Ofev®
Ukraina
Spis treści
INSTRUKCJA DO STOSOWANIA MEDYCZNEGO LĘKU Ofev® (Ofev®)
Skład:
substancja czynna: nintedanib;
1 kapsułka zawiera 100 mg lub 150 mg nintedanibu (w postaci ezylatu);
substancje pomocnicze: trójglicerydy o średniej długości łańcucha, tłuszcz stały, lecytyna (sojowa) (E 322);
otoczka kapsułki: żelatyna, glikol 85%, dwutlenek tytanu (E 171), tlenek żelaza czerwony (E 172), tlenek żelaza żółty (E 172);
farba czarna do oznakowania kapsułek: lak, etanol, glikol propylenowy (E 1520), tlenek żelaza czarny (E 172).
Postać leku. Kapsułki miękkie.
Główne właściwości fizykochemiczne:
Ofev, kapsułki miękkie 100 mg
Wydłużone, nieprzezroczyste, miękkie kapsułki żelatynowe o barwie brzoskwiniowej, z jednej strony oznaczone czarnym atramentem logo firmy „Boehringer Ingelheim” oraz oznaczenie „100”.
Kapsułki zawierają lepką zawiesinę jasnożółtego koloru.
Ofev, kapsułki miękkie 150 mg
Wydłużone, nieprzezroczyste, miękkie kapsułki żelatynowe o barwie brązowej, z jednej strony oznaczone czarnym atramentem logo firmy „Boehringer Ingelheim” oraz oznaczenie „150”.
Kapsułki zawierają lepką zawiesinę jasnożółtego koloru.
Grupa farmakoterapeutyczna. Leki przeciwnowotworowe. Inhibitory kinaz białkowych.
Kod ATC L01E X09.
Właściwości farmakologiczne.
Farmakodynamika.
Mechanizm działania
Nintedanib jest małocząsteczkowym inhibitorem tyrozynokinaz, który blokuje receptory, w tym receptor czynnika wzrostu płytek krwi (PDGFR) α i β, receptor czynnika wzrostu fibroblastów (FGFR) 1–3 oraz receptor czynnika wzrostu śródbłonka naczyń (VEGFR) 1–3. Ponadto nintedanib hamuje kinazy Lck (limfocyt-specyficzną białkową kinazę tyrozynową), Lyn (białkową kinazę tyrozynową), Src (protoonkogenową białkową kinazę tyrozynową) oraz CSF1R (receptor czynnika stymulującego kolonie 1). Nintedanib konkurencyjnie wiąże się z miejscem wiązania adenozynotrifosforanu (ATP) tych kinaz i blokuje wewnątrzkomorowy przekaz sygnału kaskadowego, który wykazano jako uczestniczący w patogenezie przebudowy tkanki włóknistej w chorobach interpaczkowych płuc.
Skutki farmakodynamiczne
W badaniach in vitro z wykorzystaniem komórek ludzkich stwierdzono, że nintedanib hamuje procesy zaangażowane w inicjację patogenezy włóknienia, uwalnianie mediatorów profibrotycznych z monocytarnych komórek krwi obwodowej oraz polaryzację makrofagów do alternatywnie aktywowanych makrofagów. Wykazano, że nintedanib hamuje podstawowe procesy w włóknieniu narządów, takie jak proliferacja i migracja fibroblastów, transformacja w aktywny fenotyp miofibroblastów oraz sekrecja macierzy pozakomórkowej. W badaniach na zwierzętach w kilku modelach IPF, CT/ZP-CT, ZP, reumatoidalnym zapaleniu stawów (RA) spowodowanym ZP oraz włóknieniu innych narządów nintedanib wykazał działanie przeciwzapalne i przeciwfibryczne w płucach, skórze, sercu, nerkach i wątrobie. Nintedanib wykazuje również aktywność naczyniową. Zmniejszał apoptozę śródbłonka nacyniowego w mikronaczyniach skórnych oraz osłabiał przebudowę naczyń płucnych poprzez zmniejszenie proliferacji komórek mięśni gładkich naczyń, grubości ścian naczyń płucnych oraz odsetka zamkniętych naczyń płucnych.
Skuteczność i bezpieczeństwo kliniczne
Idiopatyczne włóknienie płuc (IPF)
Skuteczność kliniczną nintedanibu badano u pacjentów z IPF w ramach dwóch randomizowanych, podwójnie ślepych, kontrolowanych placebo badań klinicznych fazy III o tym samym schemacie (INPULSIS-1 (1199,32) oraz INPULSIS-2 (1199,34)). Pacjenci z wyjściowym obliczeniowym wartością FVC < 50 % lub zdolnością dyfuzyjną do tlenku węgla (DLCO) skorygowaną do hemoglobiny) < 30 %, obliczoną na poziomie wyjściowym, byli wykluczeni z badania. Pacjenci zostali randomizowani w stosunku 3:2 do grupy leku Ofev® 150 mg lub grupy placebo, przyjmując lek dwa razy dziennie przez 52 tygodnie.
Pierwotnym punktem końcowym był roczny wskaźnik spadku pojemności życiowej płuc (FVC). Kluczowymi wtórnymi punktami końcowymi były zmiana ogólnej liczby punktów w kwestionariuszu szpitala św. Jerzego oceny czynności oddechowej (SGRQ) w 52. tygodniu w porównaniu z wartościami wyjściowymi oraz czas do pierwszego zaostrzenia IPF.
Roczny wskaźnik spadku FVC
Roczny wskaźnik spadku FVC (w ml) wykazał istotne zmniejszenie u pacjentów otrzymujących nintedanib w porównaniu z pacjentami otrzymującymi placebo. Efekt terapeutyczny był podobny w obu badaniach (patrz tabela 1).
Tabela 1
Roczny wskaźnik spadku FVC w badaniach INPULSIS-1, INPULSIS-2 oraz dane zsumowane w populacji pacjentów poddanych leczeniu
| Badanie |
INPULSIS-1 |
INPULSIS-2 |
INPULSIS-1 i INPULSIS-2, dane zestawione |
|||
| Leczenie |
Placebo |
Ofev® 150 mg dwukrotnie na dobę |
Placebo |
Ofev® 150 mg dwukrotnie na dobę |
Placebo |
Ofev® 150 mg dwukrotnie na dobę |
| Liczba pacjentów, których dane poddano analizie |
204 |
309 |
219 |
329 |
423 |
638 |
| Wskaźnik1 (SE) zmniejszenia w ciągu 52 tygodni |
−239,9 |
−114,7 |
−207,3 |
−113,6 |
−223,5 |
−113,6 |
| (18,71) |
(15,33) |
(19,31) |
(15,73) |
(13,45) |
(10,98) |
|
| Porównanie z placebo |
||||||
| Różnica1 |
125,3 |
93,7 |
109,9 |
|||
| 95 % CI |
(77,7, |
(44,8, |
(75,9, |
|||
| 172,8) |
142,7) |
144,0) |
||||
| Wartość p |
< 0,0001 |
0,0002 |
< 0,0001 |
|||
| 1 Oceniono na podstawie modelu regresji z losowymi współczynnikami. CI – przedział ufności. SE – błąd standardowy. |
W analizie wrażliwości, która zakładała, że u pacjentów, u których brak danych w 52. tygodniu, zmniejszenie FVC po ostatnim zarejestrowanym pomiarze jest takie samo jak u wszystkich pacjentów przyjmujących placebo, skorygowana różnica w rocznym wskaźniku zmniejszenia FVC pomiędzy grupą leczoną nintedanibem a grupą placebo wynosiła 113,9 ml/rok (95 % CI 69,2, 158,5) w badaniu INPULSIS-1 oraz 83,3 ml/rok (95 % CI 37,6, 129,0) w badaniu INPULSIS-2.
Na rys. 1 przedstawiono przebieg zmian w czasie w odniesieniu do wartości wyjściowej w obu grupach leczenia, oparty na uogólnionej analizie danych pochodzących z badań INPULSIS-1 i INPULSIS-2.
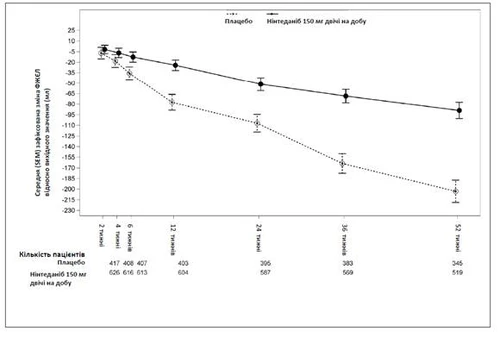
Rys. 1. Średnia (SEM) zarejestrowana zmiana FVC w czasie w odniesieniu do wartości wyjściowej (ml), dane złączone z badań INPULSIS-1 i INPULSIS-2.
Analiza danych pacjentów odpowiadających na leczenie pod względem wskaźnika FVC
W obu badaniach INPULSIS odsetek pacjentów odpowiadających na leczenie pod względem wskaźnika FVC — do kategorii tej zaliczano pacjentów, u których obliczone bezwzględne zmniejszenie FVC w % nie przekraczało 5% (wartość progowa wskazująca na zwiększone ryzyko śmiertelności w IPF) — był istotnie wyższy w grupie nintedanibu niż w grupie placebo. Podobne wyniki zaobserwowano w analizie z wykorzystaniem tradycyjnej wartości progowej 10% (patrz tabela 2).
Tabela 2
Odsetek pacjentów odpowiadających na leczenie pod względem wskaźnika FVC w 52. tygodniu w badaniach INPULSIS-1, INPULSIS-2 oraz dane złączone w populacji pacjentów poddanych leczeniu
| Badanie |
INPULSIS-1 |
INPULSIS-2 |
INPULSIS-1 i INPULSIS-2, dane zestawione |
|||
| Leczenie |
Placebo |
Ofev®, 150 mg dwa razy dziennie |
Placebo |
Ofev®, 150 mg dwa razy dziennie |
Placebo |
Ofev®, 150 mg dwa razy dziennie |
| Liczba pacjentów, których dane poddano analizie |
204 |
309 |
219 |
329 |
423 |
638 |
| Granica 5 % |
||||||
| Liczba (%) pacjentów odpowiadających na leczenie pod względem wartości FVC |
78 (38,2) |
163 (52,8) |
86 (39,3) |
175 (53,2) |
164 (38,8) |
338 (53,0) |
| W porównaniu z placebo |
||||||
| Stosunek szans |
1,85 |
1,79 |
1,84 |
|||
| 95 % CI |
(1,28; 2,66) |
(1,26; 2,55) |
(1,43; 2,36) |
|||
| Wartość p2 |
0,0010 |
0,0011 |
<0,0001 |
|||
| Granica 10 % |
||||||
| Liczba (%) pacjentów odpowiadających na leczenie pod względem wartości FVC |
116 (56,9) |
218 (70,6) |
140 (63,9) |
229 (69,6) |
256 (60,5) |
447 (70,1) |
| W porównaniu z placebo |
||||||
| Stosunek szans |
1,91 |
1,29 |
1,58 |
|||
| 95 % CI |
(1,32; 2,79) |
(0,89; 1,86) |
(1,21; 2,05) |
|||
| Wartość p2 |
0,0007 |
0,1833 |
0,0007 |
|||
1Pacjenci odpowiadający na leczenie to osoby z bezwzględnym spadkiem FVC nie większym niż o 5 lub 10 % wartości przewidywanej FVC w %, w zależności od wartości progowej, z oceną FVC w 52. tygodniu.
2Na podstawie regresji logistycznej.
Czas do postępowania choroby (bezwzględne zmniejszenie przewidywanej wartości FVC w % o ≥ 10 % lub zgon)
W obu badaniach INPULSIS wykazano klinicznie istotne zmniejszenie ryzyka postępowania choroby u pacjentów leczonych nintedanibem w porównaniu z pacjentami otrzymującymi placebo. W analizie uogólnionej współczynnik ryzyka wyniósł 0,60, co wskazuje na 40 % zmniejszenie ryzyka postępowania choroby u pacjentów leczonych nintedanibem w porównaniu z pacjentami otrzymującymi placebo.
