Novoseven®
Ukraina
Spis treści
INSTRUKCJA dotyczÄ cza stosowania leku NOVOSEVEN® (NOVOSEVEN®)
SkÅ ad:
substancja czynna: eptakog alfa (aktywowany) (rFVIIa);
1 fiolka zawiera 2 mg (100 IU) lub 5 mg (250 IU) eptakogu alfa (aktywowanego);
substancje pomocnicze: chloro sodu; chlorek wapnia, dwuwodny; glicyloglicyna; polisorbat 80; metionina; sacharoza; manitol (E421).
Po rozcieczynieniu produkt zawiera 1 mg/ml eptakogu alfa (aktywowanego) po odzyskaniu za pomocÄ rozpuszczalnika.
Rozpuszczalnik: histydyna, woda do wstrzykiwań.
PostaÄ leku. Proszek liofilizowany do sporzÄ dzenia roztworu do wstrzykiwaÅ„.
GÅ ówne wÅ aÅ›ciwoÅ›ci fizykochemiczne: biaÅ‚y proszek liofilizowany.
Grupa farmakoterapeutyczna. Å›rodki hemostatyczne. Czynniki krzepniÄ cia krwi.
Kod ATC B02B D08.
Właściwości farmakologiczne.
Farmakodynamika.
Składem leku Novoseven® jest rekombinowany aktywowany czynnik krzepnięcia VII o masie cząsteczkowej ok. 50000 Daltonów, wytwarzany metodą inżynierii genetycznej z wykorzystaniem komórek nerek noworodków chomika (komórki BHK) jako komórek gospodarza.
Mechanizm działania
Mechanizm działania polega na wiązaniu się czynnika VIIa z czynnikiem tkankowym. Ten kompleks aktywuje czynniki IX i X do postaci aktywnej – IXa i Xa, co prowadzi do przekształcenia niewielkich ilości protrombiny w trombinę. Trombina w miejscu uszkodzenia aktywuje płytki krwi oraz czynniki V i VIII, co powoduje przekształcenie fibrynogenu w fibrynę i tworzenie skrzepliny hemostatycznej. Lek Novoseven® w dawkach farmakologicznych, niezależnie od czynnika tkankowego, bezpośrednio aktywuje czynnik X na powierzchni aktywowanych płytek krwi znajdujących się w strefie uszkodzenia. To prowadzi do przekształcenia dużej ilości protrombiny w trombinę bez udziału czynnika tkankowego.
Działanie farmakodynamiczne.
W związku z tym działanie farmakodynamiczne czynnika VIIa polega na lokalnym zwiększeniu tworzenia się czynnika Xa, trombiny i fibryny.
Czas osiągnięcia maksymalnej aktywności krzepnięcia po podaniu leku Novoseven® wynosił ok. 10 minut u zdrowych ochotników oraz u pacjentów z hemofilią.
Teoretycznie nie można całkowicie wykluczyć uogólnionej aktywacji układu krzepnięcia krwi u pacjentów z chorobami sprzyjającymi rozwojowi rozsianego wewnątrznaczyniowego krzepnięcia (DIC).
Skuteczność i bezpieczeństwo kliniczne
Wrodzony deficyt FVII
W rejestrze obserwacyjnym (F7HAEM-3578) pacjentów z wrodzonym deficytem czynnika VII średnia dawka stosowana w długotrwałej profilaktyce krwawień u 22 dzieci (do 12 roku życia) z deficytem czynnika VII i ciężkim fenotypem klinicznym wynosiła 30 μg/kg (zakres od 17 do 200 μg/kg; u 10 pacjentów najczęściej stosowana dawka wynosiła 30 μg/kg) przy średniej częstotliwości podawania leku 3 dawki tygodniowo (zakres od 1 do 7; u 13 pacjentów najczęściej zgłaszana częstotliwość podawania wynosiła 3 razy w tygodniu).
W tym samym rejestrze u 3 z 91 pacjentów po zabiegach chirurgicznych zaobserwowano zjawiska tromboemboliczne.
Trombastenia Glanzmanna
Rejestr obserwacyjny (F7HAEM-3521) zawiera dane 133 pacjentów z trombastenią Glanzmanna leczonych lekiem Novoseven®. Średnia dawka na jedno podanie leku w leczeniu 333 epizodów krwawień wynosiła 90 μg/kg (zakres od 28 do 450 μg/kg). Lek Novoseven® stosowano w 157 zabiegach chirurgicznych w średniej dawce 92 μg/kg (do 270 μg/kg). Leczenie lekiem Novoseven®, samodzielnie lub w połączeniu z lekami antyfibrynolitycznymi i/lub płytkami krwi, uznawano za skuteczne, jeśli krwawienie udało się zatrzymać co najmniej na 6 godzin. Wskaźnik skuteczności wynosił odpowiednio 81% i 82% u pacjentów z dodatnim lub ujemnym wynikiem odporności na przetaczanie płytek krwi oraz 77% i 85% odpowiednio u pacjentów z dodatnim lub ujemnym wynikiem przeciwciał przeciwko płytkom krwi. Dodatni status oznacza obecność co najmniej jednego dodatniego wyniku podczas podawania leku pacjentowi.
Ciężka krwawica poporodowa
Skuteczność i bezpieczeństwo stosowania leku Novoseven® oceniano u 84 kobiet z ciężką krwawicą poporodową w ramach wieloośrodkowego, otwartego badania klinicznego. Pacjentki zostały zrandomizowane i losowo przydzielone do dwóch grup: jedna grupa otrzymywała pojedynczą dawkę 60 μg/kg leku Novoseven® (w połączeniu z terapią standardową; N = 42), a druga grupa – terapię porównawczą (tylko terapia standardowa; N = 42) po nieskutecznym zastosowaniu leków uterotonicznych (sulprostonu). Grupy terapeutyczne były dobrze zrównoważone pod względem cech demograficznych i historii wcześniejszego leczenia krwawień poporodowych przed randomizacją. Standardowe leczenie obejmowało fibrynogen i kwas traneksamowy. Informacje dotyczące stosowania fibrynogenu/kwasu traneksamowego były dostępne dla ok. 57% pacjentek w grupie leku Novoseven® i 43% pacjentek w grupie porównawczej. Z tej liczby ok. 40% pacjentek w obu grupach otrzymywało fibrynogen i/lub kwas traneksamowy. Krwawienie uznawano za zatrzymane (czyli leczenie uznawano za skuteczne), jeśli w ciągu 30 minut po randomizacji oszacowany objętość krwawienia zmniejszyła się do mniej niż 50 ml w ciągu 10 minut. Jeśli krwawienie stawało się niekontrolowane lub nieodpowiedzialne na leczenie, rozważano zastosowanie procedur inwazyjnych.
Wyniki analizy pierwotnej wykazały, że liczba kobiet, które przeszły co najmniej jedną embolizację i/lub procedurę ligatury w grupie leku Novoseven®, była mniejsza niż w grupie porównawczej (21 kontra 35), co odpowiada statystycznie istotnemu względnemu zmniejszeniu ryzyka o 40% dla grupy leku Novoseven® w porównaniu z grupą kontrolną (względne ryzyko = 0,60 (95% przedział ufności: 0,43–0,84, p = 0,0012)).
W grupie porównawczej 8 z 42 pacjentek otrzymało później lek Novoseven® jako leczenie z motywów humanitarnych w celu uniknięcia histerektomii z powodów życiowych, co zakończyło się sukcesem w 2 przypadkach.
Farmakokinetyka.
Zdrowi ochotnicy
Rozkład, wydalanie i liniowość
Farmakokinetykę leku Novoseven® badano u 35 zdrowych ochotników pochodzenia europejskiego i japońskiego w warunkach eskalacji dawek z wykorzystaniem analizy koagulacyjnego czynnika FVII. Uczestnicy byli podzieleni według płci i pochodzenia etnicznego i otrzymywali dawki 40, 80 oraz 160 μg leku Novoseven® na 1 kg masy ciała i/lub placebo (3 dawki). Farmakokinetyka nie zależała od płci ani grupy etnicznej. Średni objętość rozkładu w stanie równowagi wynosiła od 130 do 165 ml/kg, średnie wartości klirensu – od 33,3 do 37,2 ml/godz × kg. Średni okres półwylugowania końcowego – od 3,9 do 6 godzin.
Profile farmakokinetyczne były proporcjonalne do dawki.
Hemofilia A i B z obecnością inhibitorów
Rozkład, eliminacja i liniowość
Właściwości farmakokinetyczne leku Novoseven® z wykorzystaniem analizy FVII badano u 12 dzieci (2–12 lat) i 5 dorosłych pacjentów bez obecności krwawienia. Średni objętość rozkładu w stanie spoczynku wynosił 196 ml/kg u dzieci w porównaniu z 159 ml/kg u dorosłych. Średni klirens był o ok. 50% wyższy u dzieci niż u dorosłych (78 vs. 53 ml/godz × kg), natomiast średni okres półwylugowania końcowego wynosił 2,3 godziny w obu grupach. Wydaje się, że klirens jest związany z wiekiem, dlatego u młodszych pacjentów może wzrastać o więcej niż 50%.
Proporcjonalność dawki badano u dzieci dla dawek badawczych 90 i 180 μg/kg masy ciała, zgodnie z danymi uzyskanymi wcześniej przy stosowaniu niższych dawek (17,5–70 μg/kg rFVIIa).
Deficyt czynnika VII
Rozkład i eliminacja
Farmakokinetyka pojedynczego podania leku Novoseven® w dawkach 15 i 30 μg/kg masy ciała istotnie się nie różniła pod względem wartości niezależnych od dawki parametrów takich jak całkowity klirens – 70,8–79,1 ml/godz × kg, objętość rozkładu w stanie ustalonym – 280–290 ml/kg, średnia długość przebywania w krwiobiegu – 3,75–3,80 godziny, czas półwylugowania – 2,82–3,11 godziny. Średnia wartość biodostępności in vivo w osoczu wynosiła ok. 20% podanej dawki.
Trombastenia Glanzmanna
Farmakokinetyki leku Novoseven® u pacjentów z trombastenią Glanzmanna nie badano, jednak można założyć, że istotnie się nie różni od farmakokinetyki w przypadku hemofilii A i B.
Ciężka krwawica poporodowa
Farmakokinetyki leku Novoseven® u pacjentek z ciężkimi krwawieniami poporodowymi nie badano.
Dane dotyczące bezpieczeństwa przedklinicznego.
Wszystkie dane uzyskane w badaniach przedklinicznych dotyczących bezpieczeństwa były związane z efektem farmakologicznym rFVIIa.
Potencjalny efekt synergistyczny w leczeniu skojarzonym z zastosowaniem czynnika rFXIII i czynnika rFVIIa wykazano w doświadczalnym modelu sercowo-naczyniowym u makaków, co doprowadziło do istotnego efektu farmakologicznego (trombozy i śmierci) przy zastosowaniu niższych dawek niż przy podawaniu tych leków oddzielnie.
Właściwości kliniczne.
Wskazania.
Leczenie krwawień i ich zapobieganie podczas zabiegów chirurgicznych lub innych procedur inwazyjnych u pacjentów z następującymi chorobami:
- hemofilia wrodzona z poziomem inhibitorów czynnika krzepnięcia VIII lub IX > 5 BU (jednostek Bethesdy);
- hemofilia wrodzona z wyraźną reakcją na podanie czynników VIII lub IX w wywiadzie;
- hemofilia nabyte;
- wrodzony deficyt czynnika VII;
- trombastenia Glanzmanna z opornością na przetaczanie płytek krwi w przeszłości lub obecnie lub z niedoborem płytek krwi.
Krwawienia poporodowe ciężkie
Lek Novoseven® wskazany jest w leczeniu ciężkich krwawień poporodowych, gdy środki uterotoniczne nie są wystarczające do osiągnięcia hemostazy.
Przeciwwskazania.
Podwyższona wrażliwość na substancję czynną lub którykolwiek z substancji pomocniczych, a także na białka myszy, chomików lub krów.
Interakcje z innymi lekami i inne rodzaje interakcji.
Ryzyko interakcji leku Novoseven® z koncentratami czynników krzepnięcia jest nieznane. Należy unikać jednoczesnego stosowania koncentratów kompleksu protrombinowego (aktywowanych lub nieaktywowanych).
Podawanie leków antyfibrynolitycznych chorym na hemofilię zmniejsza utratę krwi podczas zabiegów chirurgicznych, szczególnie ortopedycznych lub w obszarach o wysokiej aktywności fibrynolitycznej, np. w jamie ustnej. Leki antyfibrynolityczne stosuje się również w celu zmniejszenia utraty krwi u kobiet z krwawieniem poporodowym. Doświadczenie dotyczące jednoczesnego podawania leków antyfibrynolitycznych i leku Novoseven® jest jednak ograniczone.
Zgodnie z danymi badań przedklinicznych (patrz sekcja „Dane dotyczące bezpieczeństwa przedklinicznego”), nie zaleca się jednoczesnego stosowania czynnika VIIa i czynnika XIII.
Brak danych klinicznych dotyczących interakcji między czynnikiem VIIa a czynnikiem XIII.
Szczególne ostrzeżenia i środki ostrożności dotyczące stosowania
Śledzenie
W celu poprawienia śledzenia produktów biologicznych, nazwa i numer serii podanego leku powinny być dokładnie udokumentowane.
W przypadku stanów patologicznych, w których czynnik tkankowy jest wyrażany intensywniej niż w warunkach normalnych, istnieje ryzyko wystąpienia zakrzepicy lub zespołu DIC po podaniu leku Novoseven®. Podobna sytuacja może wystąpić u pacjentów z zaawansowanym miażdżycą, zespołem zgniotu, sepsą lub zespołem DIC.
Ze względu na ryzyko powikłań zakrzepowo-zatorowych, lek Novoseven® należy stosować z ostrożnością u pacjentów z wywiadem choroby niedokrwiennej serca, u chorych z chorobami wątroby, po zabiegach operacyjnych, u kobiet w ciąży lub tuż przed porodem, u noworodków oraz u pacjentów z ryzykiem zdarzeń zakrzepowo-zatorowych lub zespołem DIC. W każdym z tych przypadków potencjalna korzyść z leczenia lekiem Novoseven® powinna być porównywana z ryzykiem tych powikłań.
Wiadomo, że ciężkie krwawienie poporodowe oraz ciąża, a także stany kliniczne (poród, ciężkie krwawienie, przetaczanie krwi, zespół DIC, zabiegi chirurgiczne/inwazyjne procedury oraz zaburzenia krzepnięcia) mogą zwiększać ryzyko zdarzeń zakrzepowo-zatorowych, w szczególności ryzyko zatorowości żył, co może być związane z podaniem Novoseven® (patrz sekcja „Działania niepożądane”).
Ponieważ lek rekombinowanego czynnika krzepnięcia krwi VIIa Novoseven® może zawierać śladowe ilości immunoglobulin G myszy i krów, a także innych białek pochodzących z hodowli (białka surowicy chomika i krów), u pacjentów otrzymujących ten lek może w późniejszym czasie rozwinąć się podwyższona wrażliwość na te białka. W takich przypadkach należy rozważyć stosowność leczenia przeciwhistaminowego dożylnego.
W przypadku wystąpienia reakcji alergicznych lub anafilaktycznych należy natychmiast przerwać stosowanie leku. W przypadku rozwoju wstrząsu należy zastosować standardowe leczenie wspierające. Pacjenta należy poinformować o wczesnych objawach reakcji nadwrażliwości. W przypadku wystąpienia takich objawów pacjent powinien natychmiast przerwać stosowanie leku i skontaktować się z lekarzem.
W przypadku ciężkich krwawień lek ten należy podawać w placówkach medycznych specjalizujących się w leczeniu pacjentów z hemofilią z obecnością inhibitorów czynnika VIII lub IX, a jeśli taka możliwość nie istnieje – we współpracy z lekarzem specjalistą zajmującym się leczeniem hemofilii.
Jeśli nie uda się zatrzymać krwawienia, pacjent musi zostać hospitalizowany. Pacjenci lub osoby, które im pomagały, powinni jak najszybciej poinformować personel medyczny o wszystkich wstrzyknięciach leku Novoseven®. U pacjentów z niedoborem czynnika VII należy oznaczyć czas protrombinowy oraz aktywność czynnika VII krzepnięcia przed i po podaniu leku Novoseven®. Jeśli aktywność czynnika VIIa nie osiąga oczekiwanego poziomu lub krwawienie nie ustępuje mimo podania zalecanych dawek, należy podejrzewać powstanie przeciwciał. W takich przypadkach konieczne jest oznaczenie ich stężenia.
Zgłaszano przypadki zakrzepicy u pacjentów z niedoborem czynnika VII, którzy otrzymywali lek Novoseven® w trakcie zabiegów chirurgicznych, jednakże stopień ryzyka wystąpienia tego zjawiska nie został ustalony (patrz sekcja „Farmakodynamika”).
Zawartość sodu
Novoseven® zawiera mniej niż 1 mmol sodu (23 mg), dlatego lek ten można uznać za preparat wolny od sodu.
Stosowanie w czasie ciąży lub karmienia piersią.
Ciąża
Jako środek ostrożności zaleca się unikanie stosowania leku Novoseven® w czasie ciąży. Ograniczone dane dotyczące stosowania leku w czasie ciąży w zatwierdzonych wskazaniach nie wskazują na szkodliwy wpływ rFVIIa na przebieg ciąży ani na zdrowie płodu/noworodka. Obecnie nie ma innych odpowiednich danych epidemiologicznych. Badania na zwierzętach nie wykazały bezpośredniego ani pośredniego negatywnego wpływu na ciążę, rozwój embrionalny/plodowy, poród czy rozwój poporodowy.
Karmienie piersią
Nie wiadomo, czy rFVIIa przechodzi do mleka matki. Wydalenie rFVIIa z mlekiem u zwierząt nie było badane. Decyzję o kontynuowaniu karmienia piersią lub o leczeniu lekiem Novoseven® należy podjąć, biorąc pod uwagę korzyści z karmienia piersią dla dziecka oraz korzyści z terapii lekiem Novoseven® dla kobiety.
Plodność
Dane badań przedklinicznych oraz dane z okresu po wprowadzeniu na rynek nie wskazują na negatywny wpływ rFVIIa na płodność mężczyzn ani kobiet.
Wpływ na zdolność prowadzenia pojazdów i obsługiwanie maszyn.
Nie przeprowadzono badań wpływu leku na zdolność do prowadzenia pojazdów i obsługiwanie maszyn.
Sposób stosowania i dawki.
Leczenie należy rozpoczynać pod nadzorem lekarza posiadającego doświadczenie w leczeniu hemofilii i/lub krwawień.
Leczenie ciężkich krwawień połogowych należy prowadzić zespołowo. Oprócz położnych, w zespół wchodzą również anestezjolodzy, intensywiści i/lub hematolodzy. Należy kontynuować stosowanie standardowych metod leczenia zgodnie z indywidualnymi potrzebami pacjenta. Zaleca się utrzymywanie odpowiedniej stężenia fibrynogenu i liczby płytek krwi w celu optymalizacji korzyści z leczenia lekiem Novoseven®.
Reżim dawkowania
Hemofilia A lub B z obecnością inhibitorów lub nabyta hemofilia
Lek Novoseven® należy podawać jak najszybciej po wystąpieniu krwawienia. Początkową zalecaną dawkę podaje się dożylnie (bolusowo) w dawce 90 µg (4,5 JME) na 1 kg masy ciała.
Po podaniu dawki początkowej może pojawić się potrzeba podania dawek powtórnego. Czas trwania leczenia oraz odstępy między dawkami zależą od nasilenia krwawienia, rodzaju procedury inwazyjnej lub zabiegu operacyjnego.
Istniejące doświadczenie kliniczne wskazuje na brak ogólnie istotnych różnic w dawkowaniu u dorosłych i dzieci, choć dzieci charakteryzują się szybszym kliremsem niż dorośli. Z tego powodu dzieci mogą wymagać wyższych dawek rFVIIa w celu osiągnięcia stężenia w osoczu podobnego do stężenia u dorosłych pacjentów (patrz punkt „Farmakokinetyka”).
Odstępy między dawkami
Początkowo, w celu osiągnięcia hemostazy, lek podaje się powtórnie co 2–3 godziny.
W razie potrzeby kontynuowania leczenia po osiągnięciu skutecznej hemostazy, dawkowanie powtarza się co 4, 6, 8 lub 12 godzin, tak długo, jak będzie to konieczne do wyleczenia.
Łagodne lub umiarkowane krwawienia (w tym leczenie ambulatoryjne)
W warunkach ambulatoryjnych wczesne podanie leku okazuje się skuteczne w leczeniu słabych lub umiarkowanych krwawień do stawów, mięśni, skóry i błon śluzowych. Można zalecić dwie schematy podawania leku:
- podawanie od dwóch do trzech dawek leku w dawce 90 µg/kg masy ciała co 3 godziny, a następnie jedna dawka w celu utrzymania hemostazy;
2) jednorazowe podanie leku w dawce 270 µg/kg masy ciała.
Czas trwania leczenia ambulatoryjnego nie powinien przekraczać 24 godzin. Kontynuowanie leczenia w domu może być rozważane jedynie po konsultacji z ośrodkiem leczenia hemofilii.
Brak doświadczenia klinicznego w stosowaniu jednorazowej dawki 270 µg/kg masy ciała u pacjentów w podeszłym wieku.
Ciężkie krwawienia
Zaleca się podanie dawki początkowej 90 µg/kg masy ciała w trakcie transportu pacjenta do szpitala, w którym zwykle jest leczony. Wielkość kolejnych dawek zależy od rodzaju i nasilenia krwawienia. Początkowo lek podaje się co drugą godzinę do osiągnięcia poprawy stanu klinicznego pacjenta. W razie potrzeby kontynuowania leczenia odstęp między dawkami wydłuża się do 3 godzin przez 1–2 doby. Następnie w kolejnym okresie leczenia odstępy między dawkami wydłuża się sukcesywnie do 4, 6, 8 lub 12 godzin. Ciężkie krwawienia czasem wymagają leczenia przez 2–3 tygodnie lub dłużej (w zależności od stanu klinicznego pacjenta).
Procedury inwazyjne/zabiegi operacyjne
Dawkę początkową 90 µg/kg masy ciała podaje się bezpośrednio przed zabiegiem. Dawkę tę powtarza się po 2 godzinach, a następnie w ciągu pierwszych 24–48 godzin – co 2–3 godziny (w zależności od zakresu zabiegu i stanu klinicznego pacjenta). W przypadku dużych zabiegów chirurgicznych lek podaje się co 2–4 godziny przez 6–7 dni. Następnie przez 2 tygodnie odstęp między dawkami wydłuża się do 6–8 godzin. Pacjentom, którzy przeszli duże zabiegi operacyjne, lek stosuje się przez 2–3 tygodnie do czasu gojenia się rany.
Nabyta hemofilia
Dawka i odstępy między dawkami
Novoseven® należy podawać jak najszybciej po wystąpieniu krwawienia. Początkową zalecaną dawkę podaje się dożylnie (bolusowo) w dawce 90 µg (4,5 JME) na 1 kg masy ciała.
Po podaniu dawki początkowej może pojawić się potrzeba podania dawek powtórnego. Czas trwania leczenia oraz odstępy między dawkami zależą od nasilenia krwawienia, rodzaju procedury inwazyjnej lub zabiegu operacyjnego.
Początkowo lek podaje się powtórnie co 2–3 godziny. W razie potrzeby kontynuowania leczenia po osiągnięciu skutecznej hemostazy, dawkowanie powtarza się co 4, 6, 8 lub 12 godzin, tak długo, jak będzie to konieczne do wyleczenia.
Deficyt czynnika VII
Zakres dawek i odstępy między dawkami
Zalecany zakres dawek dla dzieci i dorosłych w leczeniu krwawień i ich zapobieganiu u pacjentów, u których przeprowadzane będą zabiegi operacyjne lub procedury inwazyjne, wynosi 15–30 µg/kg masy ciała co 4–6 godzin do osiągnięcia hemostazy. Dawkę i odstęp podawania dobiera się indywidualnie.
Dzieci
Połączono wyniki ograniczonego doświadczenia klinicznego z długotrwałej profilaktyki u dzieci w wieku do 12 lat z ciężkim fenotypem klinicznym (patrz sekcja „Farmakodynamika”).
Dawkę i częstotliwość wstrzykiwań w profilaktyce należy dobierać na podstawie odpowiedzi klinicznej na leczenie i indywidualnie.
Trombastenia Glanzmanna
Zakres dawek i odstępy między dawkami
Zalecana dawka w leczeniu krwawień i zapobieganiu im u pacjentów, u których przeprowadzane będą zabiegi operacyjne lub procedury inwazyjne, wynosi 90 µg (zakres 80–120 µg)/kg masy ciała co 2 godziny (1,5–2,5 godziny). W celu utrzymania skutecznej hemostazy należy podać co najmniej 3 dawki. Zaleca się stosowanie iniekcji bolusowych, ponieważ powolne wlewanie może okazać się nieskuteczne. W leczeniu trombastenii Glanzmanna u pacjentów bez oporności należy przede wszystkim podawać płytki krwi.
Ciężkie krwawienie połogowe
Zakres dawek i odstęp dawkowania
Zalecany zakres dawek w leczeniu krwawienia wynosi 60–90 µg na 1 kg masy ciała z dożylnym podaniem bolusowym. Szczytowa aktywność krzepnięcia osiągana jest około 10 minut po podaniu. Drugą dawkę można podać w zależności od klinicznej reakcji konkretnego pacjenta.
W przypadku niewystarczającej odpowiedzi hemostatycznej zaleca się podanie drugiej dawki po 30 minutach.
Sposób stosowania
Wskazówki dotyczące rozcieńczania leku przed podaniem znajdują się na odwrocie instrukcji do stosowania leku Novoseven®. Roztwór podaje się w formie dożylnej iniekcji bolusowej w ciągu 2–5 minut.
Monitorowanie leczenia — testy laboratoryjne
Monitorowanie leczenia nie jest obowiązkowe, ponieważ nasilenie krwawienia i odpowiedź kliniczna na podanie leku Novoseven® decydują o konieczności dalszego stosowania leku.
Po podaniu leku Novoseven® obserwuje się skrócenie czasu protrombinowego i aktywowanego częściowego czasu tromboplastynowego, jednak nie stwierdzono korelacji tych parametrów z kliniczną skutecznością leku.
Zalecenia dotyczące postępowania z lekiem i jego utylizacji
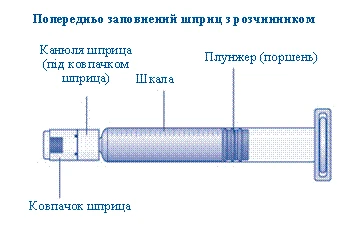
Roztwórnik do leku Novoseven® dostarczany jest w szprycie wstępnie napełnionej.
Proszek w fiolce i rozcieńczalnik w szprycie wstępnie napełnionej
Należy zawsze przestrzegać procedury aseptycznej.
Odzyskiwanie
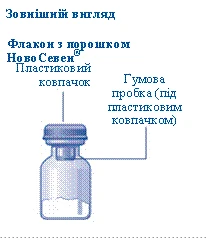
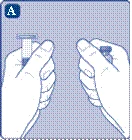
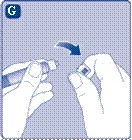
- Podczas przygotowywania fiolki z proszkiem Novoseven® i szprycy wstępnie napełnionej rozcieńczalnikiem powinny mieć temperaturę pokojową. Zdejmij plastikowy kapturzek z fiolki. Nie należy używać fiolki, jeśli kapturzek jest luźno zamknięty lub brakuje go. Przetrzyj gumową przeciwnicę fiolki sterylną serwetką alkoholową i pozostaw przeciwnicę do wyschnięcia na kilka sekund przed użyciem. Nie dotykaj gumowej przeciwnicy po przetrzeciu.
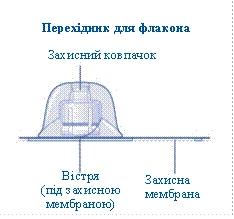
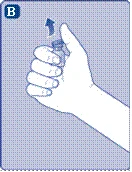
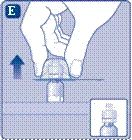
- Zdejmij ochronną membranę z przejściówki do fiolki. Nie wyjmuj przejściówki z ochronnego kapturka. Nie należy używać przejściówki do fiolki, jeśli ochronna membrana jest niepełnie uszczelniona lub uszkodzona. Odkręć ochronny kapturzek i przyłącz przejściówkę do fiolki. Delikatnie naciśnij ochronny kapturzek kciukiem i palcem wskazującym. Zdejmij ochronny kapturzek z przejściówki do fiolki.
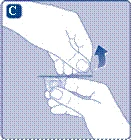
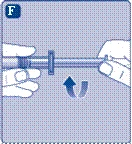
- Wkręć tłoczek strzykawki do tłoczka wewnątrz szprycy wstępnie napełnionej zgodnie z ruchem wskazówek zegara aż do oporu. Zdejmij nakrywak z szprycy, delikatnie nim potrząsając, aż do złamania perforacji. Nie dotykaj końcówki szprycy pod nakrywkiem. Nie należy używać szprycy wstępnie napełnionej, jeśli nakrywak nie przylega szczelnie do szprycy lub brakuje go.
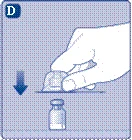
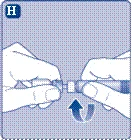
- Przyłącz szprycę wstępnie napełnioną rozcieńczalnikiem do przejściówki do fiolki, obracając szprycę zgodnie z ruchem wskazówek zegara aż do oporu. Trzymaj szprycę lekko przechyloną, fiolką do dołu. Naciśnij tłoczek, aby wprowadzić cały rozcieńczalnik do fiolki. Trzymaj tłoczek naciśnięty i delikatnie obracaj fiolkę do całkowitego rozpuszczenia proszku. Nie wstrząsaj fiolką, ponieważ może to prowadzić do powstawania piany.
Jeśli wymagana jest większa dawka, należy powtórzyć tę procedurę z dodatkowymi fiolkami, wstępnie napełnionymi szprycami i przejściówkami do fiolki.
Odzyskany roztwór leku Novoseven® jest bezbarwny. Przed zastosowaniem roztwór należy wizualnie sprawdzić pod kątem obecności zanieczyszczeń i zmiany barwy.
Zaleca się stosowanie leku Novoseven® bezpośrednio po odzyskaniu. Warunki przechowywania rozcieńczonego leku podano w sekcji „Okres ważności”.
Zastosowanie
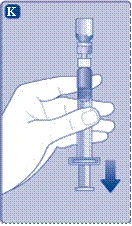
- Trzymaj tłoczek całkowicie wciskany do cylindra szprycy. Odwróć szprycę z fiolką do góry nogami. Przestań naciskać tłoczek i odczekaj, aż rozcieńczony roztwór wypełni szprycę. Delikatnie pociągnij tłoczek w dół, aby cały roztwór został wciągnięty do szprycy.
- Trzymając fiolkę do góry nogami, delikatnie postukaj po szprycy, aby wszystkie pęcherzyki powietrza uniosły się do góry. Powoli naciskaj tłoczek, aż wszystkie pęcherzyki powietrza znikną.
Jeśli nie jest potrzebna cała dawka, należy użyć skali na szprycy, aby zobaczyć, ile odzyskanego roztworu zostało pobrane.
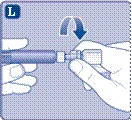
- Odejmij szprycę od przejściówki do fiolki, obracając ją przeciwnie do ruchu wskazówek zegara.
- Lek Novoseven® jest gotowy do wstrzyknięcia. Wybierz miejsce wstrzyknięcia i powoli wprowadź lek Novoseven® do żyły w ciągu 2–5 minut, nie wyciągając igły z miejsca wstrzyknięcia.
Utylizuj używane materiały zgodnie z niezbędnymi środkami ostrożności. Wszystkie nieużywane leki i odpady należy utylizować zgodnie z lokalnymi wymogami.
Procedura łączenia fiolki (możliwa tylko w warunkach szpitalnych)
W badaniach in vitro stwierdzono, że lek pozostaje chemicznie i fizycznie stabilny, gdy przechowywany jest w strzykawce polipropylenowej o pojemności 50 ml przez 24 godziny w temperaturze 25 °C. Kompatybilność z lekiem wykazano dla systemu składającego się ze strzykawki polipropylenowej o pojemności 50 ml, rurki infuzyjnej (polietylen) długości 2 m i wbudowanego filtra o wielkości porów 5 µm.
Łączenie fiolki (tylko w warunkach szpitalnych)
- Wszystkie etapy powinien wykonywać wykwalifikowany personel w kontrolowanych i certyfikowanych warunkach aseptycznych.
- Jeśli odzyskiwanie, mieszanie lub stosowanie leku przez użytkownika nie odbywa się zgodnie z instrukcją do stosowania leku, odpowiedzialność za termin i warunki przechowywania ponosi użytkownik.
- Upewnij się, że używana jest przejściówka do fiolki.
- Wykonaj odzyskiwanie leku, jak opisano powyżej w sekcji „Odzyskiwanie”. Odejmij pustą strzykawkę od przejściówki do fiolki, obracając ją przeciwnie do ruchu wskazówek zegara, i upewnij się, że przejściówka jest przyłączona do fiolki zawierającej rozcieńczony lek.
- Powtórz tę procedurę z niezbędną liczbą dodatkowych fiol, fiol z rozcieńczalnikiem / wstępnie napełnionych strzykawek i przejściówek do fiol.
- Wstrzyknij około 5 ml sterylnego powietrza do strzykawki polipropylenowej o pojemności 50 ml. Przyłącz strzykawkę do przejściówki do fiolki, obracając strzykawkę zgodnie z ruchem wskazówek zegara aż do oporu. Trzymaj strzykawkę lekko przechyloną, fiolką do dołu. Delikatnie naciśnij tłoczek, aby wprowadzić trochę powietrza do fiolki. Odwróć strzykawkę z fiolką do góry nogami i wciągnij zawartość fiolki do strzykawki. Powtórz powyższą procedurę z innymi fiolkami z odzyskanym lekiem, aby uzyskać wymagany objętość roztworu w strzykawce.
- Do podania leku należy użyć wbudowanego filtra o średnicy porów 5 µm. Przed rozpoczęciem podania upewnij się, że strzykawka, rurka infuzyjna i wbudowany filtr są wypełnione roztworem i nie zawierają pęcherzyków powietrza.
- Strzykawka z odpowiednio odzyskanym lekiem jest gotowa do użycia w pompie infuzyjnej oznaczonej CE (kompatybilnej ze strzykawką o pojemności 50 ml).
- Pompą infuzyjną może posługiwać się wyłącznie wykwalifikowany personel medyczny.
Novoseven®, instrukcja dla użytkownika
Przed zastosowaniem leku Novoseven® należy dokładnie przeczytać niniejszą instrukcję.
Lek Novoseven® dostarczany jest w postaci proszku. Przed wstrzyknięciem (podaniem) należy go rozpuścić rozcieńczalnikiem dostarczanym w strzykawce. Rozcieńczalnikiem jest roztwór histydyny. Odzyskany lek Novoseven® podaje się dożylnie (iniekcja dożylna). Zawartość tego opakowania przeznaczona jest do rozpuszczenia i podania leku Novoseven®.
Do podania dożylnego potrzebne są również zestaw infuzyjny (rurka i igła motylkowa), serwetki alkoholowe, gazy i plaster. Te wyroby medyczne nie są zawarte w opakowaniu leku Novoseven®.
Nie należy używać wyposażenia bez odpowiedniego szkolenia od lekarza lub pielęgniarki.
Przed użyciem należy zawsze umyć ręce i upewnić się, że przestrzeń wokół Ciebie jest czysta.
Podczas przygotowywania i podawania leku bezpośrednio do żyły bardzo ważne jest przestrzeganie zasad aseptyki i antyseptyki. Przy nieprawidłowej technice wykonania wstrzyknięcia możliwe jest zakażenie krwi.
Nie należy otwierać wyposażenia, dopóki nie jesteś gotowy do jego użycia.
Nie należy używać wyposażenia, jeśli upadło lub jest uszkodzone. Weź nowe opakowanie.
Nie należy używać wyposażenia, jeśli upłynął jego termin ważności. Weź nowe opakowanie. Data ważności wydrukowana jest po słowach „Przydatny do” na pudełku kartonowym, fiolce, przejściówce do fiolki i na wstępnie napełnionej strzykawce.
Nie należy używać wyposażenia, jeśli podejrzewasz jego zanieczyszczenie (kontaminację). Weź nowe opakowanie.
Nie wyrzucaj żadnej części zestawu, dopóki nie podasz przygotowanego roztworu.
Wyposażenie przeznaczone jest do jednorazowego użytku.
Zawartość opakowania :
- 1 fiolka z proszkiem Novoseven®
- 1 przejściówka do fiolki
- 1 wstępnie napełniona strzykawka z rozcieńczalnikiem
- 1 tłoczek strzykawki (znajduje się pod strzykawką)

upewnić się, że zawiera ono wymagany lek.
wstępnie wypełnioną strzykawkę z pudełka. Zostaw tłok strzykawki w pudełku.
temperatury pokojowej (nie wyższej niż 37 °C). Możesz to zrobić, trzymając je w dłoniach, aż poczujesz, że są tak samo ciepłe jak Twoje ręce.
|
|
Jeśli plastikowy kapturek luźno się trzyma lub jest uszkodzony, nie używaj fiolki.
Nie dotykaj septum gumowego palcami, ponieważ może to prowadzić do przeniesienia mikroorganizmów. |
|
Jeśli uszczelnienie ochronnej membrany jest naruszone lub membrana jest uszkodzona, nie używaj łącznika do fiolki. Nie wyjmuj łącznika do fiolki z opakowania palcami. Dotknięcie igły łącznika może prowadzić do przeniesienia mikroorganizmów z Twoich palców. |
|
|
|
i palcem wskazującym, jak pokazano na rysunku. Ściągnij ochronny kapturek z łącznika, ale w taki sposób, aby nie wyciągać łącznika z fiolki. |
|
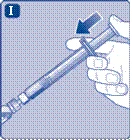
Weź tłok strzykawki za szeroki koniec i wyjmij go z pudełka. Nie dotykaj bocznych powierzchni tłoka ani jego przedniego końca. Dotknięcie bocznych powierzchni tłoka lub jego przedniego końca może prowadzić do przeniesienia mikroorganizmów z Twoich palców. Natychmiast podłącz tłok strzykawki do tłoka wewnątrz wstępnie wypełnionej strzykawki, obracając go zgodnie z ruchem wskazówek zegara do oporu. |
|
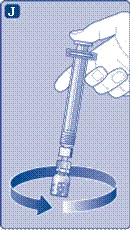
| Usuń kapturek strzykawki z wstępnie wypełnionej strzykawki, zginając go w dół, aż odłamie się wzdłuż linii perforacji. Nie dotykaj kanüli strzykawki pod kapturkiem. Jeśli dotkniesz kanüli strzykawki, może to prowadzić do przeniesienia mikroorganizmów z Twoich palców. Jeśli kapturek strzykawki luźno się trzyma lub jest uszkodzony, nie używaj tej wstępnie wypełnionej strzykawki. |
|
|
|
|
|
Nie wstrząsaj fiolki – spowoduje to powstawanie piany.
|
|
| Użyj przygotowanego roztworu leku Novoseven® natychmiast, aby uniknąć jego zakażenia. Jeśli nie użyjesz roztworu natychmiast, zobacz sekcję „Warunki przechowywania” na odwrocie ulotki. Nie przechowuj przygotowanego roztworu bez konsultacji z lekarzem lub pielęgniarką. Jeśli wymagana dawka jest większa niż 1 fiolka, powtórz kroki od 1 do 4 (rys. A–J) z dodatkowymi fiolkami, łącznikami do fiolki i wstępnie wypełnionymi strzykawkami, aż uzyskasz wymaganą dawkę. |
|
|
|
Jeśli dotkniesz kanüli strzykawki, może to prowadzić do przeniesienia mikroorganizmów z Twoich palców. |
|
| Wprowadzanie leku Novoseven® z wstępnie wypełnionej strzykawki przez połączenie beziegowe do cewnika dożylnego. Ostrzeżenie. Wstępnie wypełniona strzykawka wykonana jest ze szkła i jest kompatybilna ze standardowym połączeniem „Luer-lock”. Niektóre połączenia beziegowe z wewnętrzną igłą nie są kompatybilne z wstępnie wypełnioną strzykawką. Ta niezgodność może prowadzić do niemożności podania leku i/lub uszkodzenia połączenia beziegowego. Należy przestrzegać instrukcji obsługi połączeń beziegowych. Podczas podawania leku za pomocą połączeń beziegowych może być konieczne przetransferowanie przygotowanego roztworu do standardowej jednorazowej plastikowej strzykawki „Luer-lock” o pojemności 10 ml. Należy to zrobić natychmiast po kroku 4 (rys. J). |
|
Teraz lek Novoseven® jest gotowy do podania w formie wstrzyknięcia dożylnego.
Wstrzyknięcie roztworu przez centralny cewnik żylny lub port podskórny:
|
|
| Utylizacja
|
|
| Nie rozbieraj sprzętu przed utylizacją. Nie używaj ponownie sprzętu. |
|
Dzieci.
Lek stosuje się u dzieci (patrz sekcja „Sposób stosowania i dawki”).
Przedawkowanie .
Toksykologia dawek granicznych leku Novoseven® nie była badana w trakcie badań klinicznych.
Zgłoszono 4 przypadki przedawkowania u pacjentów z hemofilią w ciągu ostatnich 16 lat. Jedynym powikłaniem zgłoszonym w związku z przedawkowaniem było nieznaczne, przejściowe podwyższenie ciśnienia tętniczego u 16-letniego pacjenta, któremu podano 24 mg rFVIIa zamiast 5,5 mg.
Nie zgłaszano przypadków przedawkowania u pacjentów z nabytą hemofilią ani z trombastenią Glanzmanna.
U pacjentów z niedoborem czynnika VII, przy zalecanej dawce 15–30 μg/kg rFVIIa, jeden przypadek przedawkowania był związany z zjawiskiem zakrzepowym (udar okcyputalny) u starszego pacjenta płci męskiej (> 80 lat), u którego dawka przekroczyła zalecaną dawkę
10–20-krotnie. Ponadto u jednego pacjenta z niedoborem czynnika VII powstawanie przeciwciał wobec leku Novoseven® oraz FVII było powiązane z przedawkowaniem.
Stosowane dawki nie powinny przekraczać zalecanych ze względu na brak informacji o możliwym dodatkowym ryzyku.
Działania niepożądane.
Najczęściej zgłaszane działania niepożądane to: zmniejszenie odpowiedzi terapeutycznej, gorączka, wysypka, zdarzenia zakrzepowo-zatorowe żylne, świąd i pokrzywka. Reakcje te występowały rzadko (≥ 1/1000, < 1/100).
Poniżej wymieniono działania niepożądane zgłaszane podczas badań klinicznych i w okresie posprzedażowym. W każdej grupie działania niepożądane są uporządkowane według malejącej ciężkości. Częstość działań niepożądanych zgłaszanych wyłącznie w okresie posprzedażowym (nie podczas badań klinicznych) jest nieznana.
W badaniach klinicznych, w których wzięło udział 484 pacjentów (4297 epizodów leczenia) z hemofilią A i B, hemofilią nabytą, niedoborem czynnika VII lub trombastenią Glanzmanna, wykazano, że działania niepożądane związane z lekiem są częste (≥ 1/100 do <1/10). Ponieważ całkowita liczba epizodów leczenia w ramach badań klinicznych nie przekracza 10 000, najmniejsza możliwa częstość występowania działań niepożądanych, którą można wyodrębnić, to rzadko (≥ 1/10 000 do < 1/1000).
Najczęstsze działania niepożądane to gorączka i wysypka (nieczęsto: ≥ 1/1000 do <1/100), natomiast najpoważniejszymi działaniami niepożądanymi są zdarzenia zakrzepowo-zatorowe żylne (nieczęsto: ≥ 1/1000 do < 1/100) oraz zdarzenia zakrzepowo-zatorowe tęgowe (rzadko: ≥ 1/10000 do < 1/1000).
Zaburzenia układu krwiotwórczego i chłonnego
Rzadko (≥ 1/10000, < 1/1000) – rozsiane wewnątrzkrwotoczne krzepnięcie (DIC) i związane z nim odchylenia laboratoryjne, w tym podwyższenie poziomu D-dimeru i obniżenie poziomu AT-II (patrz dział „Szczególne ostrzeżenia i środki ostrożności postępowania”), koagulopatia.
Zaburzenia ze strony przewodu pokarmowego
Rzadko (≥ 1/10000, < 1/1000) – nudności.
Zaburzenia ogólne i stan w miejscu podania
Nieczeście (≥ 1/1000, < 1/100) – zmniejszenie odpowiedzi terapeutycznej, gorączka.
Rzadko (≥ 1/10000, < 1/1000) – reakcje w miejscu iniekcji, w tym ból w miejscu iniekcji.
Zaburzenia ze strony układu odpornościowego
Rzadko (≥ 1/10000, < 1/1000) – nadwrażliwość (patrz działy „Przeciwwskazania” i „Szczególne ostrzeżenia i środki ostrożności postępowania”).
Częstość nieznana – reakcje anafilaktyczne.
Badania laboratoryjne
Rzadko (≥ 1/10000, < 1/1000) – podwyższenie poziomu produktów rozpadu fibryny, podwyższenie poziomu alaninotransaminazy, fosfatazy zasadowej, dehydrogenazy mleczanowej i protrombiny.
Zaburzenia ze strony układu nerwowego
Rzadko (≥ 1/10000, < 1/1000) – ból głowy.
Zaburzenia ze strony skóry i tkanki podskórnej
Nieczeście (≥ 1/1000, < 1/100) – wysypka (w tym zapalenie skóry alergiczne i wysypki rumieniowe), świąd i pokrzywka.
Częstość nieznana – napady gorąca, obrzęk naczynioruchowy.
Zaburzenia ze strony układu naczyniowego
Nieczeście (≥ 1/1000, < 1/100) – zdarzenia zakrzepowo-zatorowe żylne (tromboza żył głębokich, tromboza w miejscu podania dożylnego, zatorowość płucna, zdarzenia zakrzepowo-zatorowe wątrobowe, w tym tromboza żyły wrotnej, tromboza żyły nerkowej, tromboflebita, tromboflebita żył powierzchownych oraz niedokrwienie jelit).
Rzadko (≥ 1/10000, < 1/1000) – zdarzenia zakrzepowo-zatorowe tęgowe (zawał mięśnia sercowego, udar niedokrwienny mózgu, niedokrwienie mózgu, okluzywna choroba naczyń mózgowych, udar, tromboza tętnicy nerkowej, niedokrwienie naczyń obwodowych, obwodowy zakrzepowy zator i niedokrwienie jelit), dławica piersiowa.
Częstość nieznana – tromboza jam serca.
Zgłaszano niewystarczającą skuteczność (zmniejszenie odpowiedzi terapeutycznej). Bardzo ważne jest, aby dawkowanie leku Novoseven® odpowiadało zaleceniom podanym w dziale „Sposób podania i dawka”.
Opis wybranych działań niepożądanych
Wytwarzanie przeciwciał inhibitorowych
W okresie posprzedażowym nie zgłaszano wykrycia przeciwciał inhibitorowych wobec leku Novoseven® ani FVII u pacjentów z hemofilią A lub B. Jednakże przypadki powstawania przeciwciał inhibitorowych wobec leku Novoseven® zgłaszano w obserwacyjnym rejestrze posprzedażowym u pacjentów z wrodzonym niedoborem czynnika VII.
W badaniach klinicznych z udziałem pacjentów z niedoborem czynnika VII, powstawanie przeciwciał wobec leku Novoseven® i czynnika VII (często: ≥ 1/100, < 1/10) było jedynym działaniem niepożądanym, o którym zgłaszano. W niektórych przypadkach przeciwciała powodowały efekt inhibitorowy in vitro. Czynniki ryzyka powstawania przeciwciał obejmują: wcześniejsze leczenie osoczem ludzkim i/lub wyizolowanym czynnikiem VII, ciężkie mutacje genu czynnika VII oraz przedawkowanie leku Novoseven®. U pacjentów z niedoborem czynnika VII leczonych lekiem Novoseven® należy stale monitorować obecność przeciwciał wobec czynnika VII (patrz dział „Szczególne ostrzeżenia i środki ostrożności postępowania”).
Zdarzenia zakrzepowo-zatorowe: tęgowe i żylne
Podczas stosowania leku Novoseven® w wskazaniach niezatwierdzonych, zdarzenia zakrzepowo-zatorowe tęgowe są częste (≥ 1/100, < 1/10). W analizie meta badania z randomizacją kontrolowaną placebo, wykazano wyższe ryzyko zdarzeń zakrzepowo-zatorowych tęgowych (patrz wyżej „Zaburzenia ze strony układu naczyniowego”) (5,6 % u pacjentów leczonych lekiem Novoseven® w porównaniu z 3,0 % u pacjentów leczonych placebo) podczas stosowania leku Novoseven® w wskazaniach niezatwierdzonych.
Zdarzenia zakrzepowo-zatorowe mogą prowadzić do zatrzymania krążenia.
Osobliwe grupy pacjentów
Pacjenci z hemofilią nabytą.
W badaniach klinicznych z udziałem 61 pacjentów z hemofilią nabytą (w tym 100 epizodów leczenia), najczęściej występujące działania niepożądane (1 % wszystkich epizodów leczenia) to: zdarzenia zakrzepowo-zatorowe tęgowe (okluzyjna choroba naczyń mózgowych, udar), zdarzenia zakrzepowo-zatorowe żylne (zatorowość płucna i tromboza żył głębokich), dławica piersiowa, nudności, gorączka, wysypki rumieniowe, podwyższenie poziomu produktów rozpadu fibryny.
Kobiety z ciężką krwawicą poporodową
W otwartym, randomizowanym badaniu klinicznym, zdarzenia zakrzepowo-zatorowe żylne zgłoszono u 2 z 51 pacjentek, które otrzymywały pojedynczą dawkę leku Novoseven® (średnia dawka 58 μg/kg); zdarzenia te nie wystąpiły u żadnej z 33 pacjentek, które nie otrzymywały leku Novoseven®; zdarzenia zakrzepowo-zatorowe tęgowe nie wystąpiły w żadnej z grup.
W 4 badaniach nieinterwencyjnych, zdarzenia zakrzepowo-zatorowe żylne wystąpiły u 3 z 358 pacjentek (0,8 %), które otrzymywały lek Novoseven® (średni zakres dawek 63–105 μg/kg), a zdarzenia zakrzepowo-zatorowe tęgowe wystąpiły u 1 pacjentki (0,3 %), która otrzymywała lek Novoseven®.
Informacje dotyczące znanych czynników zwiększających ryzyko zakrzepowo-zatorowe związane z ciążą i ciężką krwawicą poporodową, znajdują się w dziale „Szczególne ostrzeżenia i środki ostrożności postępowania”.
Zgłaszanie podejrzewanych działań niepożądanych i braku skuteczności leku
Zgłaszanie działań niepożądanych po rejestracji leku ma istotne znaczenie. Umożliwia to monitorowanie stosunku korzyści do ryzyka stosowania tego leku. Osoby medyczne i farmaceutyczne, a także pacjenci lub ich ustawowe przedstawiciele, powinni zgłaszać wszystkie przypadki podejrzewanych działań niepożądanych i braku skuteczności leku poprzez Automatyczny System Informacyjny nadzoru farmakologicznego pod adresem https://aisf.dec.gov.ua/.
Okres ważności. Okres ważności leku opakowanego do sprzedaży wynosi 3 lata, pod warunkiem przechowywania w temperaturze poniżej 25 °C.
W fiolce
Wykazano chemiczną i fizyczną stabilność rozcieńczonego leku przez 6 godzin w temperaturze 25 °C lub 24 godziny w temperaturze 5 °C.
Z mikrobiologicznego punktu widzenia, przygotowany roztwór należy użyć natychmiast. O warunki i okres przechowywania odpowiada użytkownik. Jeżeli rozcieńczenie nie było przeprowadzone w kontrolowanych i walidowanych warunkach bezpylnych, okres przechowywania rozcieńczonego leku nie powinien przekraczać 24 godzin w temperaturze 2–8 °C. Odtworzony roztwór należy przechowywać w fiolce.
W strzykawce (polipropylenowej strzykawce 50 ml) wyłącznie w warunkach szpitalnych
Rozcieńczenie powinno być przeprowadzane przez wykwalifikowany personel w kontrolowanych i certyfikowanych warunkach bezpylnych. Lek pozostaje chemicznie i fizycznie stabilny w następujących warunkach: przechowywanie w polipropylenowej strzykawce o pojemności 50 ml przez 24 godziny w temperaturze 25 °C. Jeżeli lek nie jest stosowany natychmiast, odpowiedzialność za przestrzeganienie warunków przechowywania ponosi użytkownik. Okres przechowywania leku po otwarciu opakowania nie powinien przekraczać 24 godzin.
Warunki przechowywania.
Przechowywać w miejscu niedostępnym dla dzieci, zabezpieczonym przed światłem, w temperaturze nie wyższej niż 25 °C.
Nie zamrażać.
Warunki przechowywania rozcieńczonego leku patrz w dziale „Okres ważności”.
Niezgodność. Novoseven® nie można mieszać z roztworami do wlewu ani podawać kroplowo.
Opakowanie.
1 kartonowa puszka zawiera 1 fiolkę szklaną o pojemności 2 ml lub 12 ml z białym proszkiem do sporządzenia roztworu do wstrzykiwań, zamkniętą chlorkowo-butylową korką gumową i uszczelnioną aluminiową pokrywką z ochronną wyjmowaną pokrywką z polipropylenu, oraz 1 wstępnie wypełnioną strzykawkę szklaną typu 1 o pojemności 3 ml lub 10 ml, zawierającą 2 ml lub 5 ml rozpuszczalnika (histydyna, woda do wstrzykiwań).
Strzykawka jest zamknięta z jednej strony korkiem gumowym z bromku butylowego i polipropylenu, a z drugiej – tłokiem z gumy bromku butylowego i zatyczką z polipropylenu. Do strzykawki dołączony jest również tłok z polipropylenu. W zestawie znajduje się również przejściówka do fiolki z filtrem oczyszczającym o wielkości porów 25 μm w opakowaniu indywidualnym.
Kategoria wydania. Na receptę.
Producent/Wnioskodawca.
A/T Novo Nordisk / Novo Nordisk A/S.
Miejsce produkcji i jego adres.
Novo Allé, Bagsværd, 2880, Dania / Novo Allé, Bagsværd, 2880, Denmark.