd nie zmienił ogólnej ekspozycji ani wartości Cmax metforminy po podaniu 500 mg metforminy dwa razy dziennie przez 3,5 dnia.
Warfaryna i inne pochodne kumaryny
Szmaglutyd nie zmienił ogólnej ekspozycji ani wartości Cmax warfaryny R- i S- po podaniu jednorazowej dawki warfaryny (25 mg), a parametry farmakodynamiczne warfaryny mierzone za pomocą międzynarodowego znormalizowanego współczynnika (INR) nie uległy klinicznie istotnej zmianie. Jednakże przypadki obniżenia INR zaobserwowano przy jednoczesnym stosowaniu acenokumarylu i szmaglutydu. Na początku leczenia szmaglutydem u pacjentów przyjmujących warfarynę lub inne pochodne kumaryny zaleca się częste monitorowanie INR.
Szczególne ostrzeżenia i środki ostrożności dotyczące stosowania
Śledzenie
W celu poprawy śledzenia leków biologicznych nazwa i numer serii podanego leku powinny być wyraźnie wskazane na opakowaniu.
Aspiracja w połączeniu z zastosowaniem znieczynienia ogólnego lub głębokiej sedacji
Zgłaszano przypadki aspiracji płucnych u pacjentów otrzymujących agonisty receptorów GLP-1, którzy byli poddawani znieczynieniu ogólnemu lub głębokiej sedacji. W związku z tym przed wykonaniem procedury z zastosowaniem znieczynienia ogólnego lub głębokiej sedacji należy wziąć pod uwagę zwiększone ryzyko obecności resztek treści żołądka z powodu opóźnionego opróżniania żołądka (patrz dział „Niepożądane działania przeciwwskazania”).
Ogólne informacje
Semaglutydu nie należy stosować pacjentom z cukrzycą typu 1 ani w leczeniu kwasicy ketonowej cukrzycowej. Semaglutyd nie jest substytucją insuliny. Opisywano przypadki rozwoju kwasicy ketonowej cukrzycowej u pacjentów uzależnionych od insuliny, którzy rozpoczynali leczenie agonistą receptorów GLP-1 i szybko przestawali stosować insulę lub szybko zmniejszali jej dawkę (patrz dział „Sposób stosowania i dawki”).
Nie ma doświadczenia klinicznego w leczeniu pacjentów z niewydolnością serca klasy IV według klasyfikacji New York Heart Association (NYHA), dlatego semaglutydu nie zaleca się stosować tym pacjentom.
Wpływ na przewód pokarmowy
Stosowanie agonistów receptorów GLP-1 może wiązać się z występowaniem działań niepożądanych ze strony przewodu pokarmowego. Należy to uwzględnić przy leczeniu pacjentów z zaburzeniami funkcji nerek, ponieważ nudności, wymioty i biegunka mogą prowadzić do odwodnienia, które może pogorszyć funkcję nerek (patrz dział „Niepożądane działania”).
Ostre zapalenie trzustki
Obserwowano przypadki ostrego zapalenia trzustki podczas stosowania agonistów receptorów GLP-1.
Pacjentów należy poinformować o typowych objawach ostrego zapalenia trzustki. W przypadku podejrzenia zapalenia trzustki należy odstawić leczenie semaglutydem; jeśli zapalenie trzustki zostanie potwierdzone, nie należy ponownie wznawiać leczenia semaglutydem. Semaglutyd należy stosować z ostrożnością u pacjentów z wywiadem zapalenia trzustki.
Hipoglikemia
U pacjentów otrzymujących semaglutyd w połączeniu z pochodnymi sulfoniliem lub insulina może występować zwiększony ryzyko hipoglikemii. Ryzyko hipoglikemii można zmniejszyć poprzez zmniejszenie dawki pochodnych sulfoniliowych lub insuliny na początku leczenia semaglutydem (patrz dział „Niepożądane działania”).
Retinopatia cukrzycowa
U pacjentów z retinopatią cukrzycową otrzymujących insulę i semaglutyd obserwowano zwiększone ryzyko powikłań retinopatii cukrzycowej (patrz dział „Niepożądane działania”). Semaglutyd należy stosować z ostrożnością u pacjentów z retinopatią cukrzycową otrzymujących insulę. Pacjentów tych należy dokładnie monitorować i leczyć zgodnie z zaleceniami klinicznymi. Szybkie poprawienie kontroli poziomu glukozy było związane z tymczasowym nasileniem się retinopatii cukrzycowej, jednak nie można wykluczyć wpływu innych mechanizmów.
Zawartość sodu
Ten lek zawiera mniej niż 1 mmol sodu (23 mg) na dawkę, dlatego lek można uznać za preparat niemający znaczenia zawartości sodu.
Stosowanie w czasie ciąży lub karmienia piersią
Kobiety w wieku rozrodczym
Kobietom w wieku rozrodczym zaleca się stosowanie środków antykoncepcyjnych podczas leczenia semaglutydem.
Ciąża
Badania na zwierzętach wykazały obecność toksyczności reprodukcyjnej (patrz dział „Dane dокliniczne dotyczące bezpieczeństwa”). Dane dotyczące stosowania semaglutydu u ciężarnych kobiet są ograniczone. Dlatego nie należy stosować semaglutydu w czasie ciąży. Jeśli pacjentka planuje zajść w ciążę lub jest w ciąży, stosowanie semaglutydu należy przerwać. Ze względu na długi okres półtrwania semaglutydu jego stosowanie należy przerwać co najmniej 2 miesiące przed planowaną ciążą (patrz dział „Farmakokinetyka”).
Karmienie piersią
Podczas laktacji u szczurów semaglutyd przenikał do mleka. Ponieważ nie można wykluczyć ryzyka dla niemowlęcia karmionego piersią, nie należy stosować semaglutydu w okresie karmienia piersią.
Plodność
Wpływ semaglutydu na płodność u ludzi jest nieznany. Stosowanie semaglutydu u samców szczurów nie wpływało na płodność. U samic szczurów obserwowano wydłużenie cyklu estralnego i zmniejszenie liczby owulacji przy dawkach związanych z utratą masy ciała (patrz dział „Dane dокliniczne dotyczące bezpieczeństwa”).
Wpływ na zdolność prowadzenia pojazdów lub obsługiwanie maszyn
Semaglutyd nie wpływa lub prawie nie wpływa na zdolność prowadzenia pojazdów lub obsługiwanie maszyn. W przypadku stosowania go w połączeniu z pochodnymi sulfoniliowymi lub z insulina pacjentom należy zalecić środki ostrożności, aby uniknąć rozwoju hipoglikemii podczas prowadzenia pojazdów lub pracy z innymi mechanizmami (patrz dział „Szczególne ostrzeżenia i środki ostrożności dotyczące stosowania”).
Sposób stosowania i dawki
Dozowanie
Początkowa dawka wynosi 0,25 mg semaglutydu raz w tygodniu. Po 4 tygodniach dawkę należy zwiększyć do 0,5 mg raz w tygodniu. W celu dalszego polepszenia kontroli glikemii po stosowaniu dawki 0,5 mg raz w tygodniu przez co najmniej 4 tygodnie, dawkę można zwiększyć do 1 mg raz w tygodniu.
Dawka 0,25 mg semaglutydu nie jest dawką utrzymaniową. Nie zaleca się stosowania dawek powyżej 1 mg raz w tygodniu.
Gdy lek Ozempic® jest stosowany jako uzupełnienie do już stosowanego metforminy i/lub tiazolidynodionu lub inhibitora współtransportera glukozy zależnego od sodu typu 2 (iNATG-2), aktualna dawka metforminy i/lub tiazolidynodionu lub inhibitora NADT-2 może pozostać bez zmian.
Gdy lek Ozempic® jest stosowany jako uzupełnienie do już stosowanej sulfonylomoczniny lub insuliny, należy rozważyć zmniejszenie dawki sulfonylomoczniny lub insuliny w celu zmniejszenia ryzyka wystąpienia hipoglikemii (patrz sekcje „Efekty uboczne” oraz „Wpływ na zdolność prowadzenia pojazdów i obsługiwanie maszyn”).
Do dostosowywania dawki leku Ozempic® nie jest wymagany samokontrola poziomu glukozy we krwi. Samokontrola poziomu glukozy we krwi jest wymagana do dostosowywania dawki sulfonylomoczniny i insuliny, szczególnie na początku stosowania leku Ozempic® oraz podczas zmniejszania dawki insuliny. Zalecana jest stopniowa schemat zmniejszania dawki insuliny.
Pominięta dawka
Jeśli dawkę pominięto, należy ją podać jak najszybciej w ciągu 5 dni od pominięcia. Jeśli minęło już więcej niż pięć dni, pominiętą dawkę należy pominąć, a następną dawkę podać w zaplanowanym dniu. W ten sposób pacjent może w każdym przypadku powrócić do regularnego schematu stosowania leku raz w tygodniu.
Jeśli konieczne jest zmiana dnia tygodniowego podawania leku, można to zrobić, zachowując co najmniej 3-dniowy odstęp (>72 godziny) między dwoma dawkami. Po wybraniu nowego dnia podawania leku należy kontynuować jego stosowanie według schematu raz w tygodniu.
Grupy specjalne pacjentów
Pacjenci w podeszłym wieku
Dostosowania dawki ze względu na wiek nie wymaga się. Doświadczenie leczenia pacjentów w wieku ≥ 75 lat jest ograniczone (patrz sekcja „Farmakokinetyka”).
Naruszenie funkcji nerek
Pacjentom z łagodnym, umiarkowanym lub ciężkim naruszeniem funkcji nerek nie wymaga się dostosowania dawki. Doświadczenie stosowania semaglutydu u pacjentów z ciężkim naruszeniem funkcji nerek jest ograniczone. Nie zaleca się stosowania semaglutydu u pacjentów z nerek w stadium końcowym (patrz sekcja „Farmakokinetyka”).
Naruszenie funkcji wątroby
Pacjentom z naruszeniem funkcji wątroby nie wymaga się dostosowania dawki. Doświadczenie stosowania semaglutydu u pacjentów z ciężkim naruszeniem funkcji wątroby jest ograniczone. Należy stosować semaglutyd z ostrożnością u tych pacjentów (patrz sekcja „Farmakokinetyka”).
Dzieci
Bezpieczeństwo i skuteczność stosowania semaglutydu u dzieci (do 18. roku życia) nie zostały ustalone. Brak danych.
Sposób podania
Stosować podskórnie.
Ozempic® podaje się podskórnie w okolicy przedniej ściany brzucha, uda lub ramienia. Miejsce wstrzyknięcia można zmieniać bez dostosowywania dawki. Ozempic® nie można podawać dożylnie ani do mięśnia.
Ozempic® należy stosować raz w tygodniu w dowolnym czasie dnia niezależnie od posiłku.
Aby uzyskać dalsze informacje dotyczące podania leku, patrz sekcja „Zalecenia dotyczące postępowania z lekiem i jego utylizacji”.
Zalecenia dotyczące postępowania z lekiem i jego utylizacji
Należy zalecić pacjentowi, aby po każdej iniekcji usuwał igłę i przechowywał strzykawkę-ручkę bez założonej igły. Może to zapobiec zablokowaniu igły, zanieczyszczeniu, zakażeniu, wyciekowi roztworu oraz niedokładnemu dawkowaniu. Igrzecze i inne odpady należy utylizować zgodnie z lokalnymi przepisami. Strzykawka-ручка jest przeznaczona wyłącznie do użytku indywidualnego. Nie należy stosować leku Ozempic®, jeśli nie wygląda na przezroczysty i bezbarwny lub prawie bezbarwny. Nie stosować po zamrożeniu. Strzykawka-ручka jest przeznaczona do użytku z jednorazowymi igłami NovoFine® lub NovoTwist® o długości do 8 mm. Igrzecze NovoFine® Plus są dołączone do opakowania produktu.
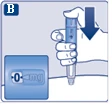
| Instrukcja obsługi dozownika wstrzykowego Ozempic® 0,25 mg, 0,5 mg (roztwór do wstrzykiwania w dozowniku wstępnie wypełnionym) |
|
| Przed użyciem dozownika wstrzykowego Ozempic® należy zapoznać się z instrukcją dla lekarzy. Należy porozmawiać z lekarzem, pielęgniarką lub farmaceutą o właściwym sposobie podawania leku Ozempic®. Przed zastosowaniem leku należy sprawdzić dozownik wstrzykowy, aby upewnić się, że zawiera on Ozempic® w dawce 0,25 mg lub 0,5 mg. Następnie należy obejrzeć poniższe rysunki, aby zapoznać się z poszczególnymi częściami dozownika wstrzykowego i igły. Jeśli pacjent nie widzi wcale lub ma słabe widzenie i nie jest w stanie odczytać wskaźnika dawki na dozowniku wstrzykowym, nie powinien korzystać z dozownika bez pomocy innej osoby. Pomagać powinna osoba o dobrym wzroku, która wie, jak korzystać z wstępnie napełnionego dozownika wstrzykowego Ozempic®. Dozownik wstrzykowy jest wstępnie napełniony i wyposażony w mechanizm do doboru dawki. Zawiera 2 mg semaglutydu. Można ustawić dawki 0,25 mg lub 0,5 mg. Jeden nowy dozownik wstrzykowy przed użyciem zawiera:
Użyj tabeli na wieczku tekturowego opakowania, aby śledzić liczbę wykonanych wstrzyknięć i ich datę: Należy wskazać dzień tygodnia wybrany na podanie
|
|
|
|
0,5 mg na dawkę. Jest to szczególnie ważne, jeśli stosuje się różne leki do wstrzykiwania. Użycie niewłaściwego leku może być szkodliwe dla zdrowia.
|
|
|
|
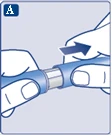
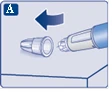
Sprawdź, czy papierowa membrana i zewnętrzna osłonka igły nie są uszkodzone, ponieważ może to oznaczać brak sterylności. W przypadku jakichkolwiek uszkodzeń należy użyć nowej igły.
|
|
| Upewnij się, że igła jest prawidłowo zamocowana.
|
|
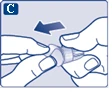
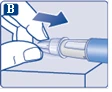
| Na igłę są założone dwie osłonki. Należy zdjąć obie osłonki. Jeśli zapomnisz zdjąć obie osłonki, nie wstrzykniesz roztworu.
|
|
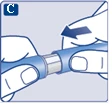
Na końcówce igły może pojawić się kropla roztworu. Jest to normalne zjawisko, jednak należy sprawdzić przepływ leku przy pierwszym użyciu nowego dozownika wstrzykowego. Zobacz punkt 2 „Sprawdzenie przepływu roztworu przy każdym nowym dozowniku wstrzykowym”. Nie zakładaj nowej igły, dopóki nie będziesz gotowy do wykonania wstrzyknięcia. |
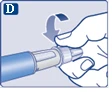
|
| Do każdej iniekcji należy zawsze używać nowej igły. To może zapobiegać zatamowaniu igły, zanieczyszczeniu, zakażeniu oraz podaniu niewłaściwej dawki leku. |
|
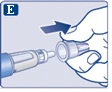
| Nigdy nie używaj igły, jeśli jest wygięta lub uszkodzona. |
|
|
|
|
|
Naciśnij i przytrzymaj przycisk dawki, aż wskaźnik dawki powróci do „0”. Wartość „0” musi pokrywać się z wskaźnikiem dawki. Na końcówce igły powinna pojawić się kropla roztworu. |
|
| Na końcówce igły może pozostać mała kropla, ale nie zostanie ona wstrzyknięta. Jeśli kropla się nie pojawiła, powtórz czynności opisane w punkcie 2 „Sprawdzenie przepływu roztworu przy nowym dozowniku wstrzykowym” do 6 razy. Jeśli kropla nadal się nie pojawi, zmień igłę i ponownie wykonaj czynności opisane w punkcie 2 „Sprawdzenie przepływu roztworu przy nowym dozowniku wstrzykowym”. Jeśli kropla nadal się nie pojawi, usuń dozownik wstrzykowy i użyj nowego. |
|
| Zawsze należy upewnić się, że kropla pojawia się na końcówce igły przed pierwszym użyciem nowego dozownika. Gwarantuje to, że roztwór zostanie wstrzyknięty. Jeśli kropla się nie pojawi, nie używaj dozownika wstrzykowego, nawet jeśli wskaźnik dawki zmienia wartość. Może to wskazywać na zatkana lub uszkodzoną igłę. Jeśli nie sprawdzisz przepływu przed pierwszą iniekcją nowego dozownika, możesz nie otrzymać wymaganej dawki roztworu, co wpłynie na oczekiwany efekt działania leku Ozempic®. |
|
|
|
Jeśli wybrałeś niewłaściwą dawkę, możesz obrócić pokrętło dawki do przodu lub do tyłu, aby ustawić poprawną dawkę. Maksymalna dawka, którą można ustawić dozownikiem wstrzykowym, wynosi 0,5 mg. |
|
| Dozownik umożliwia zmianę dawki. Tylko wskaźnik dawki i wskaźnik dawki pokażą, ile miligramów przygotowano do wstrzyknięcia. Można wybrać tylko 0,5 mg leku na dawkę. Jeśli dozownik wstrzykowy zawiera mniej niż 0,5 mg leku, wskaźnik dawki zatrzyma się przed osiągnięciem wartości 0,5. Podczas obracania pokrętła dawki do przodu, do tyłu lub gdy obracasz je dalej poza wskaźnik wybranej dawki, pokrętło dawki wydaje różne dźwięki. Nie należy liczyć liczby kliknięć dozownika wstrzykowego. |
|
| Zawsze należy korzystać z wskaźnika dawki i wskaźnika dawki, aby upewnić się, że wybrano właściwą dawkę przed podaniem leku. Nie należy liczyć liczby kliknięć dozownika wstrzykowego. Można wybrać tylko dawki 0,25 mg lub 0,5 mg za pomocą pokrętła dawki. Wybrana dawka musi dokładnie pokrywać się z wskaźnikiem dawki, aby zapewnić właściwą dawkę. |
|
| Pozostałość roztworu w dozowniku wstrzykowym |
|
|
|
| Jeśli roztworu jest za mało do podania dawki, nie należy stosować dozownika wstrzykowego. Użyj nowego dozownika wstrzykowego Ozempic®. |
|
|
|
|
|
„0” musi pokrywać się z wskaźnikiem dawki. Następnie możesz usłyszeć lub poczuć kliknięcie.
|
|
|
|
Jeśli w miejscu iniekcji pojawi się kropla krwi, delikatnie przyciśnij to miejsce. |
|
| Może pojawić się kropla roztworu na końcówce igły po wstrzyknięciu. Jest to normalne i nie wpływa na objętość podanej dawki. |
|
| Zawsze należy patrzeć na wskaźnik dawki, aby zobaczyć, ile miligramów roztworu zostało podanych. Przytrzymuj naciśnięty przycisk startowy, aż wskaźnik dawki ponownie pokaże „0”. Jak wykryć zatkana lub uszkodzoną igłę.
Jak postępować przy zatkanej igle. Zdejmij igłę, jak opisano w punkcie 5 „Po iniekcji”, i powtórz czynności opisane we wszystkich punktach, począwszy od punktu 1 „Przygotowanie dozownika wstrzykowego z nową igłą do użycia”. Upewnij się, że wybrano wymaganą dawkę. Nigdy nie dotykaj wskaźnika podczas podawania leku. Może to przerwać iniekcję. |
|
|
|
| Zawsze usuwaj igłę po każdej iniekcji, aby zapewnić wygodę następnej iniekcji i zapobiec użyciu zatkanej igły. Jeśli igła jest zatkana, nie będzie można podać leku.
|
|
|
|
|
|
| Gdy dozownik jest pusty, należy go wyrzucić bez igły zgodnie z instrukcjami od lekarza, pielęgniarki, farmaceuty lub zgodnie z lokalnymi przepisami. |
|
| Nigdy nie próbuj ponownie założyć wewnętrznej osłonki igły na igłę. Możesz się poranić. Zawsze usuwaj igłę z dozownika po każdej iniekcji. Może to zapobiegać zatamowaniu igły, zanieczyszczeniu, zakażeniu, wyciekowi roztworu oraz nieprawidłowej dawkowaniu. |
|
| Dodatkowe ważne informacje. |
|
|
|
| Opieka nad dozownikiem wstrzykowym. |
|
| Należy ostrożnie obchodzić się z dozownikiem wstrzykowym. Nieostrożne postępowanie lub niewłaściwe użycie może prowadzić do podania niewłaściwej dawki. Jeśli dojdzie do takiej sytuacji, może nie uzyskać się oczekiwanego efektu działania leku. |
|
|
|





Dzieci
Bezpieczeństwo i skuteczność stosowania semaglutydu u dzieci (do 18. roku życia) nie zostały ustalone. Brak danych.
Europejski Urząd ds. Leków odroczył obowiązek przedłożenia wyników badań semaglutydu w jednej lub kilku podgrupach populacji dziecięcej w leczeniu cukrzycy typu 2 (patrz sekcja „Sposób stosowania i dawki”).
Przedawkowanie
W trakcie badań klinicznych zgłaszano przypadki przedawkowania do 4 mg po pojedynczym podaniu leku oraz do 4 mg po stosowaniu przez okres tygodnia. Najczęściej zgłaszano nudności. Wszyscy pacjenci wyzdrowieli bez powikłań.
Nie istnieje specyficzny antydotalny środek leczniczy w leczeniu przedawkowania semaglutydu. W przypadku przedawkowania należy prowadzić leczenie objawowe zgodnie z występującymi u pacjenta objawami klinicznymi. Ze względu na długi okres półtrwania semaglutydu, który wynosi około 1 tygodnia, może istnieć konieczność przedłużenia czasu leczenia tych objawów (patrz sekcja „Farmakokinetyka”).
Efekty uboczne
Streszczenie profilu bezpieczeństwa
W ośmiu badaniach klinicznych fazy 3 uczestniczyło 4792 pacjentów otrzymujących semaglutyd w dawce do 1 mg. W trakcie badań klinicznych najczęściej zgłaszane były efekty uboczne ze strony przewodu pokarmowego, w tym nudności (bardzo często), biegunka (bardzo często) oraz wymioty (często). Ogólnie reakcje te miały charakter łagodny lub umiarkowany i były krótkotrwałe.
Efekty uboczne
Poniżej przedstawiono efekty uboczne, które wystąpiły w badaniach klinicznych fazy 3a u pacjentów z cukrzycą typu 2 (szczegóły opisano w sekcji „Farmakodynamika”). Częstotliwość występowania efektów ubocznych określono na podstawie połączonych danych z badań klinicznych fazy 3a, z wyłączeniem badań dotyczących skutków kardiologicznych (dodatkowe informacje znajdują się poniżej).
Efekty uboczne sklasyfikowano według układów narządów i częstości występowania. Ocena częstości występowania efektów ubocznych została przeprowadzona według następującej skali: bardzo często: (≥ 1/10), często: (od ≥ 1/100 do < 1/10), rzadko: (od ≥ 1/1000 do < 1/100), bardzo rzadko: (od ≥ 1/10000 do < 1/1000), nieznane: (niemożliwe do oszacowania na podstawie dostępnych danych). W każdej grupie efekty uboczne wymieniono w kolejności malejącej ich ciężkości.
Zaburzenia układu odpornościowego: rzadko – nadwrażliwośćc; bardzo rzadko – reakcja anafilaktyczna.
Zaburzenia metabolizmu i trawienia: bardzo często – hipoglikemiaa przy jednoczesnym stosowaniu z insuliną lub pochodnymi sulfoniliomocznika; często – hipoglikemiaa przy jednoczesnym stosowaniu z innymi PZPZD, zmniejszenie apetytu.
Zaburzenia układu nerwowego: często – zawroty głowy; rzadko – dysgezja.
Zaburzenia narządu wzroku: często – nasilenie retinopatii cukrzycowejb.
Zaburzenia układu sercowo-naczyniowego: rzadko – zwiększona częstość akcji serca.
Zaburzenia układu pokarmowego: bardzo często – nudności, biegunka; często – wymioty, ból brzucha, wzdęcia, zaparcia, dyspepsja, zapalenie żołądka, choroba refluksowa przełyku, odbijanie, wzdęcia; rzadko – ostra trzustaczka, opóźnione opróżnianie żołądka; nieznane – niedrożność jelitad.
Zaburzenia wątroby i dróg żółciowych: często – kamica żółciowa.
Zaburzenia skóry i tkanki podskórnej: nieznane – obrzęk naczynioruchowyd.
Ogólne zaburzenia i reakcje w miejscu wstrzyknięcia: często – zmęczenie; rzadko – reakcje w miejscu wstrzyknięcia.
Badania laboratoryjne: często – podwyższony poziom lipazy, podwyższony poziom amylazy, spadek masy ciała.
a Hipoglikemia była definiowana jako stan ciężki (wymagający pomocy innej osoby) lub stan objawowy równocześnie z poziomem glukozy we krwi < 3,1 mmol/l.
b Nasilenie retinopatii cukrzycowej obejmuje następujące cechy: fotokoagulacja siatkówki, leczenie za pomocą iniekcji wewnątrzwitkowej, krwotok do ciała szklistego, ślepotę spowodowaną cukrzycą (rzadko). Częstotliwość przypadków określono na podstawie danych z badania skutków kardiologicznych.
c Ogólny termin obejmujący również niepożądane zjawiska związane z nadwrażliwością, takie jak wysypka i pokrzywka.
d Dane zgłoszone w okresie pogoni.
Badanie skutków kardiologicznych i badanie bezpieczeństwa trwające 2 lata
U pacjentów z wysokim ryzykiem wystąpienia chorób układu sercowo-naczyniowego profil efektów ubocznych był podobny do profilu obserwowanego w innych badaniach fazy 3a (opisanych w sekcji „Farmakodynamika”).
Opis wybranych efektów ubocznych
Hipoglikemia
Podczas stosowania semaglutydu jako monoterapii nie obserwowano epizodów ciężkiej hipoglikemii. Ciężka hipoglikemia występowała głównie wtedy, gdy semaglutyd stosowano razem z pochodnymi sulfoniliomocznika (1,2 % pacjentów; 0,03 przypadki na pacjenta-rok) lub z insuliną (1,5 % pacjentów; 0,02 przypadki na pacjenta-rok). Kilka epizodów (0,1 % pacjentów; 0,001 przypadki na pacjenta-rok) zaobserwowano przy stosowaniu semaglutydu w połączeniu z doustnymi lekami przeciwcukrzycowymi, innymi niż pochodne sulfoniliomocznika.
Amerykańskie Stowarzyszenie Diabetologiczne sklasyfikowało hipoglikemię u 11,3 % pacjentów (0,3 przypadki na pacjenta-rok) przy stosowaniu semaglutydu 1,0 mg w połączeniu z iNHTG-2 w badaniu SUSTAIN 9, w porównaniu do 2,0 % pacjentów (0,04 przypadki na pacjenta-rok) przyjmujących placebo. O ciężkiej hipoglikemii zgłaszano odpowiednio u 0,7 % (0,01 zdarzenia na rok pacjenta) i 0 % pacjentów.
Reakcje ze strony układu pokarmowego
Podczas stosowania semaglutydu w dawkach 0,5 mg i 1 mg nudności występowały odpowiednio u 17,0 % i 19,9 % pacjentów, biegunka – u 12,2 % i 13,3 %, a wymioty – u 6,4 % i 8,4 %. Większość przypadków miała charakter od lekkiego do umiarkowanego i była nietrwała. Reakcje doprowadziły do przerwania leczenia u odpowiednio 3,9 % i 5 % pacjentów. Reakcje najczęściej pojawiały się w pierwszych miesiącach leczenia. U pacjentów z niską masą ciała stosowanie semaglutydu może powodować większą liczbę zaburzeń ze strony układu pokarmowego. W badaniu SUSTAIN 9 przy jednoczesnym stosowaniu iNHTG-2 zaobserwowano zaparcia i chorobę refluksową przełyku odpowiednio u 6,7 % i 4 % pacjentów przyjmujących semaglutyd 1,0 mg, w porównaniu do braku zdarzeń u pacjentów przyjmujących placebo. Częstość tych zdarzeń z czasem nie zmniejszała się.
Ostra trzustaczka
Częstotliwość potwierdzonych przypadków ostrej trzustaczki zarejestrowanych podczas badań klinicznych fazy 3a wynosiła 0,3 % przy stosowaniu semaglutydu i 0,2 % przy stosowaniu leku porównawczego. W trakcie badania reakcji kardiologicznych trwającego
2 lata częstość potwierdzonych przez ekspertów przypadków ostrej trzustaczki wynosiła 0,5 % przy stosowaniu semaglutydu i 0,6 % przy stosowaniu placebo (patrz sekcja „Szczególne ostrzeżenia i środki ostrożności stosowania”).
