Novoseven®
Ucraina
Indice
ISTRUZIONI PER L'USO MEDICINALE DEL FARMACO NOVOSEVEN® (NOVOSEVEN®)
Composizione:
sostanza attiva: eptacog alfa (attivato) (rFVIIa);
1 flaconcino contiene 2 mg (100 UI) oppure 5 mg (250 UI) di eptacog alfa (attivato);
sostanze ausiliarie: sodio cloruro; calcio cloride diidrato; glicilglicina; polisorbato 80; metionina; saccarosio; mannitolo (E421).
Dopo ricostituzione, il prodotto contiene 1 mg/ml di eptacog alfa (attivato) dopo ricostituzione con il solvente.
Solvente: istidina, acqua per preparazioni iniettabili.
Forma farmaceutica. Polvere liofilizzata per soluzione iniettabile.
Principali caratteristiche fisico-chimiche: polvere liofilizzata di colore bianco.
Gruppo farmacoterapeutico. Agenti emostatici. Fattori della coagulazione.
Codice ATC B02BD08.
Proprietà farmacologiche.
Farmacodinamica.
Il prodotto Novoseven® contiene il fattore ricombinante VII attivato, con un peso molecolare di circa 50.000 Dalton, prodotto mediante tecnologia del DNA ricombinante utilizzando cellule renali di criceto neonato (cellule BHK) come cellule ospiti.
Meccanismo d'azione.
Il meccanismo d'azione consiste nel legame del fattore VIIa con il fattore tissutale. Questo complesso attiva i fattori IX e X in forma attiva – IXa e Xa, determinando la trasformazione di piccole quantità di protrombina in trombina. La trombina, a livello del sito di lesione, attiva le piastrine e i fattori V e VIII, determinando la trasformazione del fibrinogeno in fibrina e la formazione del coagulo emostatico. Il prodotto Novoseven®, alle dosi farmacologiche, indipendentemente dal fattore tissutare, attiva direttamente il fattore X sulla superficie delle piastrine attivate presenti nella zona di danno. Ciò determina la trasformazione di una grande quantità di protrombina in trombina, senza il coinvolgimento del fattore tissutale.
Effetto farmacodinamico.
Di conseguenza, l'effetto farmacodinamico del fattore VIIa consiste nell'aumento locale della formazione del fattore Xa, della trombina e della fibrina.
Il tempo per raggiungere la massima attività coagulante dopo somministrazione di Novoseven® è stato di circa 10 minuti in volontari sani e in pazienti con emofilia.
Teoricamente, non si può escludere completamente un'attivazione generalizzata del sistema di coagulazione ematica in pazienti affetti da patologie predisponenti alla coagulazione intravascolare disseminata (CID).
Efficacia e sicurezza clinica
Deficit congenito di FVII
Nel registro osservazionale (F7HAEM-3578), la dose media per la profilassi a lungo termine delle emorragie in 22 bambini (età fino a 12 anni) con deficit del fattore VII e fenotipo clinico grave è stata di 30 µg/kg (intervallo da 17 µg/kg a 200 µg/kg; nei 10 pazienti, la dose più frequentemente utilizzata è stata di 30 µg/kg), con una frequenza media di somministrazione di 3 dosi alla settimana (intervallo da 1 a 7; nei 13 pazienti, la frequenza più riportata è stata di 3 volte alla settimana).
Nello stesso registro, in 3 su 91 pazienti sono stati osservati eventi tromboembolici dopo intervento chirurgico.
Trombostatene di Glanzmann
Il registro osservazionale (F7HAEM-3521) comprende dati di 133 pazienti con trombostatene di Glanzmann trattati con Novoseven®. La dose media per somministrazione per il trattamento di 333 episodi emorragici è stata di 90 µg/kg (intervallo da 28 a 450 µg/kg). Novoseven® è stato utilizzato in 157 interventi chirurgici con una dose media di 92 µg/kg (fino a 270 µg/kg). Il trattamento con Novoseven®, singolarmente o in combinazione con agenti antifibrinolitici e/o piastrine, è stato considerato efficace se l’emorragia era arrestata per almeno 6 ore. Il tasso di efficacia è stato dell’81% e dell’82% rispettivamente nei pazienti con esito positivo o negativo alla resistenza alla trasfusione di piastrine, e del 77% e dell’85% rispettivamente nei pazienti con esito positivo o negativo alla presenza di anticorpi anti-piastrine. Lo stato positivo indica la presenza di almeno un risultato positivo durante la somministrazione del farmaco al paziente.
Emorragia post-partum grave
L’efficacia e la sicurezza di Novoseven® sono state valutate in 84 donne con emorragia post-partum grave in uno studio clinico aperto multicentrico. Le pazienti sono state randomizzate e assegnate in modo casuale a due gruppi: un gruppo ha ricevuto una dose singola di 60 µg/kg di Novoseven® (in aggiunta alla terapia standard; N = 42), mentre l’altro gruppo ha ricevuto terapia comparativa (solo terapia standard; N = 42) dopo l’inefficacia degli agenti uterotropi (sulprostone). I gruppi terapeutici erano ben bilanciati per caratteristiche demografiche e storia di trattamento precedente per emorragia post-partum prima della randomizzazione. La terapia standard includeva fibrinogeno e acido tranexamico. Informazioni sull’uso di fibrinogeno/acido tranexamico erano disponibili per circa il 57% delle pazienti nel gruppo Novoseven® e il 43% nel gruppo di confronto. Di queste, circa il 40% delle pazienti in entrambi i gruppi ha ricevuto fibrinogeno e/o acido tranexamico. L’emorragia è stata considerata arrestata (cioè il trattamento è stato considerato un successo) se entro 30 minuti dalla randomizzazione il volume ematico stimato si riduceva a meno di 50 ml in 10 minuti. Se l’emorragia diventava incontrollabile o non rispondeva al trattamento, si valutava l’opportunità di procedure invasive.
I risultati dell’analisi primaria hanno mostrato che il numero di donne che hanno subito almeno un’embolizzazione e/o legatura nel gruppo Novoseven® era inferiore rispetto al gruppo di confronto (21 contro 35), corrispondente a una riduzione relativa del rischio statisticamente significativa del 40% per il gruppo Novoseven® rispetto al gruppo di controllo (rischio relativo = 0,60 (intervallo di confidenza al 95%: 0,43–0,84, p = 0,0012)).