Tabela 3
Odsetek pacjentów z bezwzględnym zmniejszeniem przewidywanej wartości FVC w % o ≥ 10 % lub zgonem (zdarzenia) w okresie 52 tygodni oraz czas do postępowania choroby w badaniach INPULSIS-1, INPULSIS-2 i dane uogólnione w ramach populacji pacjentów poddanych leczeniu
| Badanie |
INPULSIS-1 |
INPULSIS-2 |
INPULSIS-1 i INPULSIS-2, dane zestawione |
|||||
| Leczenie |
Placebo |
Ofev® 150 mg dwa razy dziennie |
Placebo |
Ofev® 150 mg dwa razy dziennie |
Placebo |
Ofev® 150 mg dwa razy dziennie |
||
| Liczba pacjentów w grupie poddanego ryzyka |
204 |
309 |
219 |
329 |
423 |
638 |
||
| Pacjenci z objawami, N (%) |
83 |
75 |
92 |
98 |
175 |
173 |
||
| (40,7) |
(24,3) |
(42,0) |
(29,8) |
(41,4) |
(27,1) |
|||
| Porównanie z placebo1 |
||||||||
| Wartość p2 |
0,0001 |
0,0054 |
< 0,0001 |
|||||
| Stosunek ryzyka3 |
0,53 |
0,67 |
0,60 |
|||||
| 95 % CI |
(0,39, 0,72) |
(0,51, 0,89) |
(0,49, 0,74) |
|||||
| 1 Na podstawie danych zebranych w okresie do 372 dni (52 tygodnie + 7 dni). 2 Na podstawie testu logarytmicznego rankowego. 3 Na podstawie modelu regresji Coxa. |
||||||||
Zmiana ogólnego wyniku SGRQ w 52. tygodniu w stosunku do wartości wyjściowych
W analizie zbiorczej badań INPULSIS wartości wyjściowe SGRQ wynosiły 39,51 w grupie nintedanibu i 39,58 w grupie placebo. Obliczona średnia zmiana ogólnego wyniku SGRQ w 52. tygodniu w stosunku do wartości wyjściowych była mniejsza w grupie nintedanibu (3,53) niż w grupie placebo (4,96), z różnicą między grupami leczenia wynoszącą -1,43 (95 % CI: -3,09, 0,23; p = 0,0923). Ogólnie wpływ nintedanibu na zależną od zdrowia jakość życia, mierzoną ogólnym wynikiem SGRQ, był niewielki i wykazywał mniejsze pogorszenie w porównaniu z placebo.
Czas do pierwszego zaostrzenia IPF
W analizie zbiorczej badań INPULSIS u pacjentów otrzymujących nintedanib zaobserwowano liczbowo niższe ryzyko pierwszego zaostrzenia w porównaniu z osobami przyjmującymi placebo (patrz tabela 4).
Tabela 4
Odsetek pacjentów z zaostrzeniami IPF (zdarzenia) w okresie 52 tygodni oraz czas do pierwszego zaostrzenia na podstawie danych zgłoszonych przez badacza w badaniach INPULSIS-1, INPULSIS-2 i dane zbiorcze w populacji pacjentów poddanych leczeniu
| Badania |
INPULSIS-1 |
INPULSIS-2 |
INPULSIS-1 i INPULSIS-2, dane zestawione |
|||||
| Leczenie |
Placebo |
Ofev® 150 mg dwukrotnie dziennie |
Placebo |
Ofev® 150 mg dwukrotnie dziennie |
Placebo |
Ofev® 150 mg dwukrotnie dziennie |
||
| Liczba pacjentów w grupie podatnej na zdarzenia |
204 |
309 |
219 |
329 |
423 |
638 |
||
| Pacjenci z wystąpieniem zdarzeń, N (%) |
11 (5,4) |
19 (6,1) |
21 (9,6) |
12 (3,6) |
32 (7,6) |
31 (4,9) |
||
| Porównanie z placebo1 |
||||||||
| Wartość p2 |
0,6728 |
0,0050 |
0,0823 |
|||||
| Stosunek ryzyka3 ryzyka3 |
1,15 |
0,38 |
0,64 |
|||||
| 95 % CI |
(0,54, 2,42) |
(0,19, 0,77) |
(0,39, 1,05) |
|||||
| 1 Na podstawie danych zebranych w okresie do 372 dni (52 tygodnie + 7 dni). 2 Na podstawie testu logarytmicznego szacunku Coxa-Mantela. 3 Na podstawie modelu regresji Coxa. |
||||||||
Analiza czułości wykazała, że odsetek pacjentów, u których w ciągu 52 tygodni wystąpił co najmniej jeden przypadkiem nasilenia objawów potwierdzonych przez eksperta, był niższy w grupie z nintedanibem (1,9% pacjentów) niż w grupie placebo (5,7% pacjentów). W analizie czasu do potwierdzonego przez eksperta przypadku nasilenia objawów, z wykorzystaniem danych zsumowanych, wyznaczono współczynnik ryzyka (HR) na poziomie 0,32 (95% CI 0,16, 0,65; p = 0,0010). Wskazuje to, że ryzyko wystąpienia pierwszego nasilenia IPF było istotnie statystycznie niższe w grupie z nintedanibem niż w grupie placebo w dowolnym momencie czasu.
Analiza przeżycia
W zsumowanej analizie danych dotyczących przeżycia według wcześniej określonych zmiennych w badaniach INPULSIS, ogólne współczynniki zgonów w okresie 52 tygodni były niższe w grupie z nintedanibem (5,5%) niż w grupie placebo (7,8%). W analizie czasu do śmierci ustalono wartość HR na poziomie 0,70 (95% CI 0,43, 1,12; p = 0,1399). Wyniki wszystkich punktów końcowych dotyczących przeżycia (takich jak śmiertelność podczas leczenia i śmiertelność z powodu zaburzeń oddechowych) wykazały przekonujące różnice liczbowe na korzyść nintedanibu.
Tabela 5
Śmiertelność z dowolnej przyczyny (zdarzenia) w okresie 52 tygodni w badaniach INPULSIS-1,
INPULSIS-2 oraz dane zsumowane w populacji pacjentów poddanych leczeniu
| Badanie |
INPULSIS-1 |
INPULSIS-2 |
INPULSIS-1 i INPULSIS-2, dane łączone |
|||
| Leczenie |
Placebo |
Ofev® 150 mg dwa razy dziennie |
Placebo |
Ofev® 150 mg dwa razy dziennie |
Placebo |
Ofev® 150 mg dwa razy dziennie |
| Liczba pacjentów w grupie ryzyka |
204 |
309 |
219 |
329 |
423 |
638 |
| Pacjenci z objawami, N (%) |
13 (6,4) |
13 (4,2) |
20 (9,1) |
22 (6,7) |
33 (7,8) |
35 (5,5) |
| Porównanie z placebo1 |
||||||
| Wartość p2 |
0,2880 |
0,2995 |
0,1399 |
|||
| Stosunek ryzyka3 |
0,63 |
0,74 |
0,70 |
|||
| 95 % CI |
(0,29, 1,36) |
(0,40, 1,35) |
(0,43, 1,12) |
|||
| 1 Na podstawie danych zebranych w okresie do 372 dni (52 tygodnie + 7 dni). 2 Na podstawie logarytmicznego testu rangowego. 3 Na podstawie regresyjnego modelu Coxa. |
Długotrwałe leczenie lekiem Ofev® u pacjentów z IPF (INPULSIS-ON)
W otwartym badaniu rozszerzonym udział wzięło 734 pacjentów z IPF. Pacjenci, którzy ukończyli 52-tygodniowy okres leczenia w badaniu INPULSIS, kontynuowali leczenie lekiem Ofev® w ramach dalszego badania INPULSIS-ON. Mediana czasu przyjmowania leku u pacjentów, którzy otrzymywali Ofev® w obu badaniach (INPULSIS oraz INPULSIS-ON), wyniosła 44,7 miesiąca (zakres 11,9–68,3). Punkty końcowe oceny skuteczności obejmowały roczne tempo spadku FVC przez 192 tygodnie, które wyniosło –135,1 (5,8) ml/rok we wszystkich leczonych pacjentach i odpowiadało rocznemu tempu spadku FVC u pacjentów leczonych lekiem Ofev® w badaniu III fazy INPULSIS (–113,6 ml/rok). Profil bezpieczeństwa leku Ofev® w badaniu INPULSIS-ON był zgodny z profilem bezpieczeństwa stwierdzonym w badaniu III fazy INPULSIS.
Pacjenci z IPF i ciężkim zaburzeniem funkcji płucnych (INSTAGE)
INSTAGE to wieloośrodkowe, wielonarodowe, prospektywne, randomizowane, podwójnie ślepe badanie kliniczne z równoległymi grupami trwające 24 tygodnie, w którym uczestniczyli pacjenci z IPF i ciężkim zaburzeniem funkcji płucnych (DLCO ≤ 35%). 136 pacjentów otrzymywało monoterapię lekiem Ofev®. Wynik dotyczący punktu końcowego pierwotnego wykazał zmniejszenie całkowitego wyniku w kwestionariuszu St. George’s Respiratory Questionnaire (SGRQ) o –0,77 jednostki w 12. tygodniu, na podstawie skorygowanej średniej zmiany od wartości wyjściowej. Analiza a posteriori wykazała, że spadek pojemności życiowej wymuszonej (FVC) u tych pacjentów odpowiadał spadkowi FVC u pacjentów z mniej zaawansowaną chorobą, którzy otrzymywali Ofev® w badaniu INPULSIS III fazy.
Profil bezpieczeństwa i tolerancji leku Ofev® u pacjentów z IPF i ciężkim zaburzeniem funkcji płucnych był zgodny z profilem stwierdzonym w badaniu INPULSIS III fazy.
Dodatkowe dane uzyskane w badaniu IV fazy INJOURNEY, w którym stosowano Ofev® w dawce 150 mg dwa razy dziennie oraz pirfenidon jako terapię łączoną
Leczenie skojarzone nintedanibem i pirfenidone zostało zbadane w otwartym, randomizowanym badaniu poszukującym trwającym 12 tygodni, w którym porównywano nintedanib w dawce 150 mg dwa razy dziennie oraz pirfenidon jako terapię dodatkową (z dozowaniem do 801 mg trzy razy dziennie) w porównaniu do nintedanibu w dawce 150 mg dwa razy dziennie jako monoterapii u 105 pacjentów. Pierwotnym punktem końcowym była częstość występowania niepożądanych zjawisk ze strony przewodu pokarmowego po 12 tygodniach w porównaniu do wartości wyjściowej. Zjawiska te występowały często, co było zgodne z ustalonym profilem bezpieczeństwa każdego składnika. Najczęstsze niepożądane zjawiska to biegunka, nudności i wymioty u pacjentów otrzymujących pirfenidon w połączeniu z nintedanibem, w porównaniu do tych, którzy otrzymywali nintedanib jako monoterapię. Średnie zmiany absolutne FVC (pojemności życiowej wymuszonej płuc) po 12 tygodniach w porównaniu do wartości wyjściowej wyniosły –13,3 (17,4) ml u pacjentów otrzymujących nintedanib i pirfenidon jako terapię dodatkową (n = 48), w porównaniu do –40,9 (31,4) ml u pacjentów leczonych nintedanibem jako monoterapią (n = 44).
Inne przewlekłe włókniste choroby płuc (ILD) z fenotypem postępującym
Skuteczność kliniczną nintedanibu badano u pacjentów z innymi przewlekłymi włóknistymi ILD z fenotypem postępującym w ramach podwójnie ślepego, randomizowanego, kontrolowanego placebo badania III fazy (INBUILD). Pacjentów z IPF wykluczono. Pacjentów z klinicznym rozpoznaniem przewlekłej włóknistej ILD zarekrutowano, jeśli mieli odpowiedni włóknienie (charakterystyczne cechy włóknienia > 10%) w badaniu TKHRCT oraz kliniczne oznaki postępu choroby (spadek FVC o ≥ 10%, spadek FVC o ≥ 5% i < 10% wraz z nasileniem objawów lub danymi TKHRCT, albo nasilenie objawów i danych TKHRCT w ciągu 24 miesięcy przed skriningiem). Pacjenci musieli mieć FVC ≥ 45% wartości przewidywanej oraz DLCO – od 30% do < 80% wartości przewidywanej. U pacjentów musiało występować postępowanie choroby mimo leczenia, co badacze oceniali jako odpowiednie praktyce klinicznej w związku z daną ILD.
663 pacjentów zostało zrandomizowanych w stosunku 1:1 do grupy otrzymującej 150 mg leku dwa razy dziennie lub odpowiedniej dawki placebo przez co najmniej 52 tygodnie. Mediana czasu przyjmowania leku Ofev® w całym badaniu wyniosła 17,4 miesiąca, a średnia długość przyjmowania wyniosła 15,6 miesiąca. Randomizacja była stratyfikowana według wzorca włóknienia w TKHRCT na podstawie oceny głównych ekspertów. Zrandomizowano 412 pacjentów ze wzorcem włóknienia odpowiadającym zwyklej zapaleniu płuc (UIP) w TKHRCT oraz 251 pacjentów z innymi wzorcami włóknienia w TKHRCT. Do analizy w tym badaniu określono dwie populacje pierwotne: wszystkich pacjentów (populacja ogólna) oraz pacjentów ze wzorcem włóknienia odpowiadającym UIP w TKHRCT. Pacjenci z innymi wzorcami włóknienia w TKHRCT stanowili dodatkową populację.
Pierwotnym punktem końcowym był roczny wskaźnik zmniejszenia pojemności życiowej wymuszonej płuc (FVC) (ml) w ciągu 52 tygodni. Kluczowymi wtórnymi punktami końcowymi były: zmiana absolutna od wartości wyjściowej w krótkim kwestionariuszu Kinga dotyczącego objawów choroby płucnej międzypłucnej (K-BILD) w ciągu 52 tygodni, czas do pierwszego ostrego nasilenia ILD lub śmierci w ciągu 52 tygodni oraz czas do śmierci w ciągu 52 tygodni.
Pacjenci mieli średni wiek (średnie odchylenie [SD; min.–maks.]) 65,8 (9,8; 27–87) roku oraz średni wynik FVC 69,0% wartości przewidywanej (15,6; 42–137). Głównymi klinicznymi rozpoznaniami ILD w grupach reprezentowanych w badaniu były: zapalenie płuc hipersensytywne (26,1%), autoimmunologiczne ILD (25,6%), idiopatyczne niespecyficzne zapalenie płuc (18,9%), niezaklasyfikowane idiopatyczne zapalenie międzypłucne (17,2%) oraz inne ILD (12,2%).
Roczny wskaźnik zmniejszenia FVC
Roczny wskaźnik zmniejszenia FVC (ml) w ciągu 52 tygodni wykazał istotne zmniejszenie u pacjentów otrzymujących Ofev®, w porównaniu do grupy placebo (tabela 6), co odpowiada względnej skuteczności leczenia na poziomie 57,0%.
Tabela 6
Roczny wskaźnik zmniejszenia FVC (ml) w ciągu 52 tygodni
| Parametry |
Placebo |
Ofev® |
| Liczba pacjentów, których dane poddano analizie |
331 |
332 |
| Wskaźnik 1 (SD) spadku w ciągu 52 tygodni |
-187,8 (14,8) |
-80,8 (15,1) |
| Porównanie z placebo |
||
| Różnica 1 |
107,0 |
|
| 95 % CI |
(65,4; 148,5) |
|
| Wartość p |
< 0,0001 |
|
1 Na podstawie regresji losowych współczynników z ustalonym wpływem kategorialnym leczenia, wzorcem KTBRZ, ustalonym wpływem stałym czasu, wyjściowym poziomem FZEL [ml], a także z uwzględnieniem zależności leczenia od czasu i wyjściowego poziomu od czasu.
Podobne wyniki obserwowano w populacji wtórnej pacjentów z wzorcem ZIP w KTBRZ. Wpływ leczenia był spójny w dodatkowej populacji pacjentów z innymi wzorcami zmian włóknijących w KTBRZ (wartość p zależności 0,2268) (rys. 2).
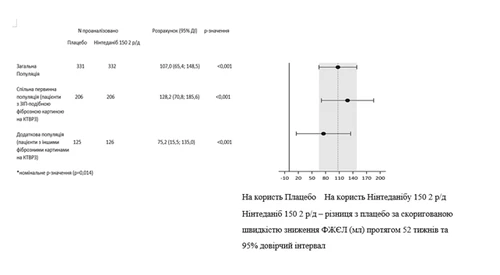
Rys. 2. Diagram rocznej szybkości spadku FZEL (ml) w okresie 52 tygodni w populacjach pacjentów.
Wpływ leku Ofev® na zmniejszenie rocznego wskaźnika spadku FZEL został potwierdzony we wszystkich wcześniej określonych analizach wrażliwości; spójne wyniki obserwowano w wcześniej zdefiniowanych podgrupach efektywności: wg płci, wieku, przynależności rasowej, wyjściowego poziomu FZEL (% wartości należnej) oraz pierwotnego głównego rozpoznania klinicznego ILD w grupach.
Na rys. 3 przedstawiono ewolucję zmiany FZEL od poziomu wyjściowego w grupach leczenia.
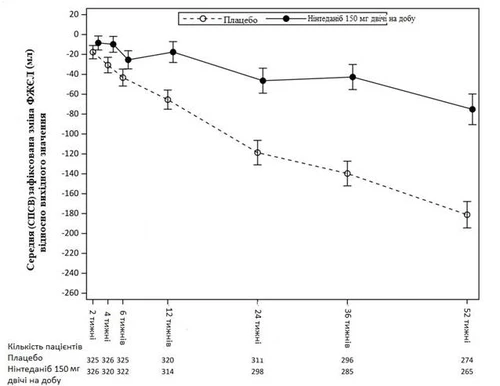
Rys. 3. Średnia SPSS (Skorygowany wskaźnik odchylenia standardowego) wykazana zmiana FZEL od poziomu wyjściowego (ml) w okresie 52 tygodni.
Ponadto, korzystne efekty leku Ofev® obserwowano w skorygowanej średniej zmianie bezwzględnej FZEL (% wartości należnej) w porównaniu z poziomem wyjściowym w 52. tygodniu. Skorygowana średnia wartość bezwzględnej zmiany FZEL (% wartości należnej) od poziomu wyjściowego do 52. tygodnia była niższa w grupie nintedanibu (-2,62 %) niż w grupie placebo (-5,86 %). Skorygowana średnia różnica między grupami leczenia wyniosła 3,24 (95 % CI: 2,09; 4,40; nominalne p < 0,0001).
Analiza pacjentów osiągających efekt terapeutyczny wg wskaźnika FZEL
Udział pacjentów osiągających efekt terapeutyczny wg wskaźnika FZEL, określony jako pacjenci z względnym spadkiem poziomu FZEL % wartości należnej nie większym niż o 5 %, był wyższy w grupie leku Ofev® w porównaniu z placebo. Analogiczne wyniki obserwowano w analizach z wykorzystaniem progu 10 % (tab. 7).
Tabela 7
Roczny wskaźnik spadku FZEL (ml) w okresie 52 tygodni
| Leczenie |
Placebo |
Ofev®, 150 mg dwa razy dziennie |
| Liczba pacjentów, których dane poddano analizie |
331 |
332 |
| 5% wartość progowa |
||
| Liczba (%) pacjentów osiągających efekt terapeutyczny według wskaźnika FVC1 |
104 (31,4) |
158 (47,6) |
| W porównaniu z placebo |
||
| Stosunek szans2 |
2,01 |
|
| 95% CI |
(1,46; 2,76) |
|
| Wartość p |
< 0,0001 |
|
| 10% wartość progowa |
||
| Liczba (%) pacjentów osiągających efekt terapeutyczny według wskaźnika FVC1 |
169 (51,1) |
197 (59,3) |
| W porównaniu z placebo |
||
| Stosunek szans2 |
1,42 |
|
| 95% CI |
(1,04; 1,94) |
|
| Wartość p |
0,0268 |
|
1 Pacjenci, którzy osiągnęli efekt terapeutyczny oceniany za pomocą FVC, to pacjenci, którzy nie wykazali spadku FVC (% wartości przewidywanej) o więcej niż 5 % lub więcej niż 10 %, w zależności od progowych wartości, oraz z oceną FVC w 52. tygodniu (pacjenci z brakującymi danymi w 52. tygodniu uznawani byli za nieosiągnęli efektu terapeutycznego).
2 Na podstawie modelu regresji logistycznej z ciągłą kowariatą wyjściowego poziomu FVC (%) oraz binarną kowariatą wzorca HRCT.
Czas do pierwszego zaostrzenia IPF lub śmierci
W całym okresie badania odsetek pacjentów, którzy doświadczyli co najmniej jednego pierwszego ostrego zaostrzenia IPF lub śmierci, wyniósł 13,9 % w grupie Ofev® i 19,6 % w grupie placebo. HR wyniósł 0,67 (95 % CI: 0,46; 0,98; nominalne p = 0,0387), co wskazuje na 33 % zmniejszenie ryzyka pierwszego zaostrzenia IPF lub śmierci u pacjentów leczonych Ofev® w porównaniu z pacjentami otrzymującymi placebo (ryc. 4).
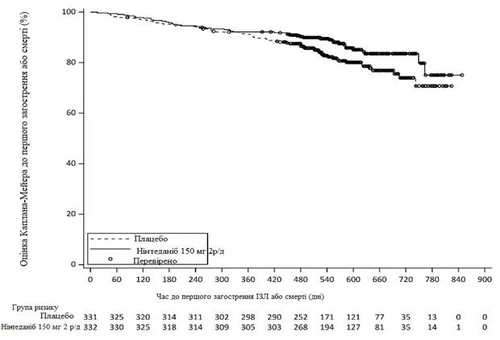
2 r/d – dwa razy dziennie
Ryc. 4. Czas do pierwszego zaostrzenia IPF lub śmierci.
Analiza przeżycia
Ryzyko śmierci było niższe w grupie leku Ofev® w porównaniu z grupą placebo. HR wyniósł 0,78 (95 % CI: 0,50; 1,21; nominalne p = 0,2594), co wskazuje na 22 % zmniejszenie ryzyka śmierci u pacjentów leczonych Ofev® w porównaniu z pacjentami otrzymującymi placebo.
Czas do progresji (bezwzględne zmniejszenie FVC (% wartości przewidywanej) o ≥ 10 %) lub śmierci
W badaniu INBUILD ryzyko progresji (bezwzględne zmniejszenie FVC (% wartości przewidywanej) o ≥ 10 %) lub śmierci było mniejsze u pacjentów leczonych Ofev®. Odsetek pacjentów z zdefiniowanym zdarzeniem wyniósł 40,4 % w grupie leku Ofev® i 54,7 % w grupie placebo. HR wyniósł 0,66 (95 % CI: 0,53; 0,83; p = 0,0003), co wskazuje na 34 % zmniejszenie ryzyka progresji (bezwzględne zmniejszenie FVC (% wartości przewidywanej) o ≥ 10 %) lub śmierci u pacjentów leczonych Ofev® w porównaniu z pacjentami otrzymującymi placebo.
Jakość życia
Skorygowana średnia zmiana od wartości wyjściowej w ogólnym wyniku K-BILD w 52. tygodniu wyniosła -0,79 jednostki w grupie placebo i 0,55 w grupie leku Ofev®. Różnica między grupami leczenia wyniosła 1,34 (95 % CI: -0,31; 2,98; nominalne p = 0,1115).
Skorygowana średnia bezwzględna zmiana od wartości wyjściowej w objawach duszności według kwestionariusza L-PF w 52. tygodniu wyniosła 4,28 w grupie leku Ofev® w porównaniu z 7,81 w grupie placebo. Skorygowana średnia różnica między grupami na korzyść leku Ofev® wyniosła -3,53 (95 % CI: -6,14; -0,92; nominalne p = 0,0081). Skorygowana średnia bezwzględna zmiana od wartości wyjściowej w objawach kaszlu według kwestionariusza L-PF w 52. tygodniu wyniosła -1,84 w grupie leku Ofev® w porównaniu z 4,25 w grupie placebo. Skorygowana średnia różnica między grupami na korzyść leku Ofev® wyniosła -6,09 (95 % CI: -9,65; -2,53; nominalne p = 0,0008).
Choroba śródmiąższowa płuc przy twardzinie systemowej (systemicznym sclerosis) (IPF-SS)
Skuteczność kliniczną nintedanibu badano u pacjentów z IPF-SS w ramach podwójnie ślepego, randomizowanego, kontrolowanego placebo badania fazy III (SENSCIS). U pacjentów rozpoznano IPF-SS na podstawie kryteriów klasyfikacyjnych SS Amerykańskiego Kolegium Reumatologii/Europejskiej Ligi Przeciw Reumatyzmowi z 2013 roku oraz tomografii komputerowej wysokiej rozdzielczości (HRCT) klatki piersiowej w ciągu ostatnich 12 miesięcy. 580 pacjentów zostało randomizowanych w stosunku 1:1 i otrzymywało albo Ofev® 150 mg dwa razy dziennie, albo placebo przez co najmniej 52 tygodnie. Z nich 576 pacjentów ukończyło leczenie. Randomizację przeprowadzono z uwzględnieniem statusu przeciwciał antytopoizomerazy (ATA). Poszczególni pacjenci byli leczeni w trybie ślepej próby do 100 tygodnia (mediana ekspozycji na Ofev® 15,4 miesiąca; średnia ekspozycja na Ofev® 14,5 miesiąca).
Pierwotnym punktem końcowym był roczny wskaźnik spadku pojemności życiowej płuc (FVC) w ciągu 52 tygodni. Kluczowymi wtórnymi punktami końcowymi były bezwzględna zmiana od wartości wyjściowej w skórze według zmodyfikowanej skali Rodnana (mRSS) w 52. tygodniu oraz bezwzględna zmiana od wartości wyjściowej w ogólnym wyniku kwestionariusza Szpitala św. Jerzego do oceny funkcji oddechowej (SGRQ) w 52. tygodniu.
W ogólnej populacji 75,2 % pacjentów stanowiły kobiety. Średni (odchylenie standardowe (SD, min.–maks.)) wiek wyniósł 54 (12,2; 20–79) lat. Ogółem 51,9 % pacjentów miało rozlężną postać skórną twardziny systemowej (SS); 48,1 % – ograniczoną postać skórną SS. Średni (SD) czas od wystąpienia pierwszego objawu „nie Raynauda” wyniósł 3,49 (1,7) roku. 49,0 % pacjentów otrzymywało stabilną terapię mykofenolanem na poziomie wyjściowym. Profil bezpieczeństwa u pacjentów, którzy otrzymywali lub nie otrzymywali mykofenolanu na poziomie wyjściowym, był podobny.
Roczny wskaźnik spadku FVC
Roczny wskaźnik spadku FVC (ml) w ciągu 52 tygodni był istotnie mniejszy o (41,0) ml u pacjentów leczonych Ofev® w porównaniu z pacjentami otrzymującymi placebo (tabela 8), co odpowiada względnej skuteczności leczenia wynoszącej 43,8 %.
Tabela 8
Roczny wskaźnik spadku FVC (ml) w ciągu 52 tygodni
| Leczenie |
Placebo |
Ofev® 150 mg dwa razy dziennie |
| Liczba pacjentów, których dane poddano analizie |
288 |
287 |
| Wskaźnik 1 (SD) spadek w ciągu 52 tygodni |
-93,3 (13,5) |
-52,4 (13,8) |
| Porównanie z placebo |
||
| Różnica 1 |
41,0 |
|
| 95 % CI |
(2,9, 79,0) |
|
| Wartość p |
< 0,05 |
1 Na podstawie regresji mieszanej z ustalonym efektem kategorii leczenia, statusu ATA, płci pacjenta, ustalonym efektem stałym czasu, wyjściową wartością FVC (ml), wiekiem, wzrostem pacjenta, a także z uwzględnieniem zależności skuteczności leczenia od czasu i zależności zmian od wyjściowego poziomu od czasu. Efekt losowy został uwzględniony dla konkretnego momentu rejestracji pacjenta oraz czasu. Błędy wewnętrzne zostały zamodelowane przy użyciu niestrukturalnej macierzy wariancji-kowariancji. Zmienność międzypacjentowa była modelowana przy użyciu macierzy wariancji-kowariancji z komponentami wariancji.
Wpływ leku Ofev® na zmniejszenie rocznego wskaźnika spadku FVC był podobny w wcześniej określonych analizach wrażliwości; nie stwierdzono heterogeniczności w wcześniej ustalonych podgrupach pacjentów (np. według wieku, płci i stosowania mykofenolanu).
Ponadto zaobserwowano podobne efekty w odniesieniu do innych punktów końcowych dotyczących funkcji płuc, w szczególności zmiany FVC w 52. tygodniu (rys. 5 i tabela 9) od poziomu wyjściowego oraz przewidywanej szybkości spadku FVC w % w ciągu 52 tygodni (tabela 10), co stanowi dodatkowe uzasadnienie działania Ofev® w zakresie spowolnienia postępu PChZ-SS. Ponadto u mniejszej liczby pacjentów w grupie Ofev® zaobserwowano bezwzględne skrócenie FVC > 5 % (20,6 % w grupie Ofev® vs 28,5 % w grupie placebo, OR = 0,65, p = 0,0287). Względne skrócenie FVC w ml > 10% było podobne w grupach (16,7 % w grupie Ofev® vs 18,1 % w grupie placebo, OR = 0,91, p = 0,6842). W tych analizach brakujące wartości FVC w 52. tygodniu były szacowane na podstawie najgorszego wyniku u pacjenta podczas leczenia.
Analiza badawcza danych do 100. tygodnia (maksymalna długość leczenia w badaniu SENSCIS) wskazuje, że wpływ leczenia lekiem Ofev® na spowolnienie postępu PChZ-SS utrzymuje się ponad 52 tygodnie.
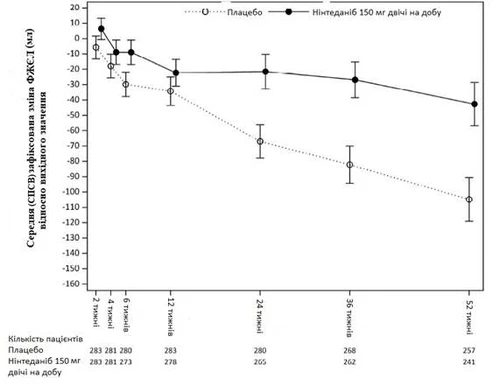
Rys. 5. Średnia (SPSW) zaobserwowana zmiana FVC od poziomu wyjściowego (ml) w ciągu 52 tygodni.
Tabela 9
Bezwzględna zmiana FVC (ml) od poziomu wyjściowego w 52. tygodniu
| Leczenie |
Placebo |
Ofev® 150 mg dwa razy dziennie |
| Liczba pacjentów, których dane poddano analizie |
288 |
288 |
| Średnia (SD) na poziomie wyjściowym |
2541,0 (815,5) |
2458,5 (735,9) |
| Średnia1 (SD) zmiana od poziomu wyjściowego w 52. tygodniu |
-101,0 (13,6) |
-54,6 (13,9) |
| Porównanie z placebo |
||
| Średnia 1 |
|
|
| 95 % CI |
(8,1; 84,7) |
|
| Wartość p |
< 0,05 |
1 Na podstawie mieszanej modeli pomiarów powtarzalnych (MMRM) z ustalonym kategorycznym wpływem statusu ATA, wizyty, interakcji leczenia według wizyty, interakcji poziomu wyjściowego według wizyty, wieku, płci i wzrostu pacjenta. Wizyta była powtarzalnym pomiarem. Błędy wewnętrzne zostały zamodelowane za pomocą niestrukturalnej macierzy wariancji-kowariancji. Skorygowana wartość średnia oparta była na liczbie wszystkich przeanalizowanych pacjentów w modelu (nie tylko tych, u których pomiary przeprowadzono na poziomie wyjściowym i w 52. tygodniu).
Tabela 10
Roczny wskaźnik spadku FVC (%) w okresie 52 tygodni
| Leczenie |
Placebo |
Ofev® 150 mg dwa razy na dobę |
| Liczba pacjentów, których dane poddano analizie |
288 |
287 |
| Wskaźnik 1 (SD) spadku w ciągu 52 tygodni |
-2,6 (0,4) |
-1,4 (0,4) |
| Porównanie z placebo |
||
| Różnica 1 |
1,15 |
|
| 95 % przedział ufności |
(0,09; 2,21) |
|
| Wartość p |
< 0,05 |
1 Na podstawie regresji mieszanej z ustalonym wpływem kategorii leczenia, statusu ATA, ustalonym stałym wpływem czasu, wyjściowym poziomem FVC (% wartości odnoszonej) oraz uwzględniającym zależność skuteczności leczenia od czasu i zależność zmian od wyjściowego poziomu od czasu. Efekt losowy został uwzględniony dla konkretnego momentu rejestracji pacjenta oraz czasu. Błędy wewnętrzne zostały zamodelowane za pomocą niestrukturalnej macierzy wariancji-kowariancji. Zmienność międzypacjentowa była modelowana za pomocą macierzy wariancji-kowariancji z komponentami wariancji.
Zmiana od wartości wyjściowej skóry według zmodyfikowanej skali Rodnana (mRSS) po 52 tygodniach
Skorygowana średnia bezwzględna zmiana od wartości wyjściowej według mRSS po 52 tygodniach była podobna w grupie leku Ofev® (-2,17 (95 % CI -2,69, -1,65)) i grupie placebo (-1,96 (95 % CI -2,48, -1,45)). Skorygowana średnia różnica między grupami leczenia wyniosła -0,21 (95 % CI -0,94, 0,53; p = 0,5785).
Zmiana od wartości wyjściowej całkowitego wyniku kwestionariusza Szpitala św. Jerzego (SGRQ) po 52 tygodniach
Skorygowana średnia bezwzględna zmiana od wartości wyjściowej całkowitego wyniku SGRQ po 52 tygodniach była podobna w grupie leku Ofev® (0,81 (95 % CI -0,92, 2,55)) i grupie placebo (-0,88 (95 % CI -2,58, 0,82)). Skorygowana średnia różnica między grupami leczenia wyniosła 1,69 (95 % CI -0,73, 4,12; p = 0,1711).
Analiza przeżycia
Wskaźnik śmiertelności w całym okresie badania był podobny w grupie leku Ofev® (N = 10; 3,5 %) i grupie placebo (N = 9; 3,1 %). W analizie czasu do śmierci w całym okresie badania określono wartość HR 1,16 (95 % CI 0,47, 2,84; p = 0,7535).
Odcinek QT
W ramach specjalnego badania z udziałem pacjentów z rakiem komórkowym nerek dokonano pomiarów odcinka QT/kompleksu QT; wyniki tych pomiarów wykazały, że pojedyncza dawka doustna 200 mg nintedanibu oraz wielokrotne dawki doustne 200 mg nintedanibu podawane dwa razy dziennie przez 15 dni nie wydłużały odcinka QT skorygowanego wg Fridericia.
Dzieci.
Ofev® nie był badany w praktyce pediatrycznej w przypadku IPF.
Farmakokinetyka.
Wchłanianie
Maksymalne stężenie nintedanibu w osoczu osiągane jest około 2–4 godziny po doustnym przyjęciu leku w postaci miękkich kapsułek żelatynowych podczas jedzenia (zakres 0,5–8 godzin). Biologiczna dostępność dawki 100 mg u zdrowych ochotników wynosi 4,69 % (90 % CI 3,615–6,078). Wchłanianie i dostępność biologiczna zmniejszają się w wyniku działania przenośnika i istotnego metabolizmu presystemowego. Stwierdzono, że ekspozycja na nintedanib wzrasta proporcjonalnie do dawki (w zakresach dawek 50–450 mg raz dziennie oraz 150–300 mg dwa razy dziennie). Stałe stężenia w osoczu osiągane są maksymalnie w ciągu jednego tygodnia od rozpoczęcia przyjmowania.
Ekspozycja na nintedanib wzrasta przy przyjmowaniu po posiłku o około 20 % w porównaniu z przyjmowaniem na czczo (CI 95,3–152,5 %), a wchłanianie jest opóźnione (mediana czasu do osiągnięcia maksymalnego stężenia w osoczu na czczo (tmax) – 2,00 godziny; po posiłku – 3,98 godziny).
Rozkład
Rozkład nintedanibu odbywa się zgodnie z kinetyką dwufazową. Po wewnątrzżylnej infuzji w fazie terminalnej obserwuje się dużą objętość rozkładu (Vss): 1050 l, geometryczny współczynnik zmienności (gCV) 45,0 %).
Wiązanie nintedanibu z białkami osocza człowieka in vitro uznaje się za znaczne, frakcja związana wynosi 97,8 %. Głównym białkiem biorącym udział w wiązaniu jest albumina osocza. Nintedanib rozkłada się głównie w osoczu, stosunek krew/osocze wynosił 0,869.
Biotransformacja
Główną reakcją biorącą udział w metabolizmie nintedanibu jest hydrolityczne rozszczepienie za pomocą esteraz, prowadzące do powstania wolnego kwasowego metabolitu nintedanibu (BIBF 1202). Następnie BIBF 1202 jest glukuronidowany przez enzymy uridylo-5'-difosfato-glukuronozylotransferazy (UGT), a mianowicie UGT 1A1, UGT 1A7, UGT 1A8 oraz UGT 1A10, z tworzeniem glukuronidu BIBF 1202.
Biotransformacja nintedanibu z udziałem izoenzymów CYP zachodzi jedynie w niewielkim stopniu; główną rolę w tym procesie odgrywa izoenzym CYP 3A4. W badaniu ADME u ludzi głównego metabolitu powstającego z udziałem izoenzymów CYP nie udało się wykryć w osoczu. Według badań in vitro metabolizm zależny od CYP stanowi około 5 %, podczas gdy rozszczepienie prowadzone przez esterazy stanowi 25 %. Nintedanib, BIBF 1202 oraz glukuronid BIBF 1202 nie hamowały ani nie stymulowały izoenzymów CYP w badaniach in vitro i badaniach przedklinicznych. W związku z tym nie należy oczekiwać interakcji lekowych między nintedanibem a substratami CYP, inhibitorami CYP lub induktorami CYP.
Wydalanie
Całkowity klirens osocza po wewnątrzżylowej infuzji jest wysoki (1390 ml/min, gCV 28,8 %). Wydalanie niezmienionej substancji czynnej z moczem w ciągu 48 godzin po doustnym przyjęciu nintedanibu wynosi około 0,05 % dawki (gCV 31,5 %), a po wewnątrzżylowym podaniu – około 1,4 % (gCV 24,2 %); klirens nerkowy wynosi 20 ml/min (gCV 32,6 %). Po doustnym przyjęciu [14C]-nintedanibu materiał radioaktywny był wydalany głównie z żółcią i wykrywany w kale (93,4 % dawki, gCV 2,61 %). Część wydalania nerkowego w całkowitym klirensie jest niska (0,649 % dawki (gCV 26,3 %)). Wydalanie uznaje się za pełne (powyżej 90 %) po 4 dniach od przyjęcia. Okres półtrwania nintedanibu w fazie terminalnej wynosi od 10 do 15 godzin (gCV około 50 %).
Liniowość/nieliniowość
Można założyć, że farmakokinetyka (PK) nintedanibu jest liniowa w czasie (czyli dane dotyczące jednorazowej dawki mogą być ekstrapolowane na dane dotyczące wielokrotnego stosowania). Wartości Cmax wynikające z akumulacji po wielokrotnym stosowaniu przekraczają wartość Cmax jednorazowej dawki 1,04-krotnie, a wartości AUCτ – 1,38-krotnie. Minimalne stężenia resztkowe nintedanibu pozostają stabilne przez ponad rok.
Transport
Nintedanib jest substratem glikoproteiny P (P-gp). Zobacz sekcję „Interakcje z innymi lekami i inne formy interakcji” w celu uzyskania informacji dotyczących możliwych interakcji nintedanibu z tym przenośnikiem. Wykazano, że nintedanib in vitro nie jest substratem ani inhibitorem OATP-1B1, OATP-1B3, OATP-2B1, OCT-2 ani MRP-2. Nintedanib nie jest również substratem BCRP. In vitro stwierdzono, że nintedanib wykazuje słabe działanie inhibitorowe wobec OCT-1, BCRP i P-gp, co uznaje się za mające niewielkie znaczenie kliniczne. Taki sam wniosek uczyniono w odniesieniu do nintedanibu jako substratu OCT-1.
Farmakokinetyka u szczególnych grup pacjentów
Właściwości farmakokinetyczne nintedanibu były porównywalne u zdrowych ochotników, pacjentów z IPF, pacjentów z innymi postępującymi chorobami śródmiąższowymi płuc, pacjentów z ILD-CTD oraz pacjentów z nowotworami. Na podstawie wyników analizy farmakokinetyki populacyjnej u pacjentów z IPF i rakiem płuc nieosoczastym (NSCLC) (N = 1 191) oraz badań opisowych, na działanie nintedanibu nie wpływały takie czynniki jak płeć pacjenta (z uwzględnieniem masy ciała), zaburzenia funkcji nerek o lekkim i umiarkowanym nasileniu (obliczone na podstawie klirensu kreatyniny), spożycie alkoholu ani genotyp glikoproteiny P. Analiza farmakokinetyki populacyjnej wykazała umiarkowany wpływ płci, masy ciała i pochodzenia rasowego pacjenta na działanie nintedanibu, jak opisano poniżej. Ze względu na wysoką międzyosobniczą zmienność ekspozycji, te nieistotne wpływy nie były uważane za klinicznie znaczące (zobacz sekcję „Szczególne ostrzeżenia i środki ostrożności podczas stosowania”).
Wiek
Ekspozycja na nintedanib liniowo wzrasta z wiekiem. U pacjentów 45-letnich wartość AUCτ,ss była niższa o 16 %, a u pacjentów 76-letnich – wyższa o 13 % w porównaniu z pacjentami o medianie wieku 62 lat. Zakres wieku oceniany w analizie wynosił 29–85 lat; wiek powyżej 75 lat występował u około 5 % populacji pacjentów. Na podstawie modelu analizy farmakokinetyki populacyjnej stwierdzono, że u pacjentów w wieku powyżej 75 lat obserwowano wzrost ekspozycji na nintedanib o około 20–25 % w porównaniu z pacjentami w wieku do 65 lat.
Analogiczne badania z udziałem dzieci nie były prowadzone.
Masa ciała
Obserwuje się odwrotną korelację między masą ciała a ekspozycją na nintedanib. U pacjentów o masie ciała 50 kg (5. percentyl) wartość AUCτ,ss wzrastała o 25 %, a u pacjentów o masie ciała 100 kg (95. percentyl) – zmniejszała się o 19 % w porównaniu z pacjentami o medianie masy ciała 71,5 kg.
Pochodzenie rasowe
Średnia ekspozycja na nintedanib była wyższa o 33–50 % u Chińczyków, mieszkańców Tajwanu i Hindusów oraz o 16 % wyższa u Japończyków, a u Koreańczyków – o 16–22 % niższa niż u pacjentów rasy kaukaskiej (z uwzględnieniem masy ciała). Dane dotyczące pacjentów rasy czarnej są bardzo ograniczone; zakres tych danych jest podobny do takiego u pacjentów rasy kaukaskiej.
Zaburzenia funkcji wątroby
W specjalistycznym badaniu fazy I u ochotników z łagodnymi zaburzeniami funkcji wątroby (klasa A wg skali Childa-Pugh) ekspozycja na podstawie Cmax i AUC była 2,2-krotnie wyższa niż u zdrowych ochotników (90 % CI 1,3–3,7 dla Cmax oraz 90 % CI 1,2–3,8 dla AUC odpowiednio). U ochotników z umiarkowanymi zaburzeniami funkcji wątroby (klasa B wg skali Childa-Pugh) w porównaniu ze zdrowymi ochotnikami ekspozycja była 7,6-krotnie wyższa na podstawie Cmax (90 % CI 4,4–13,2) i 8,7-krotnie wyższa na podstawie AUC (90 % CI 5,7–13,1) odpowiednio. Badania z udziałem pacjentów z ciężkimi zaburzeniami funkcji wątroby (klasa C wg skali Childa-Pugh) nie były prowadzone.
Terapia współistniejąca z pirfenidonem
W specjalnym badaniu farmakokinetycznym badano współistniejące stosowanie nintedanibu i pirfenidonu u pacjentów z IPF (idioaptycznym włóknieniem płuc). Grupa 1 otrzymała pojedynczą dawkę 150 mg nintedanibu przed i po zwiększeniu dawki pirfenidonu do 801 mg trzy razy dziennie w stanie równowagi (N = 20 leczonych pacjentów). Grupa 2 otrzymała leczenie w stanie równowagi z użyciem 801 mg pirfenidonu trzy razy dziennie i wzięła udział w określaniu parametrów PK przed i po 7 dniach współistniejącego leczenia z nintedanibem w dawce 150 mg dwa razy dziennie (N = 17 leczonych pacjentów). W grupie 1 wartość skorygowanego stosunku średnich geometrycznych [90% przedział ufności (CI)] Cmax i AUC0-tz nintedanibu wyniosła odpowiednio 93 % (57–151 %) i 96 % (70–131 %) (n = 12 dla porównania wewnątrzsubjektowego). W grupie 2 wartość skorygowanego stosunku średnich geometrycznych (90 % CI) wyniosła odpowiednio 97 % (86–110 %) i 95 % (86–106 %) dla Cmax,ss i AUCτ,ss pirfenidonu (n = 12 dla porównania międzypacjentowego).
To badanie nie wykazało żadnych dowodów istotnej farmakokinetycznej interakcji lekowej między nintedanibem a pirfenidonom przy stosowaniu w kombinacji (zobacz sekcję „Szczególne ostrzeżenia i środki ostrożności podczas stosowania”).
Współistniejące leczenie z bosentanem
W specjalnym badaniu farmakokinetyki badano współistniejące stosowanie leku Ofev® z bosentanem u zdrowych ochotników. Pacjenci otrzymywali jedną dawkę leku Ofev® 150 mg przed i po kilku dawkach bosentanu 125 mg dwa razy dziennie w warunkach szpitalnych. Skorygowane średnie geometryczne stosunki (90 % przedział ufności (CI)) wyniosły odpowiednio 103 % (86 – 124 %) i 99 % (91 – 107 %) dla Cmax i AUC0-tz nintedanibu (n=13), co wskazuje, że współistniejące stosowanie nintedanibu z bosentanem nie zmienia farmakokinetyki nintedanibu.
Współistniejące stosowanie doustnych środków antykoncepcyjnych hormonalnych
W specjalnym badaniu farmakokinetycznym pacjentki z ILD-CTD otrzymywały pojedynczą dawkę kombinacji 30 µg etyniloestradiolu i 150 µg lewonorgestrelu przed i po przyjęciu 150 mg nintedanibu dwa razy dziennie przez co najmniej 10 dni. Skorygowane średnie geometryczne stosunki (90 % przedział ufności (CI)) wyniosły odpowiednio 117 % (108–127 %; Cmax) i 101 % (93–111 %; AUC0–tz) dla etyniloestradiolu oraz 101 % (90–113 %; Cmax) i 96 % (91–102 %; AUC0–tz) dla lewonorgestrelu (n = 15), co wskazuje, że jednoczesne stosowanie nintedanibu nie wywiera istotnego wpływu na poziom etyniloestradiolu i lewonorgestrelu w osoczu.
Współczynnik ekspozycja–odpowiedź
Analizy współczynnika ekspozycja–odpowiedź u pacjentów z IPF i innymi przewlekłymi włóknosejącymi ILD z postępującym fenotypem wykazały słabe powiązanie między poziomem nintedanibu w osoczu a podwyższeniem poziomu ALT i/lub AST. Rzeczywiście podana dawka może być lepszym predyktorem ryzyka wystąpienia biegunki o dowolnym nasileniu, nawet jeśli nie można wykluczyć poziomu nintedanibu w osoczu jako czynnika determinującego ryzyko (zobacz sekcję „Szczególne ostrzeżenia i środki ostrożności podczas stosowania”).
Właściwości kliniczne.
Wskazania.
Ofev® jest wskazany w leczeniu idiopatycznego włóknienia płuc (IWP) u dorosłych.
Ofev® jest również wskazany w leczeniu innych przewlekłych włóknikowych chorób śródmiąższowych płuc (ChŚP) z postacią postępującą u dorosłych (patrz sekcja „Właściwości farmakodynamiczne. Farmakodynamika”).
Ofev® jest wskazany w leczeniu choroby śródmiąższowej płuc związanej ze stwardnieniem układowym (zespołem sclerodermii układowej) (ChŚP-SU).
Przeciwwskazania.
Ciąża (patrz sekcja „Stosowanie w czasie ciąży lub karmienia piersią**”**).
Podwyższona wrażliwość na nintedanib, orzechy ziemne, soję lub którykolwiek z substancji pomocniczych preparatu.
Interakcje z innymi lekami i inne rodzaje interakcji.
Białko P-glikoproteinowe (P-gp)
Nintedanib jest substancją oczekującą na działanie białka P-gp (patrz sekcja „Właściwości farmakokinetyczne. Farmakokinetyka”). W specjalnym badaniu interakcji leków stwierdzono, że jednoczesne stosowanie z silnym inhibitorem P-gp – ketokonazolem – zwiększa ekspozycję na nintedanib według wskaźnika AUC 1,61-krotnie, a według wskaźnika Cmax 1,83-krotnie. Specjalne badanie interakcji leków wykazało, że jednoczesne stosowanie ryfampicyny (silnego induktora P-gp) prowadzi do zmniejszenia ekspozycji na nintedanib według wskaźnika AUC o 50,3%, a według wskaźnika Cmax o 60,3% (w porównaniu z monoterapią nintedanibem). Silne inhibitory P-gp (np. ketokonazol, erytromycyna lub cyklosporyna) mogą zwiększać ekspozycję na nintedanib w przypadku jednoczesnego stosowania z Ofev®. Dlatego należy dokładnie monitorować tolerancję nintedanibu u pacjentów. W przypadku wystąpienia działań niepożądanych może być konieczne przerwanie leczenia, zmniejszenie dawki lub odstawienie leku Ofev® (patrz sekcja „Sposób stosowania i dawki”).
Silne induktory P-gp (np. ryfampicyna, karbamazepina, fenytoina oraz preparaty z zioła świętojaństwia zwyczajnego) mogą zmniejszać ekspozycję na nintedanib. Zaleca się dobór alternatywnej terapii wspomagającej bez działania indukcyjnego lub o minimalnym działaniu indukcyjnym na układ P-gp.
Izoenzym cytochromu (CYP)
Izoenzymy CYP odgrywają jedynie niewielką rolę w biotransformacji nintedanibu. W badaniach przedklinicznych nintedanib oraz jego metabolity (BIBF 1202 – wolny kwasowy metabolit nintedanibu i jego glukuronid BIBF 1202) nie hamowały ani nie indukowały izoenzymów CYP (patrz sekcja „Właściwości farmakokinetyczne. Farmakokinetyka”). Dlatego prawdopodobieństwo interakcji lekowych związanych z metabolizmem CYP jest uważane za niskie.
Jednoczesne stosowanie z innymi lekami
Jednoczesne stosowanie nintedanibu z doustnymi środkami antykoncepcyjnymi nie zmieniało istotnie farmakokinetyki doustnych środków antykoncepcyjnych (patrz sekcja „Właściwości farmakodynamiczne. Farmakokinetyka”).
Jednoczesne stosowanie nintedanibu z bозентanem nie zmienia farmakokinetyki nintedanibu (patrz sekcja „Właściwości farmakodynamiczne. Farmakokinetyka”).
Szczególne wytyczne dotyczące stosowania.
Zaburzenia ze strony przewodu pokarmowego
Diareia
W badaniach klinicznych (patrz sekcja „Właściwości farmakodynamiczne”) diareia była najczęściej występującym niepożądaniem ze strony przewodu pokarmowego (patrz sekcja „Reakcje niepożądane”). U większości pacjentów objawy te miały charakter łagodny lub umiarkowany i występowały w pierwszych 3 miesiącach leczenia.
W okresie po rejestracji leku zgłaszano poważne przypadki diarei prowadzącej do odwodnienia i zaburzeń elektrolitowych. Leczenie diarei (odpowiednie nawadnianie oraz leki przeciwdiareiczne, np. loperamid) należy rozpocząć przy pierwszych jej objawach. W przypadku wystąpienia diarei może być konieczna redukcja dawki lub przerwanie leczenia. Leczenie lekiem Ofev® można wznowić w dawce zmniejszonej (100 mg dwa razy dziennie) lub pełnej dawce (150 mg dwa razy dziennie). W przypadku utrzymywania się ciężkiej diarei mimo leczenia objawowego, terapię lekiem Ofev® należy odstawić.
Światłowstręt i wymioty
Światłowstręt i wymioty były często zgłaszanymi niepożądanymi objawami ze strony przewodu pokarmowego (patrz sekcja „Reakcje niepożądane”). U większości pacjentów występowały one w formie łagodnej lub umiarkowanej. W badaniach częstość światłowstrętu i wymiotów prowadzących do zaprzestania leczenia lekiem Ofev® wynosiła odpowiednio 2,1% i 1,4%.
Jeśli objawy nie ustępują pomimo odpowiedniego leczenia objawowego (w tym stosowania leków przeciwwymiotnych), może być konieczna redukcja dawki leku lub przerwanie leczenia. Leczenie można wznowić w dawce zmniejszonej (100 mg dwa razy dziennie) lub pełnej dawce (150 mg dwa razy dziennie). Jeśli ciężkie objawy nie ustępują, terapię lekiem Ofev® należy odstawić.
Zaburzenia funkcji wątroby
Bezpieczeństwo i skuteczność stosowania leku Ofev® u pacjentów z zaburzeniami funkcji wątroby umiarkowanego (klasa B wg skali Childa-Pugh) i ciężkiego (klasa C wg skali Childa-Pugh) stopnia nie były badane. Dlatego leczenie tych pacjentów lekiem Ofev® nie jest zalecane (patrz sekcje „Sposób stosowania i dawki”). Z uwagi na zwiększoną ekspozycję na lek, ryzyko wystąpienia reakcji niepożądanych może wzrosnąć u pacjentów z łagodnymi zaburzeniami funkcji wątroby (klasa A wg skali Childa-Pugh). Pacjentom z łagodnymi zaburzeniami funkcji wątroby (klasa A wg skali Childa-Pugh) należy podawać leczenie w zmniejszonej dawce leku Ofev® (patrz sekcje „Sposób stosowania i dawki” oraz „Właściwości farmakokinetyczne”).
Podczas leczenia nintedanibem obserwowano przypadki uszkodzenia wątroby wywołanego lekiem, w tym ciężkie uszkodzenie wątroby zakończone śmiercią. Większość zaburzeń wątrobowych pojawia się w pierwszych trzech miesiącach leczenia. Dlatego poziomy transaminaz wątrobowych i bilirubiny należy oznaczać przed rozpoczęciem leczenia oraz w pierwszym miesiącu leczenia lekiem Ofev®. Następnie należy regularnie kontrolować te parametry w ciągu kolejnych dwóch miesięcy leczenia i okresowo później, np. przy każdej wizycie pacjenta lub zgodnie z wskazaniami klinicznymi.
W większości przypadków po redukcji dawki lub przerwaniu leczenia wzrost stężenia enzymów wątrobowych [ALT, AST, fosfatazy alkalicznej, gammaglutamylotransferazy (GGT), patrz sekcja „Reakcje niepożądane”] oraz bilirubiny był odwracalny.
W przypadku wzrostu poziomu transaminaz (AST lub ALT) powyżej 3-krotnej górnej granicy normy zaleca się zmniejszenie dawki lub przerwanie terapii lekiem Ofev® oraz dokładne monitorowanie stanu pacjenta. Gdy poziomy transaminaz powrócą do wartości wyjściowych, leczenie lekiem Ofev® można wznowić w pełnej dawce (150 mg dwa razy dziennie) lub w dawce zmniejszonej (100 mg dwa razy dziennie), którą następnie można zwiększyć do pełnej dawki (patrz sekcja „Sposób stosowania i dawki”). Jeśli wzrost któregokolwiek z parametrów funkcji wątroby wiąże się z objawami klinicznymi uszkodzenia wątroby, np. żółtaczką, leczenie lekiem Ofev® należy trwale przerwać. Należy rozważyć inne przyczyny wzrostu stężenia enzymów wątrobowych.
Pacjenci o niskiej masie ciała (do 65 kg), pochodzenia azjatyckiego oraz kobiety należą do grupy zwiększonego ryzyka wzrostu stężenia enzymów wątrobowych. Ekspozycja na nintedanib wzrasta liniowo wraz z wiekiem pacjenta, co również zwiększa ryzyko wzrostu stężenia enzymów wątrobowych (patrz sekcja „Właściwości farmakokinetyczne”). U pacjentów z takimi czynnikami ryzyka zaleca się dokładne monitorowanie stanu.
Funkcja nerek
Podczas leczenia nintedanibem obserwowano przypadki zaburzeń funkcji nerek/niewydolności nerek, niektóre z nich miały charakter śmiertelny (patrz sekcja „Reakcje niepożądane”).
Podczas terapii nintedanibem zaleca się kontrolowanie stanu pacjentów, szczególnie tych z czynnikami ryzyka zaburzeń funkcji nerek/niewydolności nerek. W przypadku zaburzeń funkcji nerek/niewydolności nerek należy rozważyć konieczność dostosowania dawki (patrz sekcja „Sposób stosowania i dawki. Dostosowanie dawki”).
Krwiaki
Hamowanie receptora czynnika wzrostu śródbłonka naczyniowego (VEGFR) może być związane ze zwiększonym ryzykiem krwawień.
W badaniach klinicznych nie włączano pacjentów z znanym ryzykiem krwawień, w tym pacjentów z genetyczną skłonnością do krwawień lub pacjentów otrzymujących terapię przeciwzakrzepową w wysokich dawkach. W okresie po rejestracji leku zgłaszano przypadki krwawień łagodnych i ciężkich, niektóre z nich miały charakter śmiertelny (w tym przypadki u pacjentów otrzymujących terapię przeciwzakrzepową lub inne leki mogące powodować krwawienie, jak i u pacjentów bez terapii przeciwzakrzepowej). Dlatego leczenie tych pacjentów lekiem Ofev® można rozważyć tylko wtedy, gdy oczekiwana korzyść terapii przewyższa potencjalne ryzyko.
Zatorowość tętnicza
W badaniach klinicznych nie uczestniczyli pacjenci z recentym zawałem mięśnia sercowego lub udarem mózgu w wywiadzie. W badaniach przypadki zatorowości tętniczej występowały rzadko (2,5% w grupie leku Ofev® w porównaniu z 0,7% w grupie placebo w badaniu INPULSIS; 0,9% w grupie leku Ofev® w porównaniu z 0,9% w grupie placebo w badaniu INBUILD; 0,7% w grupie leku Ofev® w porównaniu z 0,7% w grupie placebo w badaniu SENSCIS). W badaniach INPULSIS częściej występował zawał mięśnia sercowego w grupie leku Ofev® (1,6%) niż w grupie placebo (0,5%), natomiast reakcje niepożądane odzwierciedlające choroby serca niedokrwienne były porównywalne w grupach leku Ofev® i placebo. W badaniu INBUILD zawał mięśnia sercowego występował rzadko: 0,9% w grupie leku Ofev® w porównaniu z 0,9% w grupie placebo. W badaniu SENSCIS przypadki zatorowości tętniczej występowały rzadko: u 0,7% pacjentów otrzymujących placebo i 0,7% pacjentów w grupie Ofev®. Zawał mięśnia sercowego występował rzadko w grupie placebo (0,7%) i nie występował w grupie Ofev®. Należy zachować ostrożność podczas leczenia pacjentów z wysokim ryzykiem sercowo-naczyniowym, w tym znaną chorobą tętnic wieńcowych. Należy rozważyć możliwość przerwania leczenia u pacjentów, u których wystąpiły objawy ostrej niedokrwienności mięśnia sercowego.
Aneurysmy i rozwarstwienie tętnic
Stosowanie inhibitorów czynnika wzrostu śródbłonka naczyniowego (VEGF) u pacjentów z nadciśnieniem tętniczym lub bez niego może prowadzić do powstawania aneurysm i/lub rozwarstwienia tętnic. Przed rozpoczęciem stosowania leku Ofev® należy dokładnie ocenić ryzyko u pacjentów z takimi czynnikami ryzyka, jak nadciśnienie tętnicze lub aneurysma w wywiadzie.
Zatorowość żylna
W badaniach klinicznych nie zaobserwowano zwiększonego ryzyka powikłań zakrzepowo-zatorowych żylnych u pacjentów przyjmujących nintedanib. Jednak ze względu na mechanizm działania nintedanibu możliwe jest zwiększone ryzyko wystąpienia zjawisk zakrzepowo-zatorowych.
Przebicia przewodu pokarmowego (GI) i niedokrwienny zapalenie okrężnicy
W badaniach klinicznych liczba pacjentów z przebiciem wynosiła 0,3% w obu grupach leczenia. Jednak ze względu na specyfikę mechanizmu działania nintedanibu u pacjentów może wzrastać ryzyko przebitek przewodu pokarmowego. W okresie po rejestracji leku zgłaszano przypadki przebitek przewodu pokarmowego i niedokrwennego zapalenia okrężnicy, niektóre z nich miały charakter śmiertelny. Szczególną uwagę należy zwrócić na leczenie pacjentów, u których wcześniej przeprowadzono operacje brzuszne, u których występuje wrzód trawienny i choroba Divertikula w wywiadzie, lub w przypadku jednoczesnego stosowania kortykosteroidów lub NLPZ. Z tego powodu lek Ofev® można stosować najwcześniej 4 tygodnie po operacjach brzusznych. W przypadku wystąpienia przebicia przewodu pokarmowego lub niedokrwennego zapalenia okrężnicy terapię lekiem Ofev® należy przerwać. W wyjątkowych przypadkach lek Ofev® można ponownie zastosować po pełnym wyleczeniu niedokrwennego zapalenia okrężnicy i dokładnej ocenie stanu pacjenta oraz innych czynników ryzyka.
Proteinuria nefrotyczna i mikroangiopatia zakrzepowa
W okresie pogwarancyjnym zgłaszano bardzo rzadkie przypadki proteinurii nefrotycznej z zaburzeniem funkcji nerek lub bez niego. Dane histologiczne w pojedynczych przypadkach odpowiadały mikroangiopatii kłębuszkowej z zakrzepami w nerkach lub bez nich. Objawy ustępowały po odstawieniu leku Ofev®, w niektórych przypadkach z resztkową proteinurią. Należy rozważyć możliwość odstawienia leczenia u pacjentów z objawami lub objawami zespołu nerczycowego.
Stosowanie inhibitorów VEGF (czynnik wzrostu śródbłonka naczyniowego) wiązano z mikroangiopatią zakrzepową, w tym z bardzo małą liczbą zgłoszeń takich przypadków przy stosowaniu nintedanibu. Jeśli dane laboratoryjne lub kliniczne wskazują na mikroangiopatię zakrzepową u pacjentów przyjmujących nintedanib, leczenie nintedanibem należy przerwać i dokładnie ocenić mikroangiopatię zakrzepową.
Nadciśnienie tętnicze
Stosowanie leku Ofev® może prowadzić do podwyższenia ciśnienia tętniczego, dlatego należy okresowo i zgodnie z wskazaniami klinicznymi mierzyć ciśnienie tętnicze.
Nadciśnienie płucne
Dane dotyczące stosowania leku Ofev® u pacjentów z nadciśnieniem płucnym są ograniczone.
Pacjenci z ciężkim nadciśnieniem płucnym (indeks sercowy ≤ 2 l/min/m², epoprostenol/treprostinil dożylne lub znaczna niewydolność prawej komory serca) byli wykluczeni z badań INBUILD i SENSCIS. Zaleca się dokładne monitorowanie pacjentów z nadciśnieniem płucnym.
Zaburzenia gojenia się ran
W badaniach klinicznych nie zaobserwowano zwiększonej częstości zaburzeń gojenia się ran. Ze względu na mechanizm działania nintedanibu substancja ta może negatywnie wpływać na gojenie się ran. Nie przeprowadzono specjalnych badań wpływu nintedanibu na gojenie się ran. Dlatego leczenie lekiem Ofev® należy rozpoczynać lub wznowić (jeśli było przerwane z powodu zabiegu chirurgicznego) z uwzględnieniem klinicznej oceny skutecznego gojenia się rany.
Leczenie pacjentów z bardzo rzadkimi postępującymi chorobami tkanki łącznej (CTD)
Badanie INBUILD nie miało odpowiedniego projektu ani mocy, aby dostarczyć dowodów na korzyści z nintedanibu w podgrupach z rozpoznaniem CTD. Skuteczność sekwencyjną wykazano w podgrupach na podstawie rozpoznań CTD (patrz sekcja „Właściwości farmakodynamiczne”). Doświadczenie w stosowaniu nintedanibu w bardzo rzadkich postępujących włóknieniowych CTD jest ograniczone. Kryteria kliniczne postępowania stosowane w badaniu INBUILD opisano w sekcji „Właściwości farmakodynamiczne”.
Jednoczesna terapia z pirfenidonem
W specjalnym badaniu farmakokinetycznym oceniano jednoczesne leczenie nintedanibem i pirfenidonom u pacjentów z IPF (włóknienie płuc idiopatyczne). Badanie to nie wykazało dowodów na istotne farmakokinetyczne oddziaływanie leków między nintedanibem a pirfenidonom przy ich stosowaniu w połączeniu (patrz sekcja „Właściwości farmakokinetyczne”). Ze względu na podobieństwo profili bezpieczeństwa obu leków można spodziewać się wystąpienia addytywnych reakcji niepożądanych, w tym objawów ze strony przewodu pokarmowego i wątroby. Stosunek korzyści do ryzyka jednoczesnego leczenia pirfenidonom nie został ustalony.
Wpływ na interwał QT
W ramach programu badań klinicznych nie wykryto żadnych oznak wydłużenia interwału QT przy stosowaniu nintedanibu (patrz sekcja „Właściwości farmakodynamiczne”). Ponieważ wiadomo, że niektóre inne inhibitory tyrozynokinaz wpływają na QT, należy zachować ostrożność przy przepisywaniu nintedanibu pacjentom z ryzykiem wydłużenia interwału QT.
Reakcje alergiczne
Wiadomo, że produkty żywienia terapeutycznego zawierające soję mogą powodować reakcje alergiczne, w tym ciężki wstrząs anafilaktyczny, u osób uczulonych na soję. Pacjenci z znaną alergią na białko orzechów ziemnych należą do grupy ryzyka ciężkich reakcji na leki zawierające soję. 1 kapsułka o zawartości 100 mg zawiera lecytynę z soi 1,2 mg; 1 kapsułka o zawartości 150 mg zawiera lecytynę z soi 1,8 mg.
Stosowanie w czasie ciąży lub karmienia piersią.
Kobiety w wieku rozrodczym
Nintedanib może negatywnie wpływać na płód człowieka. Kobiety w wieku rozrodczym powinny podczas leczenia lekiem Ofev® stosować środki zapobiegające zajściu w ciążę i używać skutecznych metod antykoncepcji podczas stosowania leku, na początku stosowania leku oraz przez co najmniej 3 miesiące po podaniu ostatniej dawki leku Ofev®. Nintedanib nie wpływa istotnie na stężenie etynylestradiolu i lewonorgestrelu w osoczu (patrz sekcja „Właściwości farmakokinetyczne”). Skuteczność doustnych hormonalnych środków antykoncepcyjnych może zmniejszać się w przypadku wymiotów i/lub diarei lub innych stanów, w których możliwy jest wpływ na wchłanianie. Kobietom przyjmującym doustne hormonalne środki antykoncepcyjne i mającym takie stany należy doradzić stosowanie alternatywnych, skutecznych środków antykoncepcji.
Ciąża
Nie przeprowadzono specjalnych badań dotyczących stosowania leku Ofev® u kobiet w ciąży, jednak w badaniach przedklinicznych na zwierzętach stwierdzono toksyczność rozrodczą tej substancji czynnej. Ponieważ nintedanib może wywierać działanie embriotoksyczne u człowieka, nie należy go stosować w czasie ciąży (patrz sekcja „Przeciwwskazania”), przed rozpoczęciem terapii lekiem Ofev® należy wykonać test ciążowy oraz odpowiednio podczas leczenia.
Pacjentki powinny natychmiast powiadomić lekarza o zajściu w ciążę podczas terapii lekiem Ofev®.
Jeśli w czasie terapii lekiem Ofev® wystąpi ciąża, leczenie należy przerwać i poinformować pacjentkę o potencjalnym ryzyku embriotoksycznego działania leku.
Karmienie piersią
Brak danych dotyczących wydzielania nintedanibu i jego metabolitów w mleko matki. W badaniach przedklinicznych wykazano, że u zwierząt w okresie laktacji do mleka przechodzi niewielka ilość nintedanibu i jego metabolitów (≤ 0,5% dawki podanej). Dlatego nie można wykluczyć ryzyka dla noworodków i niemowląt. W czasie leczenia lekiem Ofev® należy przerwać karmienie piersią.
Płodność
W badaniach przedklinicznych nie stwierdzono zaburzeń płodności u samców. W badaniach toksyczności podostrej i chronicznej, w których poziom działania systemowego leku był porównywalny z poziomem osiąganym przy stosowaniu maksymalnej zalecanej dawki u człowieka (150 mg dwa razy dziennie), nie stwierdzono zaburzeń płodności u samic zwierząt.
Wpływ na zdolność prowadzenia pojazdów lub obsługi mechanizmów.
Ofev® ma nieznaczny wpływ na zdolność prowadzenia pojazdów lub obsługi mechanizmów. Podczas stosowania leku Ofev® pacjentom należy zalecać zachowanie ostrożności przy prowadzeniu pojazdów lub obsługi mechanizmów.
Sposób stosowania i dawki.
Leczenie lekiem powinien rozpoczynać lekarz doświadczony w leczeniu pacjentów z chorobami, w przypadku których zatwierdzono stosowanie leku Ofev®.
Dawki
Zalecana dawka leku wynosi 150 mg dwa razy dziennie, w odstępach około 12 godzin. Dawka 100 mg dwa razy dziennie zalecana jest wyłącznie pacjentom, którzy źle tolerują dawkę 150 mg dwa razy dziennie.
Jeśli pominięto jakąkolwiek dawkę leku, należy kontynuować stosowanie leku w zalecanej dawce zgodnie z harmonogramem następnego przyjęcia. Jeśli dawka została pominięta, pacjent nie powinien przyjmować dodatkowej dawki leku. Maksymalna dawka dzienna wynosi 300 mg.
Korekta dawki
W przypadku wystąpienia niepożądanych reakcji na lek Ofev (patrz rozdziały „Szczególne wskazania dotyczące stosowania”, „Działania niepożądane”) oprócz terapii objawowej, w razie potrzeby, zaleca się zmniejszenie dawki lub tymczasowe przerwanie leczenia, aż do ustąpienia niepożądanej reakcji do poziomu umożliwiającego wznowienie terapii. Leczenie lekiem Ofev może być wznowione w pełnej dawce (150 mg dwa razy dziennie) lub w zmniejszonej dawce (100 mg dwa razy dziennie). Jeśli pacjent nie toleruje dawki leku 100 mg dwa razy dziennie, leczenie lekiem Ofev należy przerwać.
Jeśli biegunka, nudności i/lub wymioty nie ustępują pomimo odpowiedniej terapii wspomagającej (w tym terapii przeciwwymiotnej), może być konieczne zmniejszenie dawki lub przerwanie leczenia. Leczenie lekiem Ofev może być wznowione w zmniejszonej dawce (100 mg dwa razy dziennie) lub w pełnej dawce (150 mg dwa razy dziennie). W przypadku utrzymywania się ciężkiej biegunki pomimo leczenia objawowego, terapię lekiem Ofev należy odstawić (patrz rozdział „Szczególne wskazania dotyczące stosowania”).
W przypadku wzrostu stężenia aspataminotransferazy (AST) lub alaninotransferazy (ALT) powyżej 3-krotnej górnej granicy normy zaleca się przerwanie terapii lekiem Ofev. Gdy wartości te powrócą do normy, leczenie lekiem Ofev może być wznowione w zmniejszonej dawce (100 mg dwa razy dziennie), którą następnie można zwiększyć do pełnej dawki (150 mg dwa razy dziennie) (patrz rozdziały „Szczególne wskazania dotyczące stosowania”, „Działania niepożądane”).
Grupy specjalne pacjentów
Pacjenci w wieku podeszłym (˃ 65 lat)
Nie zaobserwowano ogólnych różnic pod względem bezpieczeństwa i skuteczności stosowania leku u pacjentów w wieku podeszłym. Korekty dawki leku w zależności od wieku pacjenta nie należy stosować. Pacjentom w wieku 75 lat i starszym może być konieczne zmniejszenie dawki w celu zminimalizowania działań niepożądanych (patrz rozdział „Właściwości farmakologiczne. Farmakokinetyka”).
Zaburzenia funkcji nerek
U pacjentów z łagodnym lub umiarkowanym zaburzeniem funkcji nerek nie należy korygować dawki początkowej. U pacjentów z ciężkim zaburzeniem funkcji nerek (klirens kreatyniny < 30 ml/min) bezpieczeństwo, skuteczność oraz farmakokinetyka nintedanibu nie były badane.
Zaburzenia funkcji wątroby
Dla pacjentów z łagodnym zaburzeniem funkcji wątroby (klasa A wg skali Childa-Pugha) zalecana dawka leku Ofev wynosi 100 mg dwa razy dziennie w odstępach około 12 godzin. U tych pacjentów należy przewidzieć możliwość przerwania lub odstawienia leczenia w celu kontroli działań niepożądanych. U pacjentów z zaburzeniem funkcji wątroby klasy B i C wg skali Childa-Pugha bezpieczeństwo i skuteczność nintedanibu nie były badane. Dlatego leczenie pacjentów z umiarkowanym (klasa B wg skali Childa-Pugha) i ciężkim (klasa C wg skali Childa-Pugha) zaburzeniem funkcji wątroby lekiem Ofev nie jest zalecane (patrz rozdział „Właściwości farmakologiczne. Farmakokinetyka”).
Dzieci
Bezpieczeństwo i skuteczność stosowania leku Ofev u dzieci (do 18. roku życia) nie były badane. Brak danych.
Sposób stosowania
Ofev przeznaczony jest do stosowania doustnego. Kapсуłki należy przyjmować podczas jedzenia, połykać całe, popijając wodą; nie należy ich żuć. Nie otwierać ani nie rozdrabniać kapsułki. W przypadku kontaktu z zawartością kapsułki należy natychmiast przemyć ręce dużą ilością wody.
Dzieci.
Lek nie stosuje się w praktyce pediatrycznej.
Przedawkowanie.
Objawy
Zanotowano przypadki przedawkowania u dwóch pacjentów uczestniczących w programie onkologicznym, którzy przyjmowali lek w maksymalnej dawce 600 mg przez osiem dni. Obserwowane działania niepożądane były zgodne z znanym profilem bezpieczeństwa nintedanibu: podwyższenie aktywności enzymów wątrobowych i zaburzenia ze strony przewodu pokarmowego. Oba przypadki skończyły się pełnym powrotem do zdrowia po ustąpieniu działań niepożądanych. W badaniach INPULSIS zanotowano jeden przypadek przypadkowego zwiększenia dawki przez pacjenta do 600 mg dziennie przez 21 dni. W okresie nieprawidłowego przyjmowania leku zanotowano wystąpienie lekkiego działania niepożądanego (nazięcie) bez rejestracji innych działań niepożądanych.
Leczenie
Nie ma specyficznego antydota. W przypadku przedawkowania należy odstawić lek i przeprowadzać terapię objawową.
Niepożądane działania.
W badaniach klinicznych oraz w okresie pogwarancyjnym najczęściej występującymi niepożądanymi działaniami związanymi z zastosowaniem nintedanibu były: biegunka, nudności i wymioty, ból brzucha, zmniejszony apetyt, spadek masy ciała oraz podwyższenie poziomu enzymów wątrobowych.
Aby uzyskać informacje dotyczące leczenia niektórych niepożądanych zjawisk, patrz sekcja „Szczególne wskazania dotyczące stosowania”.
W tabeli 11 przedstawiono niepożądane działania według klas układów narządów według MedDRA oraz częstości występowania, z wykorzystaniem następujących kryteriów oceny:
bardzo często (> 1/10); często (> 1/100 do < 1/10); rzadko (> 1/1 000 do < 1/100); pojedyncze przypadki (> 1/10 000 do < 1/1 000); rzadkie (< 1/10 000), nieznane (niemożliwe do oszacowania na podstawie dostępnych danych).
Tabela 11
| Częstotliwość |
|||
| Układ narządów |
Idiopatyczne włóknienie płuc (IWP) |
Choroba śródmiąższowa płuc przy twardzinie systemowej (zespołu sklerodermii) (ChP-SS). |
Inne przewlekłe choroby płuc śródmiąższowe o postępie włóknistym z fenotypem postępującym |
| Zaburzenia układu krwiotwórczego i limfatycznego |
|||
| Małopłytkowość |
Niekorzystne |
Niekorzystne |
Niekorzystne |
| Zaburzenia metaboliczne i odżywienia |
|||
| Spadek masy ciała |
Częste |
Częste |
Częste |
| Spadek apetytu |
Częste |
Częste |
Bardzo częste |
| Odewodnienie |
Niekorzystne |
Nieznane |
Niekorzystne |
| Zaburzenia serca |
|||
| Przewlekłe zapalenie mięśnia sercowego |
Niekorzystne |
Nieznane |
Niekorzystne |
| Zaburzenia układu krążenia |
|||
| Krwawienia (patrz dział „Szczególne ostrzeżenia i środki ostrożności”) |
Częste |
Częste |
Częste |
| Nadciśnienie |
Niekorzystne |
Częste |
Częste |
| Przerost i rozwarstwienie tętnic |
Nieznane |
Nieznane |
Nieznane |
| Zaburzenia układu pokarmowego |
|||
| Diareia |
Bardzo częste |
Bardzo częste |
Bardzo częste |
| Nudności |
Bardzo częste |
Bardzo częste |
Bardzo częste |
| Ból brzucha |
Bardzo częste |
Bardzo częste |
Bardzo częste |
| Wymioty |
Częste |
Bardzo częste |
Bardzo częste |
| Zapalenie trzustki |
Niekorzystne |
Nieznane |
Niekorzystne |
| Wolny |
Niekorzystne |
Niekorzystne |
Niekorzystne |
| Zaburzenia wątroby i dróg żółciowych |
|||
| Uszkodzenie wątroby spowodowane lekiem |
Niekorzystne |
Niekorzystne |
Częste |
| Zwiększenie poziomu enzymów wątrobowych |
Bardzo częste |
Bardzo częste |
Bardzo częste |
| Zwiększenie poziomu alaninotransaminazy (ALT) |
Częste |
Częste |
Bardzo częste |
| Zwiększenie poziomu asparaginianotransaminazy (AST) |
Częste |
Częste |
Częste |
| Zwiększenie poziomu gamma-glutamylotranspeptydazy (GGT) |
Częste |
Częste |
Częste |
| Hiperbilirubinemia |
Niekorzystne |
Nieznane |
Niekorzystne |
| Zwiększenie poziomu fosfatazy alkalicznej we krwi (FAK) |
Niekorzystne |
Częste |
Częste |
| Zaburzenia skóry i tkanek podskórnych |
|||
| Wysypka |
Częste |
Niekorzystne |
Częste |
| Zwierzenia |
Niekorzystne |
Niekorzystne |
Niekorzystne |
| Łysienie |
Niekorzystne |
Nieznane |
Niekorzystne |
| Zaburzenia nerek i dróg moczowych |
|||
| Niewydolność nerek (patrz dział „Szczególne ostrzeżenia i środki ostrożności”) |
Nieznane |
Niekorzystne |
Nieznane |
| Białkomocz |
Niekorzystne |
Nieznane |
Niekorzystne |
| Zaburzenia układu nerwowego |
|||
| Ból głowy |
Częste |
Częste |
Częste |
Opis poszczególnych działań niepożądanych
Diareia
W badaniach klinicznych (patrz sekcja „Właściwości farmakologiczne. Farmakodynamika”) biegunka była najczęstszym niepożadanym zjawiskiem ze strony przewodu pokarmowego. U większości pacjentów objawy miały łagodny lub umiarkowany stopień nasilenia. U ponad dwóch trzecich pacjentów biegunka występowała w ciągu pierwszych 3 miesięcy leczenia.
U większości pacjentów udało się przezwyciężyć niepożądane zjawiska poprzez stosowanie terapii przeciwbiegunkowej, zmniejszenie dawki lub przerwanie leczenia (patrz sekcja „Szczególne ostrzeżenia i środki ostrożności postępowania”). Przegląd przypadków biegunki w badaniach klinicznych przedstawiono w tabeli 12.
Tabela 12
Przypadki biegunki w badaniach klinicznych w ciągu 52 tygodni
| Badanie |
INPULSIS |
INBULD |
SENSCIS |
|||
| Leczenie |
Placebo |
Ofev® |
Placebo |
Ofev® |
Placebo |
Ofev® |
| Diaree |
18,4 % |
62,4 % |
23,9 % |
66,9 % |
31,6 % |
75,7 % |
| Ciężka diaree |
0,5 % |
3,3 % |
0,9 % |
2,4 % |
1,0 % |
4,2 % |
| Diaree, która spowodowała zmniejszenie dawki leku Ofev® |
0 % |
10,7 % |
0,9 % |
16,0 % |
1,0 % |
22,2 % |
| Diaree, która spowodowała przerwanie leczenia lekiem Ofev® |
0,2 % |
4,4 % |
0,3 % |
5,7 % |
0,3 % |
6,9 % |
Podwyższenie poziomu enzymów wątrobowych
W badaniach INPULSIS podwyższenie poziomu enzymów wątrobowych (patrz punkt „Szczególne ostrzeżenia i środki ostrożności podczas stosowania”) stwierdzono u 13,6 % pacjentów otrzymujących lek Ofev® w porównaniu do 2,6 % pacjentów otrzymujących placebo. W badaniu INBUILD podwyższenie poziomu enzymów wątrobowych zaobserwowano odpowiednio u 22,6 % pacjentów otrzymujących Ofev® i 5,7 % pacjentów otrzymujących placebo. W badaniu SENSCIS podwyższenie poziomu enzymów wątrobowych obserwowano u 13,2 % pacjentów otrzymujących Ofev® w porównaniu do 3,1 % pacjentów otrzymujących placebo. Podwyższenie poziomu enzymów wątrobowych miało charakter odwracalny i nie było związane z klinicznie wyrażoną chorobą wątroby. Dodatkowe informacje dotyczące szczególnych grup pacjentów, zalecanych środków ostrożności oraz dostosowania dawki w przypadku biegunki i podwyższenia poziomu enzymów wątrobowych zawarte są w punktach „Szczególne ostrzeżenia i środki ostrożności podczas stosowania” oraz „Sposób stosowania i dawki”.
Krwawienia
W badaniach klinicznych odsetek pacjentów, u których obserwowano krwawienia, był nieco wyższy lub porównywalny w grupie leku Ofev® w porównaniu z grupą placebo (10,3 % w grupie leku Ofev® w porównaniu do 7,8 % w grupie placebo w badaniu INPULSIS; 11,1 % w grupie Ofev® w porównaniu do 12,7 % w grupie placebo w badaniu INBUILD; 11,1 % w grupie Ofev® w porównaniu do 8,3 % w grupie placebo w badaniu SENSCIS). Lekkie krwawienia z nosa były najczęstszym rodzajem krwawień jako niepożądanych zjawisk. Poważne krwawienia występowały rzadko w obu grupach (1,3 % w grupie leku Ofev® w porównaniu do 1,4 % w grupie placebo w badaniu INPULSIS; 0,9 % w grupie leku Ofev® w porównaniu do 1,5 % w grupie placebo w badaniu INBUILD; 1,4 % w grupie leku Ofev® w porównaniu do 0,7 % w grupie placebo w badaniu SENSCIS).
W okresie po rejestracji leku przypadki krwawień dotyczyły przede wszystkim układu pokarmowego, oddechowego i centralnego układu nerwowego, ale nie były do nich ograniczone. Najczęściej występujące krwawienia związane były z układem pokarmowym (patrz punkt „Szczególne ostrzeżenia i środki ostrożności podczas stosowania”).
Proteinuria
W badaniach klinicznych częstość występowania proteinurii była niska i porównywalna w grupach leczenia (0,8 % w grupie leku Ofev® w porównaniu do 0,5 % w grupie placebo w badaniu INPULSIS; 1,5 % w grupie leku Ofev® w porównaniu do 1,8 % w grupie placebo w badaniu INBUILD; 1,0 % w grupie leku Ofev® w porównaniu do 0,0 % w grupie placebo w badaniu SENSCIS). W trakcie badań klinicznych nie odnotowano przypadków zespołu nerczycowego.
W okresie po wprowadzeniu leku na rynek zgłaszano bardzo rzadkie przypadki nerczycowej proteinurii z zaburzeniem funkcji nerek lub bez niego. Dane histologiczne w pojedynczych przypadkach odpowiadały mikroangiopatii naczyniowej kłębuszków nerkowych z lub bez zakrzepów nerkowych. Ustępowanie objawów obserwowano po zaprzestaniu przyjmowania leku Ofev®, w niektórych przypadkach z utrzymującą się proteinurią. Należy rozważyć możliwość przerwania leczenia u pacjentów, u których wystąpią objawy lub oznaki zespołu nerczycowego (patrz punkt „Szczególne ostrzeżenia i środki ostrożności podczas stosowania”).
Zgłaszanie niepożądanych reakcji
Zgłaszanie niepożądanych reakcji po rejestracji leku ma duże znaczenie. Umożliwia ono ciągłe monitorowanie stosunku korzyści do ryzyka stosowania leku. Prosimy pracowników medycznych o zgłaszanie wszelkich możliwych niepożądanych reakcji za pomocą krajowego systemu raportowania.
Okres ważności. 3 lata.
Warunki przechowywania.
Przechowywać w temperaturze nie wyższej niż 25 °C w oryginalnym opakowaniu w celu ochrony przed wilgocią.
Przechowywać w miejscu niedostępnym dla dzieci.
Opakowanie.
10 kapsułek w blistrze z folii aluminiowej z perforacją, 6 blistrów w opakowaniu tekturowym.
Kategoria wydania.
Na receptę.
Producent.
Boehringer Ingelheim Pharma GmbH & Co. KG.
Miejsce produkcji oraz adres siedziby.
Binger Strasse 173, 55216, Ingelheim am Rhein, Niemcy