Nasilenie retinopatii cukrzycowej
W dwuletnim badaniu klinicznym wzięło udział 3297 pacjentów z cukrzycą typu 2, wysokim ryzykiem kardiologicznym, długotrwałym przebiegiem cukrzycy i niewystarczającą kontrolą poziomu glukozy we krwi. W tym badaniu potwierdzone przypadki nasilenia retinopatii cukrzycowej występowały częściej u pacjentów otrzymujących semaglutyd (3,0 %) niż u tych, którzy otrzymywali placebo (1,8 %). Obserwacja ta dotyczyła pacjentów z potwierdzoną retinopatią cukrzycową, którzy otrzymywali insulinę. Różnica między wariantami leczenia pojawiła się wcześnie i utrzymywała się przez cały okres badania. Ocena systemowa nasilenia retinopatii cukrzycowej była przeprowadzana wyłącznie w badaniu skutków kardiologicznych. W jednorocznym badaniu klinicznym z udziałem 4807 pacjentów z cukrzycą typu 2 część pacjentów, u których zaobserwowano niepożądane reakcje związane z retinopatią cukrzycową, była podobna w grupie stosowania semaglutydu (1,7 %) i w grupie leku porównawczego (2,0 %).
Przerwanie leczenia z powodu efektów ubocznych
Częstotliwość przerwania leczenia z powodu wystąpienia niepożądanych zjawisk wynosiła 6,1 % i 8,7 % u pacjentów przyjmujących semaglutyd w dawkach 0,5 mg i 1 mg odpowiednio, w porównaniu do 1,5 % u pacjentów przyjmujących placebo. Najczęstszymi efektami ubocznymi prowadzącymi do przerwania leczenia były zaburzenia ze strony układu pokarmowego.
Reakcje w miejscu wstrzyknięcia
O wystąpieniu reakcji w miejscu wstrzyknięcia (takich jak wysypka, zaczerwienienie w miejscu iniekcji) zgłaszano u 0,6 % i 0,5 % pacjentów przyjmujących semaglutyd w dawkach 0,5 mg i 1 mg odpowiednio. Zazwyczaj reakcje te były umiarkowane.
Imunogenność
Ze względu na możliwość wystąpienia właściwości immunogennych w lekach zawierających białka lub peptydy, istnieje prawdopodobieństwo powstawania przeciwciał u pacjentów podczas leczenia semaglutydem. Odsetek pacjentów z dodatnim wynikiem badania na obecność przeciwciał anty-semaglutydowych w dowolnym czasie po rozpoczęciu leczenia był niski (1–2 %) i u żadnego z pacjentów nie stwierdzono przeciwciał neutralizujących semaglutyd ani przeciwciał z efektem neutralizującym GLP-1 do zakończenia badań klinicznych.
Zwiększenie częstości akcji serca
Podczas stosowania agonistów receptora GLP-1 obserwowano zwiększenie częstości akcji serca. W badaniach klinicznych fazy 3a u pacjentów przyjmujących lek Ozempic® obserwowano średnie zwiększenie częstości akcji serca od 1 do 6 uderzeń na minutę (ud./min) od wartości wyjściowej wynoszącej 72–76 ud./min. W długotrwałym badaniu z udziałem pacjentów z czynnikami ryzyka rozwoju chorób układu sercowo-naczyniowego po 2 latach leczenia u 16 % pacjentów przyjmujących lek Ozempic® zaobserwowano zwiększenie częstości akcji serca o > 10 ud./min w porównaniu do 11 % pacjentów przyjmujących placebo.
Zgłaszanie podejrzewanych reakcji niepożądanych
Po rejestracji leku ważne jest zgłaszanie podejrzewanych reakcji niepożądanych. Umożliwia to kontynuowanie monitorowania stosunku korzyści do ryzyka stosowania leku. Lekarze powinni zgłaszać podejrzewane reakcje niepożądane poprzez krajowy system raportowania.
Okres ważności
Do pierwszego użycia – 3 lata.
Po pierwszym użyciu – 6 tygodni.
Po pierwszym użyciu przechowywać w temperaturze poniżej 30 °C lub w lodówce (przy temperaturze 2–8 °C). Nie zamrażać.
Przechowywać strzykawkę-pen z założoną osłonką chroniącą przed światłem.
Warunki przechowywania
Do pierwszego użycia: przechowywać w lodówce (2–8 °C), nie zbyt blisko komory zamrażarki. Nie zamrażać. Nie używać po zamrożeniu.
Przechowywać strzykawkę-pen z założoną osłonką chroniącą przed światłem.
Warunki przechowywania po pierwszym otwarciu patrz w sekcji „Okres ważności”.
Przechowywać w miejscu niedostępnym dla dzieci.
Niezgodność
Ponieważ nie przeprowadzono badań niezgodności, tego leku nie wolno mieszać z innymi lekami.
Opakowanie
Wielodawkowa jednorazowa strzykawka-pen wstępnie napełniona, wykonana z polipropylenu, polioksymetylenku, poliwęglanu i akrylonitrylu butadienu styrenu, zawiera kartusz (szkło typu I), z jednej strony zamknięty tłokiem gumowym (chlorobutyl), z drugiej strony – aluminiową osłonką z laminowaną wkładką gumową (bromobutyl / poliizopren).
Strzykawka-pen zawiera 1,5 ml roztworu, umożliwiającego podanie dawek 0,25 mg lub 0,5 mg. Opakowanie zawiera 1 wstępnie napełnioną strzykawkę-pen oraz 6 jednorazowych igieł NovoFine® Plus w tekturowej pudełku.
Kategoria recepturowa. Na receptę.
Producent
A/T Novo Nordisk.
Siedziba producenta i adres miejsca prowadzenia działalności
Novo Allé
2880, Bagsværd
Dania.
Wnioskodawca
A/T Novo Nordisk.
Siedziba wnioskodawcy
Novo Allé
2880, Bagsværd
Dania.
INSTRUKCJA
do stosowania leku przez personel medyczny
OZEMPIC®
(OZEMPIC®)
Skład:
substancja czynna: semaglutyd;
1 ml roztworu zawiera 1,34 mg semaglutyd – analogu ludzkiego peptydu podobnego do glukagonu-1 (GLP-1), wyprodukowanego w Saccharomyces cerevisiae metodą rekombinacji DNA;
jeden przedwypełniony strzykacz do dawkowania zawiera 4 mg semaglutyd w 3,0 ml roztworu. Każda dawka zawiera 1 mg semaglutyd w 0,74 ml roztworu;
substancje pomocnicze: fosforan sodu, dwuwodny; glikol propylenowy; fenol; kwas chlorowodorowy (do regulacji pH); wodorotlenek sodu (do regulacji pH); woda do wstrzykiwań.
Postać farmaceutyczna. Roztwór do wstrzykiwań.
Główne właściwości fizykochemiczne: przezroczysty, bezbarwny lub prawie bezbarwny roztwór izotoniczny; pH = 7,4.
Grupa farmakoterapeutyczna. Leki stosowane w cukrzycy, analogi peptydu podobnego do glukagonu-1 (GLP-1). Kod ATC A10B J06.
Właściwości farmakodynamiczne
Farmakodynamika
Mechanizm działania
Semaglutyd jest analogiem GLP-1 o 94% homologii z GLP-1 człowieka. Semaglutyd działa jako agonista receptorów GLP-1, wiążąc się i aktywując selektywnie receptory GLP-1, które są celem działania naturalnego GLP-1.
GLP-1 jest hormonem fizjologicznym, który wpływa na wiele sposobów na regulację stężenia glukozy i apetytu, a także na układ sercowo-naczyniowy. Wpływ na stężenie glukozy i apetyt jest specyficznie pośredniczony przez receptory GLP-1 znajdujące się w trzustce i mózgu.
Semaglutyd obniża poziom glukozy we krwi działając zależnie od glukozy, poprzez stymulację sekrecji insuliny i hamowanie sekrecji glukagonu w warunkach podwyższonego stężenia glukozy we krwi. Mechanizm obniżania poziomu glukozy we krwi towarzyszy również nieznaczne opóźnienie opróżniania żołądka w wczesnej fazie pokarmowej. W przypadku hipoglikemii semaglutyd zmniejsza sekrecję insuliny i nie przeszkadza w sekrecji glukagonu.
Zastosowanie semaglutyd prowadzi do zmniejszenia masy ciała i masy tkanki tłuszczowej poprzez zmniejszenie spożycia kalorii oraz zmniejszenie apetytu. Ponadto semaglutyd zmniejsza popęd do jedzenia pokarmów o wysokiej zawartości tłuszczu.
Ekspresja receptorów GLP-1 występuje również w sercu, układzie naczyniowym, układzie odpornościowym i nerkach.
W trakcie badań klinicznych semaglutyd wykazywał pozytywny wpływ na poziomy lipidów w osoczu krwi, obniżał ciśnienie skurczowe i zmniejszał stan zapalny. W badaniach na zwierzętach semaglutyd hamował rozwój miażdżycy, zapobiegając postępowi blaszki aorty i zmniejszając stan zapalny w blaszce.
Skutki farmakodynamiczne
Wszystkie badania farmakodynamiczne przeprowadzono po 12 tygodniach leczenia (w tym okresie podwyższania dawki) w stanie równowagi po zastosowaniu semaglutyd w dawce 1 mg raz w tygodniu.
Poziom glukozy na czczo i poziom glukozy po posiłku
Semaglutyd prowadzi do zmniejszenia stężenia glukozy na czczo i stężenia glukozy po posiłku. U pacjentów z cukrzycą typu 2 leczenie semaglutydem w dawce 1 mg prowadziło do zmniejszenia stężenia glukozy w kierunku bezwzględnej zmiany od wartości wyjściowej (mmol/l) oraz względnej redukcji w porównaniu do placebo (%) dla parametrów stężenia glukozy na czczo (1,6 mmol/l; 22%), stężenia glukozy po 2 godzinach po posiłku (4,1 mmol/l; 37%), średniego dobowego stężenia glukozy (1,7 mmol/l; 22%) oraz wahań stężenia glukozy po posiłku w trakcie trzech posiłków (0,6–1,1 mmol/l) w porównaniu do placebo. Semaglutyd obniżał poziom glukozy na czczo już po pierwszym zastosowaniu.
Funkcja komórek beta i sekrecja insuliny
Semaglutyd poprawia funkcję komórek beta. W porównaniu do placebo semaglutyd poprawiał pierwszą i drugą fazę odpowiedzi insuliny z trzykrotnym i dwukrotnym wzrostem odpowiednio, a także zwiększał maksymalną aktywność sekrecyjną komórek beta u pacjentów z cukrzycą typu 2. Ponadto, w porównaniu do placebo leczenie semaglutydem prowadziło do zwiększenia stężenia insuliny na czczo.
Sekrecja glukagonu
Semaglutyd zmniejsza stężenie glukagonu na czczo i stężenie glukagonu po posiłku. U pacjentów z cukrzycą typu 2 zastosowanie semaglutyd prowadziło do względnej redukcji stężenia glukagonu w porównaniu do placebo: stężenie glukagonu na czczo (8–21%), odpowiedź glukagonu po posiłku (14–15%) oraz średnie dobowe stężenie glukagonu (12%).
Sekrecja insuliny i glukagonu zależna od glukozy
Semaglutyd obniżał wysokie stężenie glukozy we krwi poprzez stymulację sekrecji insuliny i zmniejszenie sekrecji glukagonu, działając zależnie od glukozy. Szybkość sekrecji insuliny po zastosowaniu semaglutyd u pacjentów z cukrzycą typu 2 była porównywalna do takiej u osób zdrowych.
W przypadku wywołanej hipoglikemii semaglutyd w porównaniu do placebo nie zmieniał reakcji kontrregulacyjnej podwyższenia poziomu glukagonu i nie nasilał zmniejszenia stężenia C-peptydu u pacjentów z cukrzycą typu 2.
Opróżnianie żołądka
Semaglutyd powodował nieznaczne opóźnienie wczesnego opróżniania żołądka po posiłku, tym samym zmniejszając szybkość napływu glukozy do krwi w okresie pokarmowym.
Apetyt, spożycie kalorii i wybór pokarmów
W porównaniu do placebo semaglutyd prowadził do zmniejszenia spożycia kalorii o 18–35% podczas trzech kolejnych dowolnych posiłków ad libitum. Przyczyniły się do tego wywołane przez semaglutyd hamowanie apetytu zarówno na czczo, jak i w okresie pokarmowym, poprawa kontroli nad spożyciem pokarmu, osłabienie popędu do przekąsek oraz względnie mniejszy popęd do jedzenia pokarmów o wysokiej zawartości tłuszczu.
Lipidy na czczo i w okresie pokarmowym
W porównaniu do placebo semaglutyd zmniejszał stężenia trójglicerydów na czczo i cholesterolu lipoprotein o bardzo niskiej gęstości (VLDL-C) na czczo o 12% i 21% odpowiednio. Reakcja trójglicerydów po posiłku i reakcja cholesterolu VLDL-C po jedzeniu pokarmów o wysokiej zawartości tłuszczu zmniejszały się o ponad 40%.
Elektrofizjologia serca (QTc)
Wpływ semaglutyd na proces repolaryzacji serca oceniano w starannie przeprowadzonym badaniu QTc. Semaglutyd nie wydłużał odstępów QTc w dawkach do 1,5 mg w stanie równowagi.
Skuteczność kliniczna i bezpieczeństwo
Zarówno poprawa kontroli glikemicznej, jak i zmniejszenie zachorowalności i śmiertelności sercowo-naczyniowej są integralną częścią terapii cukrzycy typu 2.
Skuteczność i bezpieczeństwo zastosowania semaglutyd w dawkach 0,5 mg i 1 mg raz w tygodniu oceniano w sześciu randomizowanych, kontrolowanych badaniach klinicznych fazy 3a z udziałem 7215 pacjentów z cukrzycą typu 2 (4107 pacjentów otrzymywało semaglutyd). Głównym celem pięciu badań klinicznych (SUSTAIN 1–5) była ocena skuteczności kontroli glikemicznej, a jednego badania (SUSTAIN 6) – skutków sercowo-naczyniowych leczenia.
Przeprowadzono dodatkowe badanie kliniczne fazy 3b (SUSTAIN 7) z udziałem 1201 pacjentów w celu porównania skuteczności i bezpieczeństwa zastosowania semaglutyd w dawkach 0,5 mg i 1 mg raz w tygodniu z dawidami dulaglutyd 0,75 mg i 1,5 mg odpowiednio raz w tygodniu. Badanie fazy 3b (SUSTAIN 9) przeprowadzono w celu zbadania skuteczności i bezpieczeństwa zastosowania semaglutyd jako uzupełnienia terapii inhibitorem symporterów sodu-glukozy typu 2 (SGLT-2).
Leczenie semaglutyd wykazało trwałe, statystycznie istotne i klinicznie znaczące zmniejszenie wartości HbA1c i masy ciała w okresie do dwóch lat w porównaniu do placebo i aktywnego leczenia kontrolnego (sitagliptyna, insulinę glarginy, egzydetyd ER i dulaglutyd).
Skuteczność semaglutyd nie zależała od wieku, płci, rasy, pochodzenia etnicznego, początkowego BMI, początkowej masy ciała (kg), długości trwania cukrzycy i stopnia zaburzenia funkcji nerek pacjenta.
Wartości osiągnęły cele badania we wszystkich randomizowanych grupach pacjentów (analizy oparte na mieszanych modelach dla powtarzanych pomiarów lub wielokrotnym warunkowym obliczaniu).
Ponadto, przeprowadzono badanie fazy 3b (SUSTAIN 11) w celu zbadania efektu semaglutyd w porównaniu z efektem insuliny aspart jako uzupełnienia do metforminy i optymalizowanej insuliny glarginy (100 J).
Szczegółowe informacje można znaleźć poniżej.
Badania SUSTAIN 1. Monoterapia
W podwójnie ślepych, kontrolowanych placebo badaniach klinicznych trwających 30 tygodni 388 pacjentów z niewystarczającą kontrolą choroby przy stosowaniu diety i aktywności fizycznej zostało zrandomizowanych do grup przyjmujących raz w tygodniu semaglutyd 0,5 mg lub 1 mg albo placebo.
Tabela 1
Badanie SUSTAIN 1: wyniki w 30. tygodniu
| Wskaźnik |
Semaglutyd 0,5 mg |
Semaglutyd 1 mg |
Placebo |
| Liczba pacjentów |
128 |
130 |
129 |
| HbA1c (%) |
|||
| Na początku (wartość średnia) |
8,1 |
8,1 |
8,0 |
| Zmiana od początku do 30. tygodnia |
-1,5 |
-1,6 |
0 |
| Różnica w porównaniu z placebo (95 % CI) |
-1,4 [-1,7, -1,1]a |
-1,5 [-1,8, -1,2]a |
- |
| Pacjenci (%), którzy osiągnęli HbA1c < 7 % |
74 |
72 |
25 |
| Glukoza we krwi na czczo (mmol/l) |
|||
| Na początku (wartość średnia) |
9,7 |
9,9 |
9,7 |
| Zmiana od początku do 30. tygodnia |
-2,5 |
-2,3 |
-0,6 |
| Masa ciała (kg) |
|||
| Na początku (wartość średnia) |
89,8 |
96,9 |
89,1 |
| Zmiana od początku do 30. tygodnia |
-3,7 |
-4,5 |
-1,0 |
| Różnica z placebo (95 % CI) |
-2,7 [-3,9, -1,6]a |
-3,6 [-4,7, -2,4]a |
- |
a p < 0,0001 (dwustronny) na rzecz.
Badanie kliniczne SUSTAIN2. Semaglutyd w porównaniu z sytagliptyną, oba w kombinacji z jednym-dwoma doustnymi lekami przeciwdiabetycznymi (metformyną i/lub tiazydyndionami)
W podwójnie ślepej, kontrolowanej aktywnie studium trwającym 56 tygodni 1231 pacjentów zostało losowo przydzielonych do grupy otrzymującej semaglutyd 0,5 mg raz w tygodniu lub 1 mg raz w tygodniu, albo sytagliptynę 100 mg raz dziennie; wszystkie w kombinacji z metformyną (94 %) i/lub tiazydyndionami (6 %).
Tabela 2
Badanie SUSTAIN 2: wyniki w 56. tygodniu
| Wskaźnik |
Semaglutyd 0,5 mg |
Semaglutyd 1 mg |
Sytagliptyna 100 mg |
| Liczba pacjentów |
409 |
409 |
407 |
| HbA1c (%) |
|||
| Na początku (średnia wartość) |
8,0 |
8,0 |
8,2 |
| Zmiana od początku do 56. tygodnia |
-1,3 |
-1,6 |
-0,5 |
| Różnica w porównaniu z sytagliptyną (95 % CI) |
-0,8 [-0,9, -0,6]a |
-1,1 [-1,2, -0,9]a |
- |
| Pacjenci (%), którzy osiągnęli HbA1c < 7 % |
69 |
78 |
36 |
| Glukoza we krwi na czczo (mmol/l) |
|||
| Na początku (średnia wartość) |
9,3 |
9,3 |
9,6 |
| Zmiana od początku do 56. tygodnia |
-2,1 |
-2,6 |
-1,1 |
| Masa ciała (kg) |
|||
| Na początku (średnia wartość) |
89,9 |
89,2 |
89,3 |
| Zmiana od początku do 56. tygodnia |
-4,3 |
-6,1 |
-1,9 |
| Różnica w porównaniu z sytagliptyną (95 % CI) |
-2,3 [-3,1, -1,6]a |
-4,2 [-4,9, -3,5]a |
- |
ap < 0,0001 (dwustronny) dla korzyści.
**
**
Rys. 1. Średnia zmiana HbA1c (%) oraz masy ciała (kg) od początku do 56. tygodnia
Badanie kliniczne SUSTAIN7. Porównanie semaglutydu z dulaglutydem, obu w połączeniu z metformyną
Uczestników otwartego badania klinicznego trwającego 40 tygodni (1201 pacjentów), którzy przyjmowali metformynę, przydzielono losowo w stosunku 1:1:1:1 do grup otrzymujących raz w tygodniu semaglutyd 0,5 mg lub dulaglutyd 0,75 mg, lub semaglutyd 1 mg, lub dulaglutyd 1,5 mg.
W badaniu porównano wyniki stosowania semaglutydu 0,5 mg z dulaglutydem 0,75 mg oraz semaglutydu 1 mg z dulaglutydem 1,5 mg.
Najczęstszymi reakcjami przeciwnymi były zaburzenia ze strony przewodu pokarmowego, które występowały u takiej samej frakcji pacjentów otrzymujących semaglutyd 0,5 mg (129 pacjentów [43 %]), semaglutyd 1 mg (133 [44 %]) oraz dulaglutyd 1,5 mg (143 [48 %]); zaburzenia te występowały rzadziej u pacjentów leczonych dulaglutydem 0,75 mg (100 [33 %]).
W 40. tygodniu zwiększenie częstości tętna pod wpływem semaglutydu (0,5 mg i 1 mg) oraz dulaglutydu (0,75 mg i 1,5 mg) wynosiło odpowiednio 2,4 i 4,0 oraz 1,6 i 2,1 uderzenia na minutę.
Tabela 3
Badanie SUSTAIN 7: wyniki w 40. tygodniu
| Wskaźnik |
Semaglutyd 0,5 mg |
Semaglutyd 1 mg |
Dulaglutyd 0,75 mg |
Dulaglutyd 1,5 mg |
| Liczba pacjentów |
301 |
300 |
299 |
299 |
| HbA1c (%) |
||||
| Na początku (średnia wartość) |
8,3 |
8,2 |
8,2 |
8,2 |
| Zmiana od początku do 40. tygodnia |
-1,5 |
-1,8 |
-1,1 |
-1,4 |
| Różnica w porównaniu z dulaglutydem (95 % CI) |
-0,4b [-0,6, -0,2)a |
-0,4c [-0,6, -0,3]a |
- |
- |
| Pacjenci (%), którzy osiągnęli HbA1c < 7 % |
68 |
79 |
52 |
67 |
| Glukoza we krwi na czczo (mmol/l) |
||||
| Na początku (średnia wartość) |
9,8 |
9,8 |
9,7 |
9,6 |
| Zmiana od początku do 40. tygodnia |
-2,2 |
-2,8 |
-1,9 |
-2,2 |
| Masa ciała (kg) |
||||
| Na początku (średnia wartość) |
96,4 |
95,5 |
95,6 |
93,4 |
| Zmiana od początku do 40. tygodnia |
-4,6 |
-6,5 |
-2,3 |
-3,0 |
| Różnica w porównaniu z dulaglutydem (95 % CI) |
-2,3b [-3,0, -1,5)a |
-3,6c [-4,3, -2,8]a |
- |
- |
a p < 0,0001 (dwustronny) na rzecz.
b Semaglutyd 0,5 mg w porównaniu z dulaglutydem 0,75 mg.
c Semaglutyd 1 mg w porównaniu z dulaglutydem 1,5 mg.
**
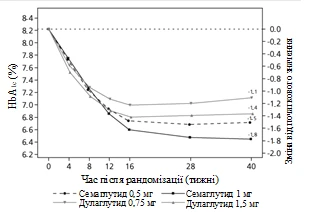
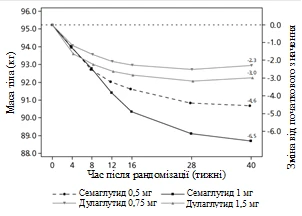
**
Rys. 2. Średnia zmiana HbA1c (%) i masy ciała (kg) od początku do 40. tygodnia
Badanie kliniczne SUSTAIN3. Semaglutyd w porównaniu z eksenatydem ER, oba w połączeniu z metforminą lub metforminą i sulfonamidem
W otwartym badaniu trwającym 56 tygodni 813 pacjentów przyjmujących wyłącznie metforminę (49%), metforminę ze sulfonamidem (45%) lub inne leki (6%) zostało losowo przydzielonych do grupy leczenia semaglutydem 1 mg lub eksenatydem ER 2 mg raz w tygodniu.
Tabela 4
Badanie SUSTAIN 3: wyniki w 56. tygodniu
| Wskaźnik |
Semaglutyd 1 mg |
Eksyenatyd ER 2 mg |
| Liczba pacjentów |
404 |
405 |
| HbA1c (%) |
||
| Na początku (wartość średnia) |
8,4 |
8,3 |
| Zmiana od początku do 56. tygodnia |
-1,5 |
-0,9 |
| Różnica w porównaniu z eksyenatydem (95 % CI) |
-0,6 [-0,8, -0,4]a |
- |
| Pacjenci (%) osiągający HbA1c < 7 % |
67 |
40 |
| Glukoza we krwi na czczo (mmol/l) |
||
| Na początku (wartość średnia) |
10,6 |
10,4 |
| Zmiana od początku do 56. tygodnia |
-2,8 |
-2,0 |
| Masa ciała (kg) |
||
| Na początku (wartość średnia) |
96,2 |
95,4 |
| Zmiana od początku do 56. tygodnia |
-5,6 |
-1,9 |
| Różnica w porównaniu z eksyenatydem (95 % CI) |
-3,8 [-4,6, -3,0]a |
- |
a p < 0,0001 (dwustronny) na korzyść.
Badanie kliniczne SUSTAIN4. Semaglutyd w porównaniu z insuliną glarginem, oba stosowane w połączeniu z jednym–dwoma doustnymi lekami przeciwdiabetycznymi (metforminą lub metforminą i związkami sulfonilomocznikowymi)
W otwartym badaniu z lekiem porównawczym trwającym 30 tygodni 1089 pacjentów zostało zrandomizowanych do grup przyjmujących semaglutyd 0,5 mg raz w tygodniu, semaglutyd 1 mg raz w tygodniu lub insulinę glarginem raz na dobę, z jednoczesnym stosowaniem metforminy (48 %) lub metforminy i związków sulfonilomocznikowych (51 %).
Tabela 5
Badanie SUSTAIN 4: wyniki w 30. tygodniu
| Wskaźnik |
Semaglutyd 0,5 mg |
Semaglutyd 1 mg |
Insulin glargina |
| Liczba pacjentów |
362 |
360 |
360 |
| HbA1c (%) |
|||
| Na początku (średnia wartość) |
8,1 |
8,2 |
8,1 |
| Zmiana od początku do 30. tygodnia |
-1,2 |
-1,6 |
-0,8 |
| Różnica w porównaniu z insuliną glarginą (95 % CI) |
-0,4 [-0,5, -0,2]a |
-0,8 [-1,0, -0,7]a |
- |
| Pacjenci (%), którzy osiągnęli HbA1c < 7 % |
57 |
73 |
38 |
| Glukoza we krwi na czczo (mmol/l) |
|||
| Na początku (średnia wartość) |
9,6 |
9,9 |
9,7 |
| Zmiana od początku do 30. tygodnia |
-2,0 |
-2,7 |
-2,1 |
| Masa ciała (kg) |
|||
| Na początku (średnia wartość) |
93,7 |
94,0 |
92,6 |
| Zmiana od początku do 30. tygodnia |
-3,5 |
-5,2 |
+1,2 |
| Różnica w porównaniu z insuliną glarginą (95 % CI) |
-4,6 [-5,3, -4,0]a |
-6,34 [-7,0, -5,7]a |
- |
a p < 0,0001 (dwustronny) na rzecz korzyści.
Badanie kliniczne SUSTAIN5. Semaglutyd w porównaniu z placebo, oba stosowane w połączeniu z insuliną bazalną
W podwójnie ślepych, kontrolowanych placebo badaniach trwających 30 tygodni 397 pacjentów z niewystarczającym kontrolowaniem leczenia insuliną bazalną bez stosowania lub ze stosowaniem metforminy zostało losowo przydzielonych do grup otrzymujących semaglutyd 0,5 mg raz w tygodniu, semaglutyd 1 mg raz w tygodniu lub placebo.
Tabela 6
Badanie SUSTAIN 5: wyniki w 30. tygodniu
| Wskaźnik |
Semaglutyd 0,5 mg |
Semaglutyd 1 mg |
Placebo |
| Liczba pacjentów |
132 |
131 |
133 |
| HbA1c (%) |
|||
| Na początku (wartość średnia) |
8,4 |
8,3 |
8,4 |
| Zmiana od początku do 30. tygodnia |
-1,4 |
-1,8 |
-0,1 |
| Różnica w porównaniu z placebo (95 % CI) |
-1,4 [-1,6, -1,1]a |
-1,8 [-2,0, -1,5]a |
- |
| Pacjenci (%), którzy osiągnęli HbA1c < 7 % |
61 |
79 |
11 |
| Glukoza we krwi na czczo (mmol/l) |
|||
| Na początku (wartość średnia) |
8,9 |
8,5 |
8,6 |
| Zmiana od początku do 30. tygodnia |
-1,6 |
-2,4 |
-0,5 |
| Masa ciała (kg) |
|||
| Na początku (wartość średnia) |
92,7 |
92,5 |
89,9 |
| Zmiana od początku do 30. tygodnia |
-3,7 |
-6,4 |
-1,4 |
| Różnica w porównaniu z placebo (95 % CI) |
-2,3 [-3,3, -1,3]a |
-5,1 [-6,1, -4,0]a |
- |
a p < 0,0001 (dwustronny) na korzyść.
Badanie kliniczne SUSTAIN9. Semaglutyd w porównaniu z placebo, oba w połączeniu z inhibitorem SGLT-2± metforminą lub sulfonylomocznikiem
W podwójnie ślepej, kontrolowanej placebo, 30-tygodniowej studium klinicznym, 302 pacjentów z niedostatecznym kontrolowaniem cukrzycy leczonych inhibitorem SGLT-2, z lub bez metforminy lub sulfonylomocznika, zostało losowo przydzielonych do grupy otrzymującej semaglutyd 1,0 mg raz w tygodniu lub do grupy placebo.
Tabela 7
Badanie SUSTAIN 9: wyniki w 30. tygodniu
| Wskaźnik |
Semaglutyd 1 mg |
Placebo |
| Liczba pacjentów |
151 |
151 |
| HbA1c (%) |
||
| Na początku (wartość średnia) |
8,0 |
8,1 |
| Zmiana od początku do 30. tygodnia |
-1,5 |
-0,1 |
| Różnica w porównaniu do placebo (95 % CI) |
-1,4 [-1,6, -1,2]a |
- |
| Pacjenci (%), którzy osiągnęli HbA1c < 7 % |
|
|
| Glukoza we krwi na czczo (mmol/l) |
||
| Na początku (wartość średnia) |
9,1 |
8,9 |
| Zmiana od początku do 30. tygodnia |
-2,2 |
|
| Masa ciała (kg) |
||
| Na początku (wartość średnia) |
89,6 |
93,8 |
| Zmiana od początku do 30. tygodnia |
-4,7 |
-0,9 |
| Różnica w porównaniu do placebo (95 % CI) |
-3,8 [-4,7, -2,9]a |
- |
a p < 0,0001 (dwustronny) na rzecz korzyści, skorygowany pod względem wielokrotności na podstawie hierarchicznego testowania wartości HbA1c i masy ciała.
Badań kliniczne SUSTAIN11 – semaglutyd w porównaniu z insuliną aspart jako dodatek do insuliny glarginy + metforminy
W 52-tygodniowym otwartym badaniu 1748 pacjentów z niedostatecznie kontrolowanym cukrzycą typu 2 po 12-tygodniowym okresie wprowadzania insuliny glarginy i metforminy podzielono losowo w stosunku 1:1 na grupę otrzymującą semaglutyd raz w tygodniu (0,5 mg lub 1,0 mg) lub insulinę aspart trzy razy dziennie. Włączona populacja miała średni czas trwania cukrzycy 13,4 roku i średni HbA1c 8,6 %, z docelowym HbA1c w zakresie 6,5–7,5 %.
Leczenie semaglutydem spowodowało obniżenie HbA1c w 52. tygodniu (-1,5 % dla semaglutydu w porównaniu do -1,2 % dla insuliny aspartatu).
Liczba epizodów ciężkiej hipoglikemii w obu grupach leczenia była niska (4 epizody przy stosowaniu semaglutydu w porównaniu do 7 epizodów przy stosowaniu insuliny aspartatu).
Średnia początkowa masa ciała zmniejszyła się przy stosowaniu semaglutydu (-4,1 kg) i wzrosła przy stosowaniu insuliny aspart (+2,8 kg), przewidywana różnica zależna od leczenia wyniosła -6,99 kg (95 % CI od -7,41 do -6,57) w 52. tygodniu.
Stosowanie w połączeniu z monoterapią sulfonylomocznikami
W badaniu klinicznym SUSTAIN 6 (patrz sekcja „Choroby sercowo-naczyniowe”) 123 pacjentów początkowo stosowało monoterapię sulfonylomocznikami. Początkowy poziom HbA1c wynosił 8,2 %, 8,4 % i 8,4 % przy stosowaniu semaglutydu 0,5 mg, semaglutydu 1 mg i placebo odpowiednio. Po 30 tygodniach poziom H0,1 % przy stosowaniu placebo.
Stosowanie w połączeniu z insuliną premiks ± jeden-dwa doustne leki obniżające poziom glukozy (PZG)
W badaniu SUSTAIN 6 (patrz sekcja „Choroby sercowo-naczyniowe”) 867 pacjentów stosowało insulinę premiks (na początku razem z PZG lub bez nich). Początkowy poziom HbA1c wynosił 8,8 %, 8,9 % i 8,9 % przy stosowaniu semaglutydu 0,5 mg, semaglutydu 1 mg i placebo odpowiednio. Po 30 tygodniach poziom HbA1c zmniejszył się o 1,3 %, 1,8 % i 0,4 % przy stosowaniu semaglutydu 0,5 mg, semaglutydu 1 mg i placebo odpowiednio.
Choroby sercowo-naczyniowe
W podwójnie ślepej próbie klinicznej trwającej 104 tygodnie (SUSTAIN 6) 3297 pacjentów z cukrzycą typu 2 i wysokim ryzykiem sercowo-naczyniowym zostało losowo przydzielonych do grupy otrzymującej semaglutyd 0,5 mg lub semaglutyd 1 mg raz w tygodniu lub placebo jako dodatek do standardowego leczenia przez następne 2 lata. Ogółem 98 % pacjentów ukończyło badanie, a na koniec dostępne były dane dotyczące funkcji życiowych dla 99,6 % z nich.
Populacja w badaniu była podzielona według wieku w następujący sposób: 1598 pacjentów (48,5 %) w wieku ≥ 65 lat, 321 (9,7 %) – w wieku ≥ 75 lat oraz 20 (0,6 %) – w wieku ≥ 85 lat. U 2358 pacjentów funkcja nerek była normalna lub z lekkim zaburzeniem, u 832 pacjentów stwierdzono umiarkowane zaburzenie funkcji nerek, a u 107 – ciężkie zaburzenie lub zaawansowaną niewydolność nerek. W populacji było 61 % mężczyzn, których średni wiek wynosił 65 lat, a średni wskaźnik BMI – 33 kg/m². Średni czas trwania cukrzycy wynosił 13,9 roku.
Pierwotnym punktem końcowym był czas od randomizacji do pierwszego wystąpienia poważnego niepożądanego zdarzenia sercowo-naczyniowego (MACE): śmiertelny skutek choroby sercowo-naczyniowej, nieśmiertelny zawał mięśnia sercowego lub nieśmiertelny udar mózgu.
Całkowity wskaźnik złożonych elementów pierwotnego punktu końcowego MACE wyniósł 254, w tym 108 (6,6 %) przy stosowaniu semaglutydu i 146 (8,9 %) przy placebo. Zobacz rysunek 4, na którym przedstawiono wyniki dotyczące pierwotnych i wtórnych punktów końcowych sercowo-naczyniowych. Leczenie semaglutydem spowodowało zmniejszenie ryzyka wystąpienia złożonego pierwotnego punktu końcowego o 26 % w odniesieniu do śmiertelnego skutku choroby sercowo-naczyniowej, nieśmiertelnego zawału mięśnia sercowego i nieśmiertelnego udaru mózgu. Całkowita liczba śmiertelnych skutków choroby sercowo-naczyniowej, nieśmiertelnego zawału mięśnia sercowego lub nieśmiertelnego udaru mózgu wyniosła odpowiednio 90, 111 i 71, w tym 44 (2,7 %), 47 (2,9 %) i 27 (1,6 %) przypadków przy stosowaniu semaglutydu (rysunek 4). Zmniejszenie ryzyka w pierwotnym złożonym punkcie końcowym było głównie spowodowane zmniejszeniem częstości występowania nieśmiertelnego udaru mózgu (39 %) i nieśmiertelnego zawału mięśnia sercowego (26 %) (rysunek 3).
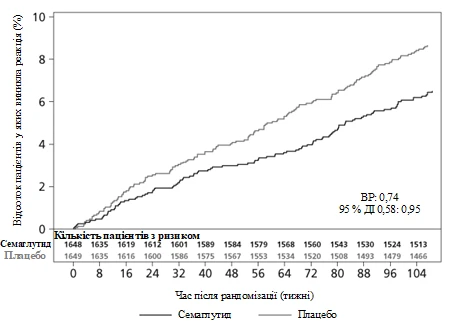
Rys. 3. Krzywa Kaplana-Meiera przedstawiająca czas do pierwszego wystąpienia złożonego zdarzenia: śmiertelny skutek choroby sercowo-naczyniowej, nieśmiertelny zawał mięśnia sercowego i nieśmiertelny udar mózgu (badanie kliniczne SUSTAIN 6).
**
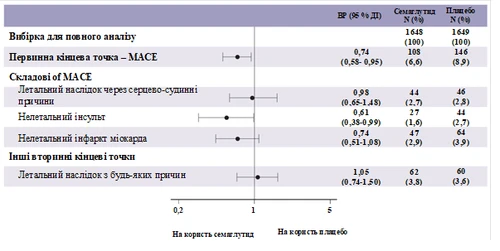
**
Rys. 4. Diagram lasu (Forest plot): analiza czasu do pierwszego wystąpienia złożonego zdarzenia, jego składowe oraz przypadki śmiertelności z dowolnej przyczyny (badanie SUSTAIN 6)
Zarejestrowano 158 przypadków nefropatii, które po raz pierwszy pojawiły się lub pogłębiły się. HR [95 % CI] czasu do wystąpienia nefropatii, zdefiniowanej jako trwała makroalbuminuria (pierwsze wystąpienie trwałej makroalbuminurii, trwałe podwojenie stężenia kreatyniny w surowicy, potrzeba ciągłej terapii zastępczej nerek i śmiertelny skutek choroby nerek), wyniósł 0,64 [0,46; 0,88].
Masa ciała
Po roku leczenia utrata masy ciała na poziomie ≥ 5 % i ≥ 10 % występowała u większej liczby pacjentów przy stosowaniu semaglutydu 0,5 mg (46 % i 13 %) i 1 mg (52–62 % i 21–24 %) niż przy stosowaniu aktywnego środka porównawczego sitagliptyny (18 % i 3 %) i ekzynatydzie ER (17 % i 4 %).
W badaniu porównawczym z dulaglutidem trwającym 40 tygodni utrata masy ciała na poziomie ≥ 5 % i ≥ 10 % została osiągnięta przez większą liczbę pacjentów przy stosowaniu semaglutydu 0,5 mg (44 % i 14 %) niż dulaglutidu 0,75 mg (23 % i 3 %), a także przy stosowaniu semaglutydu 1 mg (do 63 % i 27 %) niż dulaglutidu 1,5 mg (30 % i 8 %).
W trakcie badania klinicznego SUSTAIN 6 zaobserwowano istotne i trwałe zmniejszenie masy ciała od początku leczenia do 104. tygodnia przy stosowaniu semaglutydu 0,5 mg i 1 mg w porównaniu z placebo 0,5 mg i 1 mg jako dodatek do standardowego leczenia (-3,6 kg i -4,9 kg w porównaniu z -0,7 kg i -0,5 kg odpowiednio).
Ciśnienie tętnicze
Zaobserwowano istotne zmniejszenie średniego ciśnienia tętniczego skurczowego przy stosowaniu semaglutydu 0,5 mg (3,5–5,1 mm Hg) i 1 mg (5,4–7,3 mm Hg) w połączeniu z doustnymi lekami przeciwcukrzycowymi lub z insuliną bazalną. Nie zaobserwowano istotnej różnicy w wartości ciśnienia tętniczego rozkurczowego między leczeniem semaglutydem a leczeniem środkami porównawczymi.
Dzieci
Europejska Agencja Leków odroczyła obowiązek przedłożenia wyników badań leku Ozempic® w jednej lub kilku podgrupach pacjentów pediatrycznych w leczeniu cukrzycy typu 2 (patrz sekcja „Sposób stosowania i dawki”).
Farmakokinetyka
W porównaniu z naturalnym GLP-1 semaglutyd charakteryzuje się wydłużonym okresem półtrwania trwającym 1 tydzień, co umożliwia jego podawanie podskórne raz w tygodniu. Głównym mechanizmem takiego przedłużonego działania jest wiązanie z albuminą, które prowadzi do zmniejszenia klirensu nerkowego i ochrony przed degradacją metaboliczną. Ponadto semaglutyd jest stabilizowany przed degradacją przez enzym DPP-4.
Absorpcja
Maksymalne stężenie osiągane jest w ciągu 1–3 dni po podaniu leku. Stężenie stacjonarne osiągane jest po 4–5 tygodniach po stosowaniu leku według schematu raz w tygodniu. U pacjentów z cukrzycą typu 2 średnie stężenie stacjonarne po podaniu podskórnie 0,5 mg i 1 mg semaglutydu wynosiło odpowiednio około 16 nmol/l i 30 nmol/l. Przy dawkach 0,5 mg i 1 mg ekspozycja na semaglutyd wzrastała w sposób zależny od dawki. Podobna ekspozycja została osiągnięta po podaniu podskórny semaglutydu w okolicy przedniej ściany brzucha, uda lub ramienia. Bezpośrednia biodostępność semaglutydu po podaniu podskórny wynosiła 89 %.
Rozkład
Średni objętość rozkładu semaglutydu po podaniu podskórny u pacjentów z cukrzycą typu 2 wynosił około 12,5 l. Semaglutyd w znacznym stopniu wiązał się z albuminą osocza krwi (> 99 %).
Metabolizm
Przed wydaleniem semaglutyd jest aktywnie metabolizowany poprzez proteolityczne rozszczepienie peptydowego szkieletu białka i kolejne beta-oksydowanie bocznego łańcucha kwasów tłuszczowych. Przypuszcza się, że w metabolizmie semaglutydu uczestniczy enzym neutralnej endopeptydazy (NEP).
Wydalanie
W badaniu z jednorazową dawką semaglutydu podawaną podskórnie i znakowaną izotopem radioaktywnym stwierdzono, że substancja związana z semaglutydem wydala się głównie z moczem i kałem; około 2/3 substancji związanej z semaglutydem wydalało się z moczem i około 1/3 – z kałem. Z moczem wydalało się około 3 % podanej dawki w postaci niezmienionego semaglutydu. U pacjentów z cukrzycą typu 2 klirens semaglutydu wynosił około 0,05 l/h. Przy okresie półtrwania trwającym około 1 tydzień semaglutyd obecny jest w krążeniu przez około 5 tygodni po podaniu ostatniej dawki.
Osobliwe grupy pacjentów
Pacjenci w podeszłym wieku
Na podstawie danych uzyskanych w trakcie badań fazy 3a z udziałem pacjentów w wieku 20–86 lat wiek nie wpływa na farmakokinetykę semaglutydu.
Płeć, rasa i pochodzenie etniczne
Płeć, rasa (europejska, negroidalna lub mongoloidalna) i pochodzenie etniczne (hiszpańskie lub latynoamerykańskie, niehiszpańskie lub nielatynoamerykańskie) nie wpływały na farmakokinetykę semaglutydu.
Masa ciała
Masa ciała wpływała na ekspozycję na semaglutyd. Wyższa masa ciała prowadzi do niższej ekspozycji; różnica masy ciała poszczególnych pacjentów na poziomie 20 % prowadzi do prawie 16 % różnicy w ekspozycji. Dawki semaglutydu 0,5 mg i 1 mg zapewniają wystarczającą ekspozycję systemową przy masie ciała w zakresie 40–198 kg.
Zaburzenia funkcji nerek
Zaburzenia funkcji nerek nie wpływały klinicznie istotnie na farmakokinetykę semaglutydu. To zostało wykazane po podaniu jednorazowej dawki 0,5 mg semaglutydu u pacjentów z zaburzeniami funkcji nerek różnego stopnia ciężkości (łagodnym, umiarkowanym, ciężkim i u pacjentów na dializie) w porównaniu z pacjentami z normalną funkcją nerek. To zostało również potwierdzone danymi z klinicznych badań fazy 3a u pacjentów z cukrzycą typu 2 i zaburzeniami funkcji nerek, choć doświadczenie w stosowaniu u pacjentów z zaawansowaną niewydolnością nerek było ograniczone.
Zaburzenia funkcji wątroby
Zaburzenia funkcji wątroby nie wpływały w żaden sposób na ekspozycję na semaglutyd. W badaniach z podaniem jednorazowej dawki 0,5 mg semaglutydu oceniano farmakokinetykę semaglutydu u pacjentów z różnym stopniem zaburzeń funkcji wątroby (łagodnym, umiarkowanym, ciężkim) w porównaniu z pacjentami z normalną funkcją wątroby.
Dzieci
Badania stosowania semaglutydu u pacjentów pediatrycznych nie przeprowadzono.
Dane przedkliniczne dotyczące bezpieczeństwa
Dane przedkliniczne oparte na badaniach farmakologicznej bezpieczeństwa, toksyczności powtarzanych dawek i genotoksyczności nie wykazały żadnego ryzyka dla człowieka.
Nieśmiertelne guzy pochodzące z komórek C tarczycy, obserwowane u gryzoni, należą do efektów charakterystycznych dla klasy agonistów receptorów GLP-1. W badaniu kancerogennym trwającym 2 lata u szczurów i myszy semaglutyd powodował wystąpienie guzów komórek C tarczycy przy klinicznie istotnych poziomach ekspozycji. Nie obserwowano żadnych innych nowotworów, których pojawienie się mogłoby być związane z leczeniem. Guzy u gryzoni są spowodowane niegenotoksycznym, specyficznym dla receptora GLP-1 mechanizmem pośredniczonym przez receptory GLP-1, do którego częściowo są wrażliwe gryzonie. Znaczenie tego mechanizmu dla człowieka jest wystarczająco niskie, ale nie może być całkowicie wykluczone.
W badaniach dotyczących płodności u szczurów semaglutyd nie wpływał na skuteczność kojarzenia ani na płodność u samców. U samic szczurów zaobserwowano wydłużenie cyklu estralnego i niewielkie zmniejszenie liczby ciał żółtych (owulacji) przy dawkach towarzyszących utracie masy ciała u samic.
W badaniach rozwoju embrionów i płodów u szczurów semaglutyd wywoływał działanie embriotoksyczne przy ekspozycji niższej niż klinicznie istotne poziomy. Semaglutyd powodował znaczne zmniejszenie masy ciała u samic i zmniejszenie wskaźników przeżycia i wzrostu embrionów. U płodów zaobserwowano duże wady kostne i wisceralne, w tym zmiany w kościach długich, żebrach, kręgosłupie, kościach ogona, naczyniach krwionośnych i komorach mózgu. Ocena mechanistyczna wykazała, że efekt embriotoksyczny obejmuje pośrednio pośredniczony przez receptory GLP-1 zaburzony dopływ substancji odżywczych do embrionu wzdłuż worka żółtkowego u szczurów. Ze względu na różnice anatomiczne budowy worka żółtkowego i jego funkcji u różnych gatunków szczurów oraz brak ekspresji receptorów GLP-1 w worku żółtkowym u naczłowieków ten mechanizm uznaje się za mało prawdopodobny dla organizmu człowieka. Jednakże nie można wykluczyć bezpośredniego wpływu semaglutydu na płód.
W badaniach toksycznego wpływu leku na rozwój u królików i makaków jawajskich przy klinicznie istotnych poziomach ekspozycji zaobserwowano zwiększenie częstości poronień i pewne zwiększenie częstości wad płodu. Takie wyniki pokrywały się ze znaczną utratą masy ciała u samic dochodzącą do 16 %. Nie wiadomo, czy takie efekty są związane ze zmniejszeniem spożycia pokarmu przez samice w wyniku bezpośredniego wpływu GLP-1.
Postnatalny wzrost i rozwój oceniano u jamajskich makaków jawajskich. Noworodki były nieco mniejsze, ale ich masa ciała znormalizowała się w okresie karmienia piersią.
U młodych szczurów semaglutyd powodował opóźnienie dojrzewania płciowego zarówno u samców, jak i samic. Takie opóźnienie nie wpływało ani na płodność i zdolność rozrodczą obu płci, ani na zdolność samic do utrzymania ciąży.
Właściwości kliniczne
Wskazania
Lek Ozempic® stosuje się w przypadku niekontrolowanego cukrzycy typu 2 jako uzupełnienie diety i aktywności fizycznej:
- jako monoterapię, gdy metformina jest uznawana za niewskazaną z powodu nietolerancji lub przeciwwskazań;
- jako uzupełnienie innych leków stosowanych w leczeniu cukrzycy.
Informacje dotyczące wyników badań stosowania kombinacji leków, wpływu na kontrolę glikemii i zdarzenia sercowo-naczyniowe, jak również dane dotyczące populacji badanych pacjentów, znajdują się w rozdziałach „Szczególne ostrzeżenia i środki ostrożności podczas stosowania”, „Współdziałanie z innymi lekami i inne formy oddziaływania” oraz „Farmakodynamika”.
Przeciwwskazania
Podwyższona wrażliwość na substancję czynną lub na którykolwiek z pozostałych składników leku (patrz rozdział „Skład”).
Współdziałanie z innymi lekami i inne formy oddziaływania
Semaglutyd opóźnia opróżnianie żołądka i może wpływać na szybkość absorpcji doustnych leków stosowanych jednocześnie. Semaglutyd należy stosować z ostrożnością u pacjentów przyjmujących leki doustne, które wymagają szybkiego wchłaniania w przewodzie pokarmowym.
Paracetamol
Na podstawie oceny farmakokinetyki paracetamolu w trakcie standaryzowanego testu z posiłkiem, semaglutyd opóźnia szybkość opróżniania żołądka. Po jednoczesnym podaniu paracetamolu i semaglutyd w dawce 1 mg wartości AUC0-60min i Cmax paracetamolu zmniejszyły się odpowiednio o 27% i 23%. Całkowite narażenie na paracetamol (AUC0–5h) nie uległo zmianie. W przypadku jednoczesnego stosowania paracetamolu i semaglutyd nie jest wymagana korekta dawki.
Antykoncepcja doustna
Nie oczekuje się, że semaglutyd obniży skuteczność doustnej antykoncepcji. Po jednoczesnym podaniu semaglutyd z kombinowanym doustnym środkiem antykoncepcyjnym (etyniloestradiol 0,03 mg / lewonorgestrel 0,15 mg) semaglutyd nie wywierał klinicznie istotnego wpływu na całkowite narażenie na etyniloestradiol i lewonorgestrel. Narażenie na etyniloestradiol nie było zaburzone; narażenie na lewonorgestrel zwiększyło się o 20% w stanie równowagi. Wartości Cmax żadnej z tych substancji nie uległy zmianie.
Atorwastatyna
Semaglutyd nie zmienił całkowitego narażenia na atorwastatynę po podaniu pojedynczej dawki atorwastatyny (40 mg). Wartość Cmax atorwastatyny zmniejszyła się o 38%. Uważa się to za klinicznie nieistotne.
Digoksyna
Semaglutyd nie zmienił całkowitego narażenia ani wartości Cmax digoksyny po podaniu pojedynczej dawki digoksyny (0,5 mg).
Metformina
Semaglutyd nie zmienił całkowitego narażenia ani wartości Cmax metforminy po podaniu metforminy w dawce 500 mg dwa razy dziennie przez 3,5 dnia.
Warczaryna i inne pochodne kumaryny
Semaglutyd nie zmienił całkowitego narażenia ani wartości Cmax R- i S-warczaryny po podaniu pojedynczej dawki warczaryny (25 mg), a parametry farmakodynamiczne warczaryny, mierzone za pomocą międzynarodowego znormalizowanego stosunku (INR), nie uległy klinicznie istotnej zmianie. Jednakże zarejestrowano przypadki obniżenia INR przy jednoczesnym stosowaniu akenokumarolu i semaglutyd. Na początku leczenia semaglutydem u pacjentów przyjmujących warczarynę lub inne pochodne kumaryny zaleca się częste monitorowanie INR.
Szczególne wskazania dotyczące stosowania
Śledzenie
W celu poprawy śledzenia leków biologicznych nazwa i numer serii podawanego leku powinny być wyraźnie wskazane na opakowaniu.
Aspiracja w połączeniu z znieczuleniem ogólnym lub głęboką sedacją
Zgłaszano przypadki aspiracji płucnej u pacjentów stosujących agonisty receptora GLP-1, którzy byli pod znieczuleniem ogólnym lub głęboką sedacją. W związku z tym przed wykonaniem procedury z zastosowaniem znieczulenia ogólnego lub głębokiej sedacji należy wziąć pod uwagę zwiększony ryzyko obecności resztek treści żołądka z powodu opóźnionego opróżniania żołądka (patrz dział „Efekty uboczne”).
Informacje ogólne
Semaglutyd nie powinien być stosowany u pacjentów z cukrzycą typu 1 ani w leczeniu ketoacydozy cukrzycowej. Semaglutyd nie jest zamiennikiem insuliny. Opisywano przypadki rozwoju ketoacydozy cukrzycowej u pacjentów uzależnionych od insuliny, którzy rozpoczynali leczenie agonistą receptora GLP-1 i szybko przerywali stosowanie insuliny lub szybko zmniejszali jej dawkę (patrz dział „Sposób dawkowania i stosowania”).
Nie ma doświadczeń klinicznych w leczeniu pacjentów z niewydolnością serca klasy IV według klasyfikacji New York Heart Association (NYHA), dlatego semaglutyd nie jest zalecany tym pacjentom.
Wpływ na przewód pokarmowy
Stosowanie agonistów receptora GLP-1 może wiązać się z występowaniem działań niepożądanych ze strony przewodu pokarmowego. Należy to uwzględnić przy leczeniu pacjentów z zaburzeniami funkcji nerek, ponieważ nudności, wymioty i biegunka mogą prowadzić do odwodnienia, które może pogorszyć funkcję nerek (patrz dział „Efekty uboczne”).
Ostre zapalenie trzustki
Obserwowano przypadki ostrego zapalenia trzustki podczas stosowania agonistów receptora GLP-1.
Pacjentów należy poinformować o charakterystycznych objawach ostrego zapalenia trzustki. W przypadku podejrzenia zapalenia trzustki należy przerwać leczenie semaglutydem; jeśli zapalenie trzustki zostanie potwierdzone, nie należy ponownie rozpoczynać leczenia semaglutydem. Semaglutyd należy stosować z ostrożnością u pacjentów z wywiadem zapalenia trzustki.
Hipoglikemia
U pacjentów stosujących semaglutyd w połączeniu z sulfonamidami lub insuliną może występować zwiększony ryzyko hipoglikemii. Ryzyko hipoglikemii można zmniejszyć poprzez zmniejszenie dawki sulfonamidów lub insuliny na początku leczenia semaglutydem (patrz dział „Efekty uboczne”).
Retinopatia cukrzycowa
U pacjentów z retinopatią cukrzycową, którzy otrzymują insulinę i semaglutyd, obserwowano zwiększony ryzyko powikłań retinopatii cukrzycowej (patrz dział „Efekty uboczne”). Semaglutyd należy stosować z ostrożnością u pacjentów z retinopatią cukrzycową, którzy otrzymują insulinę. Pacjenci ci powinni być dokładnie monitorowani i leczeni zgodnie z zaleceniami klinicznymi. Szybkie poprawienie kontroli poziomu glukozy było związane z tymczasowym nasileniem się retinopatii cukrzycowej, jednak nie można wykluczyć wpływu innych mechanizmów.
Zawartość sodu
Ten lek zawiera mniej niż 1 mmol sodu (23 mg) na dawkę, dlatego lek można uważać za pozbawiony sodu.
Stosowanie w okresie ciąży lub karmienia piersią
Kobiety w wieku rozrodczym
Kobietom w wieku rozrodczym zaleca się stosowanie środków antykoncepcyjnych podczas leczenia semaglutydem.
Ciąża
Badania na zwierzętach wykazały obecność toksyczności rozrodczej (patrz dział „Dane przedkliniczne dotyczące bezpieczeństwa”). Dane dotyczące stosowania semaglutydu u ciężarnych kobiet są ograniczone. Dlatego semaglutyd nie powinien być stosowany w okresie ciąży. Jeśli pacjentka planuje zajście w ciążę lub jest w ciąży, należy przerwać leczenie semaglutydem. Ze względu na długi okres półtrwania semaglutydu należy przerwać jego stosowanie co najmniej 2 miesiące przed planowaną ciążą (patrz dział „Farmakokinetyka”).
Karmienie piersią
Podczas laktacji u szczurów semaglutyd przenikał do mleka. Ponieważ nie można wykluczyć ryzyka dla niemowlęcia karmionego piersią, semaglutyd nie powinien być stosowany w okresie karmienia piersią.
Plodność
Wpływ semaglutydu na płodność u ludzi jest nieznany. Stosowanie semaglutydu u samców szczurów nie wpływało na płodność. U samic szczurów obserwowano wydłużenie cyklu estralnego i zmniejszenie liczby owulacji przy dawkach związanych z utratą masy ciała (patrz dział „Dane przedkliniczne dotyczące bezpieczeństwa”).
Wpływ na zdolność prowadzenia pojazdów lub obsługiwania maszyn
Semaglutyd nie wpływa lub prawie nie wpływa na zdolność prowadzenia pojazdów lub obsługiwanie maszyn. W przypadku stosowania semaglutydu w połączeniu z sulfonamidami lub insuliną pacjentom należy zalecić środki ostrożności, aby uniknąć rozwoju hipoglikemii podczas prowadzenia pojazdów lub pracy z innymi mechanizmami (patrz dział „Szczególne wskazania dotyczące stosowania”).
Sposób stosowania i dawki
Dozowanie
Początkowa dawka to 0,25 mg semaglutydu 1 raz w tygodniu. Po 4 tygodniach dawkę należy zwiększyć do 0,5 mg 1 raz w tygodniu. W celu dalszego poprawienia kontroli glikemii po stosowaniu dawki 0,5 mg 1 raz w tygodniu przez co najmniej 4 tygodnie, dawkę można zwiększyć do 1 mg 1 raz w tygodniu.
Dawka 0,25 mg semaglutydu nie jest dawką utrzymaną. Dawek przekraczających 1 mg semaglutydu 1 raz w tygodniu nie zaleca się.
Gdy lek Ozempic® jest stosowany dodatkowo do metformyny i/lub tiazolidynodionu lub inhibitora współprzewodzenia glukozy i sodu typu 2 (iSGLT-2), które są już stosowane, aktualne dawki metformyny i/lub tiazolidynodionu lub inhibitora SGLT-2 mogą pozostać bez zmian.
Gdy lek Ozempic® jest stosowany dodatkowo do leków sulfonylomocznikowych lub insuliny, które są już stosowane, należy rozważyć zmniejszenie dawki leku sulfonylomocznikowego lub insuliny w celu zmniejszenia ryzyka wystąpienia hipoglikemii (patrz sekcje „Działania niepożądane” oraz „Wpływ na zdolność prowadzenia pojazdów i obsługiwanie maszyn”).
Do dostosowania dawki leku Ozempic® nie jest wymagany samokontrola poziomu glukozy we krwi. Samokontrola poziomu glukozy we krwi jest wymagana do dostosowania dawki leków sulfonylomocznikowych i insuliny, szczególnie na początku stosowania leku Ozempic® oraz podczas zmniejszania dawki insuliny. Zalecana jest stopniowa procedura zmniejszania dawki insuliny.
Pominięta dawka
Jeśli dawka została pominięta, należy ją podać jak najszybciej w ciągu 5 dni od pominięcia. Jeśli upłynęło już więcej niż 5 dni, pominiętą dawkę należy pominąć i podać następną dawkę w zaplanowanym terminie. W ten sposób pacjenci mogą w każdym przypadku powrócić do regularnego schematu stosowania leku 1 raz w tygodniu.
Jeśli konieczne jest zmiana dnia tygodniowego podawania leku, należy zachować odstęp co najmniej 3 dni (>72 godziny) między dwiema dawkami. Po wybraniu nowego dnia podawania leku należy kontynuować jego stosowanie według schematu 1 raz w tygodniu.
Osobliwe grupy pacjentów
Pacjenci w wieku podeszłym
Dostosowania dawki ze względu na wiek nie należy stosować. Doświadczenie w leczeniu pacjentów w wieku ≥ 75 lat jest ograniczone (patrz sekcja „Farmakokinetyka”).
Zaburzenia funkcji nerek
U pacjentów z łagodnym, umiarkowanym lub ciężkim zaburzeniem funkcji nerek nie jest wymagane dostosowanie dawki. Doświadczenie w stosowaniu semaglutydu u pacjentów z ciężkim zaburzeniem funkcji nerek jest ograniczone. Stosowanie semaglutydu u pacjentów z nerek w stadium końcowym nie jest zalecane (patrz sekcja „Farmakokinetyka”).
Zaburzenia funkcji wątroby
U pacjentów z zaburzeniem funkcji wątroby nie jest wymagane dostosowanie dawki. Doświadczenie w stosowaniu semaglutydu u pacjentów z ciężkim zaburzeniem funkcji wątroby jest ograniczone. Semaglutyd należy stosować z ostrożnością u tych pacjentów (patrz sekcja „Farmakokinetyka”).
Dzieci
Bezpieczeństwo i skuteczność stosowania semaglutydu u dzieci (do 18 roku życia) nie zostały ustalone. Brak danych.
Sposób podania
Stosować podskórnie.
Ozempic® podaje się podskórnie w okolicę przedniej ściany brzucha, uda lub ramienia. Miejsce wstrzyknięcia można zmieniać bez konieczności dostosowania dawki. Ozempic® nie można podawać dożylnie ani domięśniowo.
Ozempic® należy stosować 1 raz w tygodniu w dowolnym czasie dnia, niezależnie od przyjmowania pokarmu.
Aby uzyskać dalsze informacje dotyczące podania leku, patrz sekcja „Zalecenia dotyczące postępowania z lekiem i jego utylizacji”.
Zalecenia dotyczące postępowania z lekiem i jego utylizacji
Należy zalecić pacjentowi, aby po każdej iniekcji usuwał igłę i przechowywał strzykawkę-wyszczotkę bez założonej igły. Zapobiegnie to zablokowaniu igły, zanieczyszczeniu, zakażeniu, wyciekowi roztworu oraz niedokładnemu dawkowaniu. Igiły i inne odpady należy utylizować zgodnie z lokalnymi przepisami. Strzykawka-wyszczotka przeznaczona jest wyłącznie do użytku indywidualnego. Leku Ozempic® nie należy stosować, jeśli nie jest przezroczysty i bezbarwny lub prawie bezbarwny. Nie stosować po zamrożeniu. Strzykawka-wyszczotka przeznaczona jest do użytku z jednorazowymi igłami NovoFine® lub NovoTwist® o długości do 8 mm. Igiły NovoFine® Plus są dołączane do opakowania leku.
| Instrukcja obsługi dozownika OZEMPIC® 1 mg (roztwór do wstrzykiwań w dozowniku wstępnie załadowanym). |
||
| Aby uzyskać szczegółowe informacje, należy zapoznać się z ulotką do leku przed użyciem dozownika Ozempic®. Należy porozmawiać z lekarzem, pielęgniarką lub farmaceutą o właściwym sposobie wstrzyknięcia leku Ozempic®. Przed użyciem dozownika należy go najpierw sprawdzić, aby upewnić się, że zawiera on lek Ozempic® w dawce 1 mg. Następnie należy zapoznać się z rysunkami poniżej, aby poznać poszczególne części dozownika i igły. Jeśli pacjent nie widzi wcale lub widzi słabo i nie jest w stanie odczytać wyświetlacza dawki na dozowniku, nie może on używać dozownika bez pomocy osoby trzeciej. Pomoc powinna świadczyć osoba o dobrym wzroku, która wie, jak korzystać z wstępnie załadowanego dozownika Ozempic®. Dozownik jest wstępnie załadowany i wyposażony w mechanizm doboru dawki. Zawiera 4 mg semaglutydu. Można ustawić wyłącznie dawkę 1 mg. Nowy dozownik przed użyciem zawiera cztery dawki po 1 mg. Użyj tabeli wewnątrz pokrywy tekturowego opakowania, aby śledzić liczbę wykonywanych wstrzyknięć i daty ich wykonania: Należy wskazać dzień tygodnia wybrany na wstrzyknięcie Ważne informacje Szczególną uwagę należy zwrócić na te uwagi, ponieważ są one istotne dla bezpiecznego użytkowania dozownika. A także: Otworzyć tutaj Podnieść tutaj |
|
|
|
||
|
|
|
|
|
|
Sprawdzić, czy papierowa membrana i zewnętrzna osłonka igły nie są uszkodzone, ponieważ może to oznaczać brak sterylności. W przypadku jakichkolwiek uszkodzeń należy użyć nowej igły.
|
|
|
| Upewnić się, że igła jest prawidłowo zamocowana.
|
|
|
| Na igłę są założone dwie osłonki. Należy zdjąć obie osłonki. Jeśli zapomni się zdjąć obu osłonek, nie uda się wykonać wstrzyknięcia roztworu.
|
|
|
Na końcówce igły może pojawić się kropla roztworu. Jest to zjawisko normalne, jednak należy sprawdzić dopływ leku przy pierwszym użyciu nowego dozownika. Zobacz punkt 2 „Sprawdzenie przepływu roztworu w każdym nowym dozowniku”. Nie przyłączać nowej igły, dopóki nie będzie się gotowym do wykonania wstrzyknięcia. |
|
|
| Do każdego wstrzyknięcia należy zawsze używać nowej igły. Może to zapobiec zatkaniu igły, zanieczyszczeniu, infekcji oraz podaniu niewłaściwej dawki leku. |
||
| Nigdy nie używać igły, jeśli jest wygięta lub uszkodzona. |
||
|
||
|
|
|
Nacisnąć i przytrzymać przycisk dawki, aż wyświetlacz dawki powróci do „0”. Wartość „0” powinna pokrywać się ze wskaźnikiem dawki. Na końcówce igły powinna pojawić się kropla roztworu. |
|
|
| Na końcówce igły może pozostać mała kropla, ale nie zostanie ona wstrzyknięta. Jeśli kropla się nie pojawi, wrócić do punktu 2 „Sprawdzenie przepływu roztworu w nowym dozowniku” do 6 razy. Jeśli kropla nadal nie pojawi się, zmienić igłę i powtórzyć czynności opisane w punkcie 2 „Sprawdzenie przepływu roztworu w nowym dozowniku” ponownie. Jeśli kropla nadal się nie pojawi, zutylizować dozownik i użyć nowego. |
||
| Zawsze należy upewnić się, że kropla pojawi się na końcówce igły przed pierwszym użyciem nowego dozownika. Gwarantuje to, że roztwór zostanie wstrzyknięty. Jeśli kropla się nie pojawi, nie należy używać dozownika, nawet jeśli wyświetlacz dawki zmienia wartość. Może to wskazywać na zatkana lub uszkodzoną igłę. Jeśli nie sprawdzi się przepływu przed pierwszym wstrzyknięciem nowego dozownika, może nie zostać podana wymagana dawka roztworu, co wpłynie na oczekiwany efekt działania leku Ozempic®. |
||
|
||
Obracać pokrętło dawki, aż wyświetlacz dawki zatrzyma się i pokaże dawkę 1 mg. |
|
|
| Tylko wyświetlacz dawki i wskaźnik dawki pokażą 1 mg, czyli dawkę, którą przygotowano do wstrzyknięcia. Można wybrać wyłącznie 1 mg leku na dawkę. Jeśli dozownik zawiera mniej niż 1 mg leku, wyświetlacz dawki zatrzyma się przed osiągnięciem wartości 1. Podczas obracania pokrętła do przodu, do tyłu lub gdy obraca się je dalej po osiągnięciu wskaźnika wybranej dawki 1 mg, pokrętło dawki wydaje różne dźwięki kliknięcia. Nie należy liczyć liczby kliknięć dozownika. |
||
| Zawsze należy korzystać z wyświetlacza dawki i wskaźnika dawki, aby upewnić się, że wybrano 1 mg przed podaniem leku. Nie należy liczyć liczby kliknięć dozownika. Dawka 1 mg musi dokładnie pokrywać się z wskaźnikiem dawki, aby zapewnić właściwą dawkę. |
||
| Zalegający roztwór w dozowniku |
||
|
|
|
| Jeśli roztworu jest za mało do podania dawki, nie należy stosować dozownika. Należy użyć nowego dozownika Ozempic®. |
||
|
||
|
|
|
Obserwować, aż wyświetlacz dawki powróci do „0”. „0” powinno pokrywać się ze wskaźnikiem dawki. Następnie może być słychać lub odczuwalne kliknięcie.
|
|
|
|
|
|
Jeśli w miejscu wstrzyknięcia pojawi się kropla krwi, delikatnie przycisnąć to miejsce. |
|
|
| Może się pojawić kropla roztworu na końcówce igły po wstrzyknięciu. Jest to normalne i nie wpływa na objętość podanej dawki. |
||
| Zawsze należy patrzeć na wskaźnik dawki, aby zobaczyć, ile miligramów roztworu zostało podanych. Trzymać naciśnięty przycisk startowy, aż wskaźnik dawki ponownie pokaże „0”. Jak wykryć zatkana lub uszkodzoną igłę.
Jak postąpić w przypadku zatkanej igły. Zdjąć igłę, jak opisano w punkcie 5 „Po wstrzyknięciu”, i powtórzyć czynności opisane we wszystkich punktach, począwszy od punktu 1 „Przygotowanie dozownika z nową igłą do użycia”. Nigdy nie dotykać wyświetlacza podczas podawania leku. Może to przerwać wstrzyknięcie. |
||
|
||
| Zawsze zutylizować igłę po każdym wstrzyknięciu, aby zapewnić wygodę wstrzyknięcia i zapobiec użyciu zatkanej igły. Jeśli igła jest zatkana, nie będzie można podać leku.
|
|
|
|
|
|
|
|
|
| Kiedy dozownik będzie pusty, należy go wyrzucić bez igły zgodnie z instrukcjami otrzymanymi od lekarza, pielęgniarki, farmaceuty lub zgodnie z lokalnymi przepisami. |
||
| Nigdy nie próbować ponownie założyć wewnętrznej osłonki igły na igłę. Istnieje ryzyko urazu igłą. Zawsze usuwać igłę z dozownika po każdym wstrzyknięciu. Może to zapobiec zatkaniu igły, zanieczyszczeniu, zakażeniu, wyciekowi roztworu oraz niedokładnemu dawkowaniu. |
||
| Dodatkowe ważne informacje. |
||
|
||
| Opieka nad dozownikiem. |
||
| Należy ostrożnie obchodzić się z dozownikiem. Nieodpowiednie postępowanie lub niewłaściwe użycie może prowadzić do podania niewłaściwej dawki. Jeśli dojdzie do takiej sytuacji, może nie uzyskać się oczekiwanego efektu działania leku. |
||
|
||

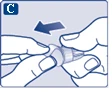
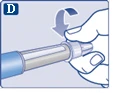
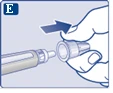
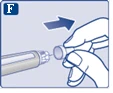
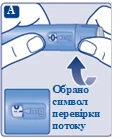
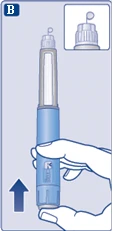
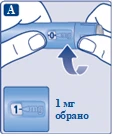
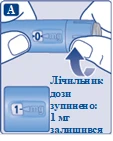
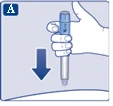
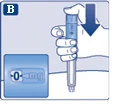
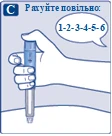
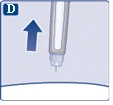
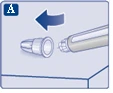
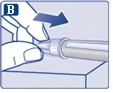
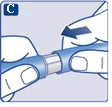
Dzieci
Bezpieczeństwo i skuteczność stosowania semaglutydu u dzieci (do 18. roku życia) nie zostały ustalone. Brak danych.
Europejska Agencja Leków zwolniła z obowiązku przedstawienia wyników badań semaglutydu w jednej lub kilku podgrupach populacji pediatrycznej w leczeniu cukrzycy typu 2 (patrz sekcja „Sposób i dawka stosowania”).
Przedawkowanie
W trakcie badań klinicznych zgłaszano przypadki przedawkowania do 4 mg przy jednorazowym stosowaniu leku oraz do 4 mg przy stosowaniu przez okres tygodnia. Najczęściej zgłaszano nudności. Wszyscy pacjenci wyzdrowieli bez powikłań.
Nie istnieje specyficzny antydota na przedawkowanie semaglutydu. W przypadku przedawkowania należy prowadzić leczenie objawowe zgodnie z występującymi u pacjenta objawami klinicznymi. Ze względu na długi okres półtrwania semaglutydu, wynoszący około 1 tygodnia, może istnieć konieczność przedłużenia czasu leczenia tych objawów (patrz sekcja „Farmakokinetyka”).
Niepożądane reakcje
Streszczenie profilu bezpieczeństwa
W ośmiu badaniach klinicznych fazy 3 uczestniczyło 4792 pacjentów otrzymujących semaglutyd w dawce do 1 mg. W trakcie badań klinicznych najczęściej zgłaszano niepożądane reakcje ze strony przewodu pokarmowego, w tym nudności (bardzo często), biegunkę (bardzo często) oraz wymioty (często). Ogólnie reakcje te miały charakter łagodny lub umiarkowany i były krótkotrwałe.
Efekty uboczne
Poniżej przedstawiono efekty uboczne wykryte we wszystkich badaniach klinicznych fazy 3a u pacjentów z cukrzycą typu 2 (opisane szczegółowo w sekcji „Farmakodynamika”). Częstość występowania efektów ubocznych określono na podstawie zestawionych danych z klinicznych badań fazy 3a, z wyłączeniem badań wyników kardiologicznych (dodatkowe informacje poniżej).
Efekty uboczne sklasyfikowano według układów narządów i częstości występowania. Ocena częstości występowania efektów ubocznych została przeprowadzona według następującej skali: bardzo często: (≥ 1/10), często: (od ≥ 1/100 do < 1/10), rzadko: (od ≥ 1/1000 do < 1/100), bardzo rzadko: (od ≥ 1/10000 do < 1/1000), nieznane: (niemożliwe do oszacowania na podstawie dostępnych danych). W każdej grupie efekty uboczne wymieniono w kolejności malejącej ich powagi.
Zaburzenia układu odpornościowego: rzadko – nadwrażliwośćc; bardzo rzadko – reakcja anafilaktyczna.
Zaburzenia metabolizmu i trawienia: bardzo często – hipoglikemiaa przy jednoczesnym stosowaniu z insuliną lub pochodnymi sulfoniliomocznika; często – hipoglikemiaa przy jednoczesnym stosowaniu z innymi doustnymi lekami przeciwcukrzycowymi, zmniejszenie apetytu.
Zaburzenia układu nerwowego: często – zawroty głowy; rzadko – dysgeuzja.
Zaburzenia oka: często – nasilenie retinopatii cukrzycowejb.
Zaburzenia serca: rzadko – zwiększona częstość skurczów serca.
Zaburzenia przewodu pokarmowego: bardzo często – nudności, biegunka; często – wymioty, ból brzucha, wzdęcia, zaparcia, niestrawność, zapalenie żołądka, choroba refluksowa przełyku, odbijanie, wzdęcia; rzadko – ostre zapalenie trzustki, opóźnione opróżnianie żołądka; nieznane – niedrożność jelitad.
Zaburzenia wątroby i dróg żółciowych: często – kamica żółciowa.
Zaburzenia skóry i tkanek podskórnych: nieznane – obrzęk naczynioruchowyd.
Ogólne zaburzenia i reakcje w miejscu wstrzyknięcia: często – zmęczenie; rzadko – reakcje w miejscu wstrzyknięcia.
Badania laboratoryjne: często – podwyższony poziom lipazy, podwyższony poziom amylazy, spadek masy ciała.
a Hipoglikemia była definiowana jako stan ciężki (wymagający pomocy innej osoby) lub objawowy stan towarzyszący stężeniu glukozy we krwi < 3,1 mmol/l.
b Nasilenie retinopatii cukrzycowej obejmuje następujące cechy: fotokoagulacja siatkówki, leczenie środkami do iniekcji wewnątrzwiotowej, krwawienie do ciała szklistego, ślepotę spowodowaną cukrzycą (rzadko). Częstość przypadków określono na podstawie danych z badania wyników kardiologicznych.
c Ogólny termin obejmujący również niepożądane zjawiska związane z nadwrażliwością, takie jak wysypka i pokrzywka.
d Dane zgłoszone w okresie posprzedażowym.
Badania wyników kardiologicznych i bezpieczeństwa trwające 2 lata
U pacjentów z wysokim ryzykiem wystąpienia chorób układu sercowo-naczyniowego profil efektów ubocznych był podobny do profilu obserwowanego w innych badaniach fazy 3a (opisanych w sekcji „Farmakodynamika”).
Opis wybranych efektów ubocznych
Hipoglikemia
Podczas stosowania semaglutydu jako monoterapii nie obserwowano epizodów ciężkiej hipoglikemii. Ciężka hipoglikemia występowała głównie wtedy, gdy semaglutyd stosowano razem z pochodnymi sulfoniliomocznika (1,2 % pacjentów; 0,03 przypadki na pacjenta-rok) lub insuliną (1,5 % pacjentów; 0,02 przypadki na pacjenta-rok). Kilka epizodów (0,1 % pacjentów; 0,001 przypadki na pacjenta-rok) wystąpiło przy stosowaniu semaglutydu w połączeniu z doustnymi lekami przeciwcukrzycowymi innymi niż pochodne sulfoniliomocznika.
Amerykańskie Towarzystwo Diabetologiczne sklasyfikowało hipoglikemię u 11,3 % pacjentów (0,3 przypadki na pacjenta-rok) przy stosowaniu semaglutydu 1,0 mg w połączeniu z iNKTG-2 w badaniu SUSTAIN 9, w porównaniu do 2,0 % pacjentów (0,04 przypadki na pacjenta-rok) stosujących placebo. O ciężkiej hipoglikemii donoszono odpowiednio u 0,7 % (0,01 zdarzenia na rok pacjenta) i 0 % pacjentów.
Reakcje ze strony przewodu pokarmowego
Podczas stosowania semaglutydu w dawce 0,5 mg i 1 mg nudności obserwowano odpowiednio u 17,0 % i 19,9 % pacjentów, biegunkę – u 12,2 % i 13,3 %, wymioty – u 6,4 % i 8,4 %. Większość przypadków miała charakter od lekkiego do umiarkowanego i była nietrwała. Reakcje doprowadziły do przerwania leczenia odpowiednio u 3,9 % i 5 % pacjentów. Reakcje najczęściej rejestrowano w pierwszych miesiącach leczenia.
U pacjentów o niskiej masie ciała leczonych semaglutydem liczba zaburzeń ze strony przewodu pokarmowego może być większa.
W badaniu SUSTAIN 9 przy jednoczesnym stosowaniu iNKTG-2 zaparcia i chorobę refluksową przełyku obserwowano odpowiednio u 6,7 % i 4 % pacjentów stosujących semaglutyd 1,0 mg, w porównaniu do braku zdarzeń u pacjentów stosujących placebo. Częstość tych zdarzeń z czasem nie malała.
Ostre zapalenie trzustki
Częstość przypadków potwierdzonego ostrego zapalenia trzustki zarejestrowanych w trakcie klinicznych badań fazy 3a wynosiła 0,3 % przy stosowaniu semaglutydu i 0,2 % przy stosowaniu leku porównawczego. W trakcie badania reakcji ze strony układu sercowo-naczyniowego trwającego 2 lata częstość przypadków ostrego zapalenia trzustki potwierdzonych przez ekspertów wynosiła 0,5 % przy stosowaniu semaglutydu i 0,6 % przy stosowaniu placebo (patrz sekcja „Szczególne ostrzeżenia i środki ostrożności”).
Nasilenie retinopatii cukrzycowej
W klinicznym badaniu trwającym 2 lata wzięło udział 3297 pacjentów z cukrzycą typu 2, wysokim ryzykiem kardiologicznym, długotrwałym przebiegiem cukrzycy i niewystarczającym kontrolowaniem poziomu glukozy we krwi. W tym badaniu potwierdzone reakcje nasilenia retinopatii cukrzycowej występowały częściej u pacjentów otrzymujących semaglutyd (3,0 %) niż u tych, którzy otrzymywali placebo (1,8 %). Obserwacja ta dotyczyła pacjentów z potwierdzoną retinopatią cukrzycową, którzy otrzymywali insulinę. Różnica między wariantami leczenia pojawiała się wcześnie i utrzymywała się przez cały okres badania. Systematyczna ocena nasilenia retinopatii cukrzycowej była przeprowadzana wyłącznie w badaniu wyników kardiologicznych. W klinicznym badaniu trwającym do jednego roku z udziałem 4807 pacjentów z cukrzycą typu 2 odsetek pacjentów, u których zaobserwowano efekty uboczne związane z retinopatią cukrzycową, był podobny w grupie stosującej semaglutyd (1,7 %) i w grupie leku porównawczego (2,0 %).
Przerwanie leczenia z powodu wystąpienia efektów ubocznych
Częstość przypadków przerwania leczenia z powodu wystąpienia niepożądanych zjawisk wynosiła 6,1 % i 8,7 % u pacjentów otrzymujących semaglutyd w dawce 0,5 mg i 1 mg odpowiednio, w porównaniu do 1,5 % u osób przyjmujących placebo. Najczęstsze reakcje uboczne prowadzące do przerwania leczenia to zaburzenia ze strony przewodu pokarmowego.
Reakcje w miejscu wstrzyknięcia
O wystąpieniu reakcji w miejscu wstrzyknięcia (takich jak wysypka, zaczerwienienie w miejscu iniekcji) donoszono u 0,6 % i 0,5 % pacjentów otrzymujących semaglutyd w dawce 0,5 mg i 1 mg odpowiednio. Zazwyczaj reakcje te były umiarkowane.
Imunogenność
Ze względu na możliwość wystąpienia właściwości immunogennych leków zawierających białka lub peptydy, istnieje prawdopodobieństwo powstawania przeciwciał u pacjentów podczas leczenia semaglutydem. Odsetek pacjentów z pozytywnym wynikiem badania na obecność przeciwciał anty-semaglutydowych w dowolnym czasie po rozpoczęciu leczenia był niski (1–2 %) i żaden z pacjentów nie miał przeciwciał neutralizujących semaglutyd ani przeciwciał z efektem neutralizującym GLP-1 do zakończenia badań klinicznych.
Zwiększenie częstości skurczów serca
Podczas stosowania agonistów receptorów GLP-1 obserwowano zwiększenie częstości skurczów serca. W klinicznych badaniach fazy 3a u pacjentów otrzymujących lek Ozempic® obserwowano średnie zwiększenie częstości skurczów serca od 1 do 6 uderzeń na minutę (ud./min) w stosunku do wartości wyjściowej 72–76 ud./min. W długotrwałym badaniu z udziałem pacjentów z czynnikami ryzyka rozwoju choroby układu sercowo-naczyniowego po 2 latach leczenia u 16 % pacjentów otrzymujących lek Ozempic® obserwowano zwiększenie częstości skurczów serca o > 10 ud./min w porównaniu do 11 % pacjentów przyjmujących placebo.
Zgłaszanie podejrzewanych reakcji niepożądanych
Po rejestracji leku ważne jest zgłaszanie podejrzewanych efektów ubocznych. Umożliwia to kontynuację monitorowania stosunku korzyści do ryzyka stosowania leku. Lekarze powinni zgłaszać podejrzewane reakcje niepożądane poprzez krajowy system raportowania.
Okres ważności
Przed pierwszym użyciem – 3 lata.
Po pierwszym użyciu – 6 tygodni.
Po pierwszym użyciu przechowywać w temperaturze poniżej 30 °C lub w lodówce (przy temperaturze 2–8 °C). Nie zamrażać.
Przechowywać długopis strzykawkowy z założoną osłonką chroniącą przed działaniem światła.
Warunki przechowywania
Przed pierwszym użyciem: przechowywać w lodówce (2–8 °C), nie zbyt blisko komory zamrażarki. Nie zamrażać. Nie używać po zamrożeniu.
Przechowywać długopis strzykawkowy z założoną osłonką chroniącą przed działaniem światła.
Warunki przechowywania po pierwszym otwarciu patrz w sekcji „Okres ważności”.
Przechowywać w miejscu niedostępnym dla dzieci.
Niezgodność
Ponieważ nie przeprowadzono badań niezgodności, tego leku nie wolno mieszać z innymi lekami.
Opakowanie
Wstępnie napełniony wielodawkowy jednorazowy długopis strzykawkowy wykonany z polipropylenu, polioksymetylenu, poliwęglanu i akrylonitrylu butadienu styrenu, zawierający kartuszek (szkło typu I), z jednej strony zamknięty gumowym (chlorobutyl) tłokiem, z drugiej strony – aluminiową osłonką z laminowaną wkładką gumową (bromobutyl / poliizopren).
Długopis strzykawkowy zawiera 3,0 ml roztworu, umożliwiającego podanie dawek po 1,0 mg. Opakowanie zawiera 1 wstępnie napełniony długopis strzykawkowy i 4 jednorazowe igły NovoFine® Plus w tekturowej puszce.
Kategoria wydania. Na receptę.
Producent
A/T Novo Nordisk.
Miejsce produkcji i adres siedziby działalności
Novo Allé
2880, Bagsværd
Dania.
Wniosek
A/T Novo Nordisk.
Siedziba wnioskodawcy
Novo Allé
2880, Bagsværd
Dania.