Nel gruppo di confronto, 8 su 42 pazienti hanno successivamente ricevuto Novoseven® come trattamento per motivi di umanità, nel tentativo di evitare l’isterectomia d’urgenza, con successo in 2 casi.
Farmacocinetica.
Volontari sani
Distribuzione, escrezione e linearità
La farmacocinetica di Novoseven® è stata studiata in 35 volontari sani di razza caucasica e giapponese in uno studio con escalation di dose, utilizzando un saggio di coagulazione per FVII. I partecipanti sono stati stratificati per sesso ed etnia e hanno ricevuto dosi di 40, 80 e 160 µg/kg di Novoseven® e/o placebo (3 dosi). La farmacocinetica non è risultata influenzata dal sesso né dal gruppo etnico. Il volume medio di distribuzione allo stato stazionario è variato da 130 a 165 ml/kg, i valori medi di clearance sono stati compresi tra 33,3 e 37,2 ml/ora × kg. Il periodo terminale medio di emivita è stato compreso tra 3,9 e 6 ore.
I profili farmacocinetici erano proporzionali alla dose.
Emofilia A e B con presenza di inibitori
Distribuzione, eliminazione e linearità
Le proprietà farmacocinetiche di Novoseven® sono state studiate mediante saggio FVII in 12 bambini (2-12 anni) e 5 adulti in assenza di emorragia. Il volume medio di distribuzione allo stato stazionario è stato di 196 ml/kg nei bambini rispetto ai 159 ml/kg negli adulti. La clearance media era circa il 50% più elevata nei bambini rispetto agli adulti (78 vs 53 ml/ora × kg), mentre il periodo terminale medio di emivita è stato di 2,3 ore in entrambi i gruppi. È evidente che la clearance è correlata all’età, pertanto nei pazienti più giovani la clearance può aumentare di oltre il 50%.
La proporzionalità alla dose è stata studiata nei bambini per le dosi indagate di 90 e 180 µg/kg, in base ai dati precedenti ottenuti con dosi più basse (17,5–70 µg/kg di rFVIIa).
Deficit del fattore VII
Distribuzione ed eliminazione
La farmacocinetica della somministrazione singola di Novoseven® alle dosi di 15 e 30 µg/kg non ha mostrato differenze significative nei parametri indipendenti dalla dose: clearance totale – 70,8-79,1 ml/ora × kg, volume di distribuzione allo stato stazionario – 280-290 ml/kg, durata media nel circolo ematico – 3,75-3,80 ore, emivita – 2,82-3,11 ore. Il recupero plasmatico in vivo medio è stato di circa il 20% della dose somministrata.
Trombostatene di Glanzmann
La farmacocinetica di Novoseven® nei pazienti con trombostatene di Glanzmann non è stata studiata, ma si può presumere che non differisca sostanzialmente da quella osservata nell’emofilia A e B.
Emorragia post-partum grave
La farmacocinetica di Novoseven® nelle pazienti con emorragia post-partum grave non è stata studiata.
Dati sulla sicurezza preclinica.
Tutti i dati ottenuti negli studi preclinici sulla sicurezza sono stati correlati all’effetto farmacologico di rFVIIa.
Un potenziale effetto sinergico con il trattamento combinato di rFXIII e rFVIIa è stato dimostrato in un modello cardiovascolare sperimentale in macachi, portando a un effetto farmacologico significativo (trombosi e morte) con dosi inferiori rispetto a quelle somministrate separatamente.
Caratteristiche cliniche.
Indicazioni
Trattamento e prevenzione delle emorragie in caso di interventi chirurgici o altre procedure invasive nei pazienti affetti da:
- emofilia congenita con livelli di inibitori dei fattori della coagulazione VIII o IX > 5 UB (unità Bethesda);
- emofilia congenita con risposta anamnestica significativa all’infusione di fattori VIII o IX;
- emofilia acquisita;
- deficit congenito del fattore VII;
- trombastenia di Glanzmann con resistenza pregressa o attuale alla trasfusione di piastrine o con piastrinopenia.
Emorragie post-parto gravi
Il medicinale NovoSeven® è indicato nel trattamento delle emorragie post-parto gravi quando gli agenti uterotropi non sono sufficienti per ottenere l’emostasi.
Controindicazioni
Ipersensibilità alla sostanza attiva o a qualsiasi eccipiente, nonché alle proteine di topo, di criceto o di bovino.
Interazioni con altri medicinali ed altre forme di interazione
L’interazione tra il medicinale NovoSeven® e i concentrati dei fattori della coagulazione non è nota. Si deve evitare l’uso contemporaneo di concentrati del complesso protrombinico (attivati o non attivati).
L’impiego di agenti anti-fibrinolitici nei pazienti emofilici riduce la perdita di sangue durante gli interventi chirurgici, in particolare ortopedici o in sedi con elevata attività fibrinolitica, come la cavità orale. Gli agenti anti-fibrinolitici vengono inoltre utilizzati per ridurre la perdita ematica nelle donne con emorragia post-parto. Tuttavia, l’esperienza relativa all’uso contemporaneo di agenti anti-fibrinolitici e del medicinale NovoSeven® è limitata.
In base ai dati degli studi preclinici (vedere paragrafo «Dati sulla sicurezza preclinica»), non si raccomanda l’uso concomitante del fattore VIIa e del fattore XIII.
Non sono disponibili dati clinici sull’interazione tra fattore VIIa e fattore XIII.
Caratteristiche particolari di impiego.
Tracciabilità
Al fine di migliorare la tracciabilità dei medicinali biologici, il nome e il numero di lotto del prodotto somministrato devono essere registrati chiaramente.
In condizioni patologiche in cui il fattore tissutale viene espresso in misura maggiore rispetto alla norma, l’uso del medicinale Novoseven® comporta un rischio di trombosi o di sindrome da DIC. Una situazione simile può verificarsi in pazienti con marcata aterosclerosi, sindrome da schiacciamento (crush syndrome), sepsi o sindrome da DIC.
A causa del rischio di complicanze tromboemboliche, il medicinale Novoseven® deve essere usato con cautela nei pazienti con anamnesi di cardiopatia ischemica, malattie epatiche, nei pazienti dopo interventi chirurgici, nelle donne in gravidanza o prossime al parto, nei neonati o nei pazienti con rischio di eventi tromboembolici o di sindrome da DIC. In ciascuna di queste situazioni, il beneficio potenziale del trattamento con Novoseven® deve essere attentamente valutato rispetto al rischio di tali complicanze.
È noto che un'emorragia post-partum grave e la gravidanza, così come alcune condizioni cliniche (parto, emorragia grave, trasfusioni di sangue, sindrome da DIC, interventi chirurgici/procedure invasive e coagulopatie) possono aumentare il rischio tromboembolico, in particolare il rischio di tromboembolia venosa, che può essere associato alla somministrazione di Novoseven® (vedere la sezione «Effetti indesiderati»).
Poiché il medicinale contenente il fattore di coagulazione ricombinante VIIa Novoseven® può contenere tracce di immunoglobuline G di topo e bovine, nonché di altre proteine di coltura (proteine sieriche di criceto e bovine), nei pazienti trattati può svilupparsi, dopo un certo periodo, ipersensibilità a queste proteine. In tali casi, si dovrà considerare l’opportunità di un trattamento antistaminico per via endovenosa.
In caso di reazioni allergiche o anafilattiche, l’uso del medicinale deve essere immediatamente interrotto. In caso di sviluppo di shock, si dovrà ricorrere al trattamento medico standard per tale condizione. Il paziente deve essere informato sui segni precoci di reazioni di ipersensibilità. Se tali sintomi si manifestano, al paziente deve essere raccomandato di interrompere immediatamente l’uso del medicinale e di consultare immediatamente un medico.
In caso di emorragie gravi, questo medicinale deve essere somministrato in strutture specializzate nella cura di pazienti affetti da emofilia con inibitori del fattore VIII o IX; in mancanza di tale possibilità, deve essere somministrato in stretta collaborazione con un medico specialista nel trattamento dell’emofilia.
Se l’emorragia non può essere arrestata, il paziente deve essere ricoverato urgentemente. I pazienti o le persone che li hanno assistiti devono informare il più rapidamente possibile il personale sanitario riguardo a tutte le iniezioni di Novoseven® effettuate. Nei pazienti con deficit del fattore VII, il tempo di protrombina e l’attività del fattore VII di coagulazione devono essere determinati prima e dopo la somministrazione di Novoseven®. Se l’attività del fattore VIIa non raggiunge il livello atteso o se l’emorragia non si arresta nonostante l’uso delle dosi raccomandate, si potrà ipotizzare la formazione di anticorpi. In questi casi, è necessario determinarne il livello.
Sono stati riportati casi di trombosi in pazienti con deficit del fattore VII che ricevevano Novoseven® durante interventi chirurgici, ma il livello di rischio associato a tale evento non è stato chiarito (vedere la sezione «Farmacodinamica»).
Contenuto di sodio
Novoseven® contiene meno di 1 mmol di sodio (23 mg); pertanto, il medicinale può considerarsi praticamente privo di sodio.
Uso durante la gravidanza o l’allattamento.
Gravidanza
Come misura precauzionale, si raccomanda di evitare l’uso del medicinale Novoseven® durante la gravidanza. I dati limitati sull’uso del medicinale durante la gravidanza per le indicazioni approvate non indicano effetti avversi di rFVIIa sul decorso della gravidanza o sulla salute del feto/neonato. Attualmente non sono disponibili altri dati epidemiologici pertinenti. Gli studi sugli animali non hanno evidenziato effetti negativi diretti o indiretti sulla gravidanza, lo sviluppo dell’embrione/feto, il parto o lo sviluppo postnatale.
Allattamento
Non è noto se rFVIIa passi nel latte materno umano. L’escrezione di rFVIIa nel latte materno degli animali non è stata studiata. La decisione di continuare o interrompere l’allattamento o il trattamento con Novoseven® deve essere presa tenendo in considerazione i benefici dell’allattamento per il neonato e quelli della terapia con Novoseven® per la madre.
Fertilità
I dati degli studi preclinici e quelli post-marketing non indicano un effetto negativo di rFVIIa sulla fertilità di uomini o donne.
Capacità di guidare veicoli o di usare macchinari.
Non sono stati effettuati studi sull’effetto del medicinale sulla capacità di guidare veicoli o di usare macchinari.
Modalità di somministrazione e dosaggio
Il trattamento deve essere iniziato sotto la supervisione di un medico esperto nella gestione dell'emofilia e/o delle emorragie.
Il trattamento delle emorragie post-parto gravi deve essere effettuato da un'équipe multidisciplinare che, oltre agli ostetrici, comprende anestesisti, medici di terapia intensiva e/o ematologi. I metodi standard di trattamento devono essere continuati in base alle esigenze individuali del paziente. Si raccomanda di mantenere concentrazioni adeguate di fibrinogeno e un numero sufficiente di piastrine per ottimizzare i benefici del trattamento con Novoseven®.
Schema posologico
Emofilia A o B con inibitori o emofilia acquisita
Novoseven® deve essere somministrato il più rapidamente possibile dopo l'inizio dell'emorragia. La dose iniziale raccomandata è di 90 µg (4,5 UOI) per kg di peso corporeo, da somministrare per via endovenosa (in bolo).
Dopo la somministrazione della dose iniziale, potrebbe rendersi necessaria una somministrazione ripetuta. La durata del trattamento e gli intervalli tra le somministrazioni variano in base alla gravità dell'emorragia, al tipo di procedura invasiva o intervento chirurgico.
L'esperienza clinica disponibile indica che in generale non vi sono differenze significative tra le posologie per adulti e bambini, anche se i bambini presentano un clearance più rapido rispetto agli adulti. Per questo motivo, nei bambini potrebbero essere necessarie dosi più elevate di rFVIIa per raggiungere una concentrazione plasmatica simile a quella degli adulti (vedere sezione «Farmacocinetica»).
Intervalli tra le somministrazioni
Inizialmente, per ottenere l'emostasi, il farmaco deve essere somministrato nuovamente ogni 2-3 ore.
Se necessario, dopo il raggiungimento di un'emostasi efficace, la somministrazione può essere ripetuta ogni 4, 6, 8 o 12 ore, per il tempo necessario al trattamento.
Emorragie lievi o moderate (incluso il trattamento ambulatoriale)
Nelle condizioni ambulatoriali, la somministrazione precoce del farmaco si è dimostrata efficace nel trattamento di emorragie lievi o moderate a livello articolare, muscolare, cutaneo e delle mucose. Possono essere raccomandati due schemi di somministrazione:
- somministrazione da due a tre dosi del farmaco a 90 µg/kg di peso corporeo ogni 3 ore, seguita da un'ulteriore dose per il mantenimento dell'emostasi;
- somministrazione singola del farmaco a 270 µg/kg di peso corporeo.
La durata del trattamento ambulatoriale non deve superare le 24 ore. Il proseguimento del trattamento a casa può essere considerato solo dopo consultazione con un centro specializzato per il trattamento dell'emofilia.
Non esiste esperienza clinica sull'uso della dose singola di 270 µg/kg di peso corporeo nei pazienti anziani.
Emorragie gravi
Si raccomanda di somministrare la dose iniziale di 90 µg/kg di peso corporeo durante il trasporto del paziente all'ospedale dove riceve abitualmente le cure. La dose successiva dipende dal tipo e dalla gravità dell'emorragia. Inizialmente, il farmaco deve essere somministrato ogni due ore fino al miglioramento delle condizioni cliniche del paziente. Se necessario, per il proseguimento del trattamento, l'intervallo tra le somministrazioni può essere aumentato a 3 ore per 1-2 giorni. Successivamente, durante il periodo successivo di trattamento, l'intervallo tra le somministrazioni può essere progressivamente aumentato a 4, 6, 8 o 12 ore. Le emorragie gravi talvolta richiedono un trattamento per 2-3 settimane o più (a seconda delle condizioni cliniche del paziente).
Procedure invasive/interventi chirurgici
La dose iniziale di 90 µg/kg di peso corporeo deve essere somministrata immediatamente prima dell'intervento. Questa dose deve essere ripetuta dopo 2 ore, quindi ogni 2-3 ore durante le prime 24-48 ore (in base all'estensione dell'intervento e alle condizioni cliniche del paziente). Nei grandi interventi chirurgici, il farmaco deve essere somministrato ogni 2-4 ore per 6-7 giorni. Successivamente, durante le 2 settimane successive, l'intervallo tra le somministrazioni deve essere aumentato a 6-8 ore. Nei pazienti sottoposti a grandi interventi chirurgici, il farmaco deve essere somministrato per 2-3 settimane fino alla completa guarigione della ferita.
Emofilia acquisita
Dosaggio e intervalli di somministrazione
Novoseven® deve essere somministrato il più rapidamente possibile dopo l'inizio dell'emorragia. La dose iniziale raccomandata è di 90 µg (4,5 UOI) per kg di peso corporeo, da somministrare per via endovenosa (in bolo).
Dopo la somministrazione della dose iniziale, potrebbe rendersi necessaria una somministrazione ripetuta. La durata del trattamento e gli intervalli tra le somministrazioni variano in base alla gravità dell'emorragia, al tipo di procedura invasiva o intervento chirurgico.
Inizialmente, il farmaco deve essere somministrato nuovamente ogni 2-3 ore. Se necessario, dopo il raggiungimento di un'emostasi efficace, la somministrazione può essere ripetuta ogni 4, 6, 8 o 12 ore, per il tempo necessario al trattamento.
Carenza del fattore VII
Intervallo di dosaggio e intervalli di somministrazione
L'intervallo di dosaggio raccomandato per adulti e bambini per il trattamento e la prevenzione delle emorragie in pazienti sottoposti a interventi chirurgici o procedure invasive è di 15-30 µg/kg di peso corporeo ogni 4-6 ore fino al raggiungimento dell'emostasi. La dose e l'intervallo di somministrazione devono essere adattati individualmente.
Bambini
I risultati di un'esperienza clinica limitata sulla profilassi a lungo termine in bambini fino a 12 anni con fenotipo clinico grave sono stati riuniti (vedere sezione «Farmacodinamica»).
La dose e la frequenza delle iniezioni per la profilassi devono basarsi sulla risposta clinica al trattamento e devono essere adattate individualmente.
Trombostenia di Glanzmann
Intervallo di dosaggio e intervalli di somministrazione
La dose raccomandata per il trattamento e la prevenzione delle emorragie in pazienti sottoposti a interventi chirurgici o procedure invasive è di 90 µg (intervallo da 80 a 120 µg)/kg di peso corporeo ogni 2 ore (1,5-2,5 ore). Per mantenere un'emostasi efficace, devono essere somministrate almeno 3 dosi. Si raccomandano iniezioni in bolo, poiché l'infusione lenta potrebbe risultare inefficace. Nei pazienti con trombostenia di Glanzmann non resistenti, la prima scelta terapeutica deve essere la somministrazione di piastrine.
Emorragia post-parto grave
Intervallo di dosaggio e intervallo di somministrazione
L'intervallo di dosaggio raccomandato per il trattamento dell'emorragia è di 60-90 µg per kg di peso corporeo, somministrato per via endovenosa in bolo. Il picco di attività coagulante si verifica entro 10 minuti dalla somministrazione. Una seconda dose può essere somministrata in base alla risposta clinica del singolo paziente.
In caso di risposta emostatica insufficiente, si raccomanda di somministrare la seconda dose entro 30 minuti.
Modalità di somministrazione
Le istruzioni per la ricostituzione del medicinale prima della somministrazione sono riportate sul retro del foglio illustrativo di Novoseven®. Il soluto deve essere somministrato come iniezione endovenosa in bolo nell'arco di 2-5 minuti.
Monitoraggio del trattamento — test di laboratorio
Il monitoraggio del trattamento non è obbligatorio, poiché la gravità dell'emorragia e la risposta clinica alla somministrazione di Novoseven® determinano la necessità di ulteriori somministrazioni.
Dopo la somministrazione di Novoseven®, si osserva una riduzione del tempo di protrombina e del tempo di tromboplastina parziale attivata, tuttavia non è stata dimostrata una correlazione tra questi parametri e l'efficacia clinica del farmaco.
Avvertenze per la manipolazione e lo smaltimento del medicinale
Il solvente per Novoseven® è fornito in una siringa preriempita.
Polvere nel flaconcino e solvente nella siringa preriempita
È sempre necessario rispettare le procedure di asepsi.
Ricostituzione
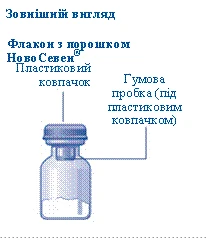
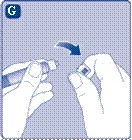
- Prima della ricostituzione, il flaconcino con la polvere di Novoseven® e la siringa preriempita con il solvente devono essere portati a temperatura ambiente. Rimuovere il tappo di plastica dal flaconcino. Non utilizzare il flaconcino se il tappo è allentato o mancante. Pulire il setto di gomma del flaconcino con un batuffolo sterile imbevuto di alcol e lasciarlo asciugare per alcuni secondi prima dell'uso. Non toccare il setto di gomma dopo la pulizia.
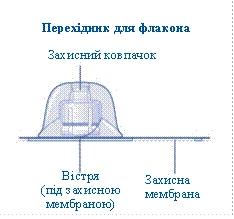
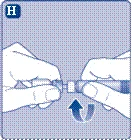
- Rimuovere la membrana protettiva dal connettore per flaconcino. Non rimuovere il connettore per flaconcino dal tappo protettivo. Non utilizzare il connettore per flaconcino se la membrana protettiva non è sigillata correttamente o è danneggiata. Ruotare il tappo protettivo e collegare il connettore per flaconcino. Premere leggermente il tappo protettivo con il pollice e l'indice. Rimuovere il tappo protettivo dal connettore per flaconcino.
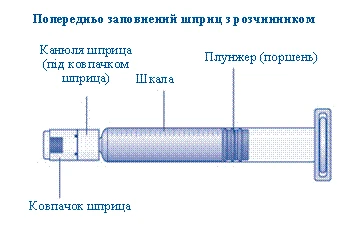
- Inserire lo stantuffo della siringa nello stantuffo interno della siringa preriempita ruotandolo in senso orario fino a quando non si blocca. Rimuovere il cappuccio della siringa agitandolo finché la perforazione non si rompe. Non toccare la punta della siringa sotto il cappuccio. Non utilizzare la siringa preriempita se il cappuccio non è ben aderente o è mancante.
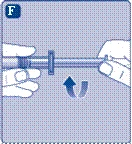
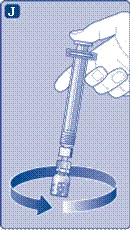
- Collegare la siringa preriempita con il solvente al connettore per flaconcino ruotando la siringa in senso orario fino a quando non si blocca. Tenere la siringa leggermente inclinata con il flaconcino rivolto verso il basso. Premere lo stantuffo per iniettare tutto il solvente nel flaconcino. Tenere lo stantuffo premuto e ruotare delicatamente il flaconcino fino a completa dissoluzione della polvere. Non agitare il flaconcino, poiché ciò potrebbe causare la formazione di schiuma.
Se è necessaria una dose maggiore, ripetere questa procedura con ulteriori flaconcini, siringhe preriempite e connettori per flaconcini.
La soluzione ricostituita di Novoseven® è incolore. Prima della somministrazione, la soluzione deve essere ispezionata visivamente per verificare la presenza di particelle estranee o variazioni di colore.
Si raccomanda di somministrare Novoseven® immediatamente dopo la ricostituzione. Le condizioni di conservazione della soluzione diluita sono descritte nella sezione «Periodo di validità».
Somministrazione
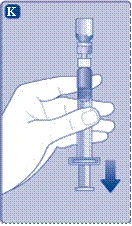
- Tenere lo stantuffo completamente inserito nel cilindro della siringa. Capovolgere la siringa con il flaconcino verso il basso. Smettere di premere lo stantuffo e attendere che la soluzione diluita riempia la siringa. Tirare leggermente lo stantuffo verso il basso per aspirare tutta la soluzione nella siringa.
- Tenendo il flaconcino capovolto, battere delicatamente sulla siringa per far risalire tutte le bolle d'aria in superficie. Premere lentamente lo stantuffo finché tutte le bolle d'aria non scompaiono.
Se non è necessaria l'intera dose, utilizzare la scala sulla siringa per verificare il volume di soluzione ricostituita prelevato.
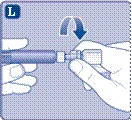
- Scollegare la siringa dal connettore per flaconcino ruotandola in senso antiorario.
- Novoseven® è pronto per l'iniezione. Scegliere il sito di iniezione e somministrare lentamente Novoseven® in vena nell'arco di 2-5 minuti, senza rimuovere l'ago dal sito di iniezione.
Smaltire i materiali utilizzati seguendo le necessarie precauzioni di sicurezza. Tutti i medicinali non utilizzati e i rifiuti devono essere smaltiti in conformità con i requisiti locali.
Procedura di unione dei flaconcini (possibile solo in ambiente ospedaliero)
Studi in vitro hanno dimostrato che il farmaco rimane chimicamente e fisicamente stabile se conservato in una siringa di polipropilene da 50 ml per 24 ore a 25 °C. È stata dimostrata la compatibilità con un sistema costituito da una siringa di polipropilene da 50 ml, un tubo di infusione (polietilene) da 2 m e un filtro integrato con pori da 5 µm.
Unione dei flaconcini (solo in ambiente ospedaliero)
- Tutte le fasi devono essere eseguite da personale qualificato in condizioni asettiche controllate e certificate.
- Se la ricostituzione, la miscelazione o l'uso del farmaco da parte dell'utilizzatore non avviene in conformità con il foglio illustrativo, la responsabilità per la durata e le condizioni di conservazione ricade sull'utilizzatore.
- Assicurarsi di utilizzare il connettore per flaconcino.
- Eseguire la ricostituzione del farmaco come descritto nella sezione «Ricostituzione» sopra. Scollegare la siringa vuota dal connettore per flaconcino ruotandola in senso antiorario e assicurarsi che il connettore sia collegato al flaconcino contenente il farmaco ricostituito.
- Ripetere questa procedura con il numero necessario di flaconcini aggiuntivi, solventi/flaconcini / siringhe preriempite e connettori per flaconcini.
- Aspirare circa 5 ml di aria sterile in una siringa di polipropilene da 50 ml. Collegare la siringa al connettore per flaconcino ruotando in senso orario fino a quando non si blocca. Tenere la siringa leggermente inclinata con il flaconcino rivolto verso il basso. Premere delicatamente lo stantuffo per iniettare leggermente aria nel flaconcino. Capovolgere la siringa con il flaconcino verso il basso e aspirare il contenuto del flaconcino nella siringa. Ripetere la procedura descritta sopra con gli altri flaconcini contenenti il farmaco ricostituito per ottenere il volume di soluzione desiderato nella siringa.
- Per la somministrazione del farmaco deve essere utilizzato un filtro integrato con diametro dei pori di 5 µm. Prima dell'inizio della somministrazione, assicurarsi che la siringa, il tubo di infusione e il filtro integrato siano riempiti di soluzione e privi di bolle d'aria.
- La siringa contenente il farmaco correttamente ricostituito è pronta per l'uso con una pompa per infusione CE (compatibile con siringhe da 50 ml).
- L'uso della pompa per infusione deve essere effettuato esclusivamente da personale medico qualificato.
Novoseven®, istruzioni per l'utilizzatore
Prima di utilizzare Novoseven®, leggere attentamente queste istruzioni.
Novoseven® è fornito sotto forma di polvere. Prima dell'iniezione (somministrazione), deve essere ricostituito con il solvente fornito nella siringa. Il solvente è una soluzione di istidina. Il farmaco ricostituito Novoseven® viene somministrato per via endovenosa (iniezione endovenosa). Il contenuto di questa confezione è destinato alla ricostituzione e alla somministrazione del medicinale Novoseven®.
Per la somministrazione endovenosa sono necessari anche un set per infusione (tubo e ago a farfalla), batuffoli di alcol, garze e cerotti. Questi dispositivi medici non sono inclusi nella confezione di Novoseven®.
Non utilizzare l'apparecchiatura senza un'adeguata formazione da parte di un medico o di un'infermiera.
Prima dell'uso, lavarsi sempre le mani e assicurarsi che lo spazio circostante sia pulito.
Durante la preparazione e la somministrazione del medicinale direttamente in vena, è estremamente importante rispettare le norme di asepsi e antisepsi. Un'iniezione eseguita con tecnica inadeguata può causare un'infezione del sangue.
Non aprire l'apparecchiatura finché non si è pronti a utilizzarla.
Non utilizzare l'apparecchiatura se è caduta o danneggiata. Prendere una nuova confezione.
Non utilizzare l'apparecchiatura se è scaduta. Prendere una nuova confezione. La data di scadenza è stampata dopo la dicitura «Scad.» sulla scatola di cartone, sul flaconcino, sul connettore per flaconcino e sulla siringa preriempita.
Non utilizzare l'apparecchiatura se si sospetta che sia contaminata. Prendere una nuova confezione.
Non gettare alcuna parte del kit finché non si è somministrata la soluzione preparata.
L'apparecchiatura è destinata a un uso singolo.
Contenuto della confezione:
- 1 flaconcino con polvere Novoseven®
- 1 connettore per flaconcino
- 1 siringa preriempita con solvente
- 1 stantuffo (posizionato sotto la siringa)

assicurarsi che contenga il medicinale richiesto.
la siringa pre-riempita dalla scatola. Lasciare lo stantuffo nella scatola.
temperatura ambiente (non superiore a 37 °C). È possibile farlo tenendoli in mano finché non si sentono caldi come le mani.
|
|
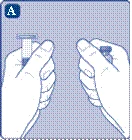
Se il tappo in plastica non è ben fissato o è assente, non utilizzare la fiala.
Non toccare il setto di gomma con le dita, poiché ciò potrebbe causare il trasferimento di microrganismi. |
|
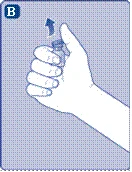
Se l’integrità della membrana protettiva è compromessa o è danneggiata, non utilizzare il dispositivo di collegamento per fiala. Non estrarre il dispositivo di collegamento per fiala dalla confezione con le dita. Se si tocca l’ago del dispositivo di collegamento, ciò potrebbe causare il trasferimento di microrganismi dalle dita. |
|
|
|
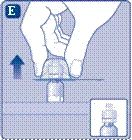
e l’indice, come mostrato nell’immagine. Rimuovere il tappo protettivo dal dispositivo di collegamento, facendo attenzione a non estrarre il dispositivo di collegamento dalla fiala. |
|
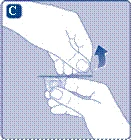
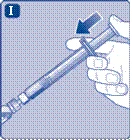
Prendere lo stantuffo dalla parte larga ed estrarlo dalla scatola. Non toccare le superfici laterali dello stantuffo né la punta anteriore dello stantuffo. Se si toccano le superfici laterali o la punta dello stantuffo, ciò potrebbe causare il trasferimento di microrganismi dalle dita. Collegare immediatamente lo stantuffo al pistone all’interno della siringa pre-riempita, ruotandolo in senso orario fino in fondo. |
|
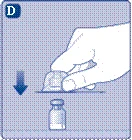
| Rimuovere il tappo della siringa dalla siringa pre-riempita, piegandolo verso il basso finché non si stacca lungo la linea di perforazione. Non toccare l’ago della siringa sotto il tappo. Se si tocca l’ago della siringa, ciò potrebbe causare il trasferimento di microrganismi dalle dita. Se il tappo sulla siringa non è ben fissato o è assente, non utilizzare questa siringa pre-riempita. |
|
|
|
|
|
Non agitare la fiala – poiché ciò causerebbe la formazione di schiuma.
|
|
| Utilizzare immediatamente la soluzione pronta del medicinale Novoseven® per evitare contaminazioni. Se non si utilizza immediatamente la soluzione, vedere la sezione «Condizioni di conservazione» sul retro del foglietto illustrativo. Non conservare la soluzione pronta senza aver prima consultato il medico o l’infermiere. Se la dose richiesta è superiore a 1 fiala, ripetere i passaggi da 1 a 4 (fig. A – J) con fiale aggiuntive, dispositivi di collegamento per fiala e siringhe pre-riempite, fino a ottenere la dose necessaria. |
|
|
|
Se si tocca l’ago della siringa, ciò potrebbe causare il trasferimento di microrganismi dalle dita. |
|
| Somministrazione del medicinale Novoseven® dalla siringa pre-riempita tramite connettore senza ago per cateteri endovenosi. Avvertenza. La siringa pre-riempita è realizzata in vetro ed è compatibile con il connettore standard «Luer-lock». Alcuni connettori senza ago con ago interno non sono compatibili con la siringa pre-riempita. Questa incompatibilità può impedire la somministrazione del medicinale e/o danneggiare il connettore senza ago. È necessario seguire le istruzioni per l’uso dei connettori senza ago. Per la somministrazione del medicinale tramite connettori senza ago, potrebbe essere necessario trasferire la soluzione pronta in una siringa di plastica monouso standard «Luer-lock» da 10 ml. Questa operazione deve essere eseguita immediatamente dopo il passaggio 4 (fig. J). |
|
Ora il medicinale Novoseven® è pronto per essere somministrato come iniezione endovenosa.
Iniezione della soluzione attraverso un catetere venoso centrale o un port sottocutaneo:
|
|
| Smaltimento
|
|
| Non smontare l’apparecchiatura prima dello smaltimento. Non riutilizzare l’apparecchiatura. |
|
Pediatria.
Il medicinale può essere utilizzato nei bambini (vedere il paragrafo «Posologia e modo di somministrazione»).
Sovradosaggio.
La tossicità dose-limitante del medicinale Novoseven® non è stata studiata durante le indagini cliniche.
Negli ultimi 16 anni sono stati riportati 4 casi di sovradosaggio in pazienti con emofilia. L’unico evento avverso riportato in relazione al sovradosaggio è stato un lieve aumento transitorio della pressione arteriosa in un paziente di 16 anni che aveva ricevuto 24 mg di rFVIIa invece di 5,5 mg.
Non sono stati riportati casi di sovradosaggio in pazienti con emofilia acquisita o trombastenia di Glanzmann.
Nei pazienti con deficit del fattore VII, alla dose raccomandata di 15–30 µg/kg di rFVIIa, un caso di sovradosaggio è stato associato a un evento trombotico (ictus occipitale) in un paziente di sesso maschile anziano (> 80 anni) che aveva superato la dose raccomandata da 10 a 20 volte. Inoltre, la formazione di anticorpi contro il medicinale Novoseven® e contro il fattore VII è stata associata a un caso di sovradosaggio in un paziente con deficit del fattore VII.
Le dosi somministrate non devono superare quelle raccomandate, a causa della mancanza di informazioni riguardo a un potenziale rischio aggiuntivo.
Effetti indesiderati.
Le reazioni avverse più comunemente riportate sono state: riduzione della risposta terapeutica, febbre, eruzioni cutanee, tromboembolia venosa, prurito e orticaria. Queste reazioni si sono verificate con frequenza rara (≥ 1/1000, < 1/100).
Le reazioni avverse riportate durante gli studi clinici e nel periodo post-marketing sono elencate di seguito. All'interno di ciascuna categoria per frequenza, le reazioni avverse sono elencate in ordine decrescente di gravità. La frequenza delle reazioni avverse riportate solo nel periodo post-marketing (e non durante gli studi clinici) è sconosciuta.
Negli studi clinici condotti su 484 pazienti (4297 episodi di trattamento) affetti da emofilia A e B, emofilia acquisita, carenza del fattore VII o trombastenia di Glanzmann, si è dimostrato che le reazioni avverse al medicinale sono comuni (≥ 1/100 fino a <1/10). Poiché il numero totale di episodi di trattamento negli studi clinici non supera le 10 000 unità, la frequenza minima possibile di reazioni avverse che può essere identificata è rara (≥ 1/10 000 fino a < 1/1000).
Le reazioni avverse più comuni al medicinale sono febbre ed eruzioni cutanee (non comuni: ≥ 1/1000 fino a <1/100), mentre le reazioni avverse più gravi associate al medicinale sono considerate gli eventi tromboembolici venosi (non comuni: ≥ 1/1000 fino a < 1/100) e gli eventi tromboembolici arteriosi (rari: ≥ 1/10 000 fino a < 1/1000).
Disturbi del sangue e del sistema linfatico
Raro (≥ 1/10 000, < 1/1000) – Coagulazione intravasale disseminata (CID) e alterazioni di laboratorio correlate, compresi aumento dei livelli di D-dimero e riduzione dei livelli di AT-III (vedere sezione «Avvertenze speciali e precauzioni di impiego»), coagulopatia.
Disturbi del sistema gastrointestinale
Raro (≥ 1/10 000, < 1/1000) – Nausea.
Disturbi generali e condizioni nel sito di somministrazione
Non comune (≥ 1/1000, < 1/100) – Riduzione della risposta terapeutica, febbre.
Raro (≥ 1/10 000, < 1/1000) – Reazioni nel sito di iniezione, compreso dolore nel sito di iniezione.
Disturbi del sistema immunitario
Raro (≥ 1/10 000, < 1/1000) – Ipersensibilità (vedere sezioni «Controindicazioni» e «Avvertenze speciali e precauzioni di impiego»).
Frequenza sconosciuta – Reazioni anafilattiche.
Esami di laboratorio
Raro (≥ 1/10 000, < 1/1000) – Aumento dei livelli dei prodotti di degradazione della fibrina, aumento dei livelli di alanina aminotransferasi, fosfatasi alcalina, lattato deidrogenasi e protrombina.
Disturbi del sistema nervoso
Raro (≥ 1/10 000, < 1/1000) – Cefalea.
Disturbi della cute e del tessuto sottocutaneo
Non comune (≥ 1/1000, < 1/100) – Eruzioni cutanee (inclusi dermatite allergica ed eruzioni eritematose), prurito e orticaria.
Frequenza sconosciuta – Vasodilatazione (flush), angioedema.
Disturbi del sistema vascolare
Non comune (≥ 1/1000, < 1/100) – Eventi tromboembolici venosi (trombosi venosa profonda, trombosi nel sito di somministrazione endovenosa, embolia polmonare, eventi tromboembolici epatici, inclusa trombosi della vena porta, trombosi della vena renale, tromboflebite, tromboflebite delle vene superficiali e ischemia intestinale).
Raro (≥ 1/10 000, < 1/1000) – Eventi tromboembolici arteriosi (infarto miocardico, infarto cerebrale, ischemia cerebrale, occlusione dei vasi cerebrali, ictus, trombosi dell'arteria renale, ischemia vascolare periferica, trombosi ischemica periferica e ischemia intestinale), angina pectoris.
Frequenza sconosciuta – Trombosi delle camere cardiache.
È stato riportato un insufficiente effetto terapeutico (riduzione della risposta terapeutica). È estremamente importante che il regime posologico del medicinale Novoseven® sia conforme a quanto raccomandato nella sezione «Modalità di somministrazione e posologia».
Descrizione di reazioni avverse specifiche
Formazione di anticorpi inibitori
Durante l’uso post-marketing, non sono stati riportati casi di rilevamento di anticorpi inibitori contro il medicinale Novoseven® o contro il FVII in pazienti affetti da emofilia A o B. Sono stati riportati casi di formazione di anticorpi inibitori contro il medicinale Novoseven® in un registro osservazionale post-marketing di pazienti con carenza congenita del fattore VII.
Negli studi clinici condotti su pazienti con carenza del fattore VII, la formazione di anticorpi contro il medicinale Novoseven® e contro il fattore VII (frequente: ≥ 1/100, < 1/10) è stata l’unica reazione avversa riportata. In alcuni casi, gli anticorpi hanno determinato un effetto inibitorio in vitro. I fattori di rischio per la formazione di anticorpi comprendono: trattamento precedente con plasma umano e/o fattore VII purificato da plasma, mutazioni gravi del gene del fattore VII e sovradosaggio del medicinale Novoseven®. Nei pazienti con carenza del fattore VII in trattamento con Novoseven®, si raccomanda un monitoraggio costante della presenza di anticorpi contro il fattore VII (vedere sezione «Avvertenze speciali e precauzioni di impiego»).
Eventi tromboembolici: arteriosi e venosi
Quando il medicinale Novoseven® viene utilizzato per indicazioni non approvate, gli eventi tromboembolici arteriosi sono comuni (≥ 1/100, < 1/10). Un rischio maggiore di eventi avversi tromboembolici arteriosi (vedere sopra «Disturbi del sistema vascolare») (5,6 % nei pazienti trattati con Novoseven® rispetto al 3,0 % nei pazienti trattati con placebo) è stato osservato in un’analisi metà di dati aggregati di studi controllati con placebo, quando il medicinale è stato utilizzato per indicazioni non approvate.
Gli eventi tromboembolici possono portare a arresto cardiaco.
Popolazioni di pazienti particolari
Pazienti con emofilia acquisita
Negli studi clinici condotti su 61 pazienti con emofilia acquisita (includenti 100 episodi di trattamento), le reazioni avverse più comuni (1 % di tutti gli episodi di trattamento) sono state: eventi tromboembolici arteriosi (occlusione dei vasi cerebrali, ictus), eventi tromboembolici venosi (embolia polmonare e trombosi venosa profonda), angina pectoris, nausea, febbre, eruzioni eritematose, aumento dei livelli dei prodotti di degradazione della fibrina.
Donne con emorragia post-partum grave
In uno studio clinico randomizzato, aperto, sono stati riportati eventi tromboembolici venosi in 2 su 51 pazienti che hanno ricevuto una singola dose del medicinale Novoseven® (dose media 58 mcg/kg); tali eventi non sono stati osservati in nessuna delle 33 pazienti che non hanno ricevuto il medicinale Novoseven®; eventi tromboembolici arteriosi non sono stati osservati in nessuno dei gruppi.
In 4 studi non interventisti, eventi tromboembolici venosi sono stati osservati in 3 su 358 pazienti (0,8 %) che hanno ricevuto il medicinale Novoseven® (intervallo medio di dosi 63–105 mcg/kg), mentre eventi tromboembolici arteriosi sono stati osservati in 1 paziente (0,3 %) che ha ricevuto il medicinale Novoseven®.
Per informazioni sui fattori noti che aumentano il rischio tromboembolico associato alla gravidanza e alle emorragie post-partum gravi, vedere la sezione «Avvertenze speciali e precauzioni di impiego».
Segnalazione di reazioni avverse sospette e mancata efficacia del medicinale
La segnalazione delle reazioni avverse dopo l’autorizzazione del medicinale è di fondamentale importanza. Permette un monitoraggio continuo del rapporto beneficio/rischio del medicinale. I professionisti sanitari, i farmacisti, nonché i pazienti o i loro rappresentanti legali devono segnalare tutti i casi sospetti di reazioni avverse e di mancata efficacia del medicinale attraverso il Sistema Informatizzato di Farmacovigilanza all’indirizzo https://aisf.dec.gov.ua/.
Durata della conservazione. La durata della conservazione del medicinale confezionato per la vendita è di 3 anni, se conservato a una temperatura inferiore a 25 °C.
Nel flacone
È stata dimostrata la stabilità chimica e fisica della soluzione ricostituita per 6 ore a 25 °C o per 24 ore a 5 °C.
Dal punto di vista microbiologico, il medicinale ricostituito deve essere utilizzato immediatamente. L’utente è responsabile del periodo e delle condizioni di conservazione. Se la ricostituzione non è stata effettuata in condizioni asettiche controllate e validate, il periodo di conservazione della soluzione ricostituita non deve superare le 24 ore a una temperatura di 2–8 °C. La soluzione ricostituita deve essere conservata nel flacone.
Nella siringa (siringa di polipropilene da 50 ml) solo in ambiente ospedaliero
La procedura di diluizione deve essere eseguita da personale qualificato in condizioni asettiche controllate e certificate. Il medicinale rimane chimicamente e fisicamente stabile nelle seguenti condizioni: conservazione in una siringa di polipropilene da 50 ml per 24 ore a 25 °C. Se il medicinale non viene utilizzato immediatamente, l’utente è responsabile del rispetto delle condizioni di conservazione. Il periodo di conservazione del medicinale dopo l’apertura della confezione non deve superare le 24 ore.
Condizioni di conservazione.
Conservare in un luogo inaccessibile ai bambini, protetto dalla luce, a una temperatura non superiore a 25 °C.
Non congelare.
Per le condizioni di conservazione del medicinale ricostituito, vedere la sezione «Durata della conservazione».
Incompatibilità. Novoseven® non deve essere mescolato con soluzioni per infusione né somministrato per via endovenosa lenta (in fleboclisi).
Confezionamento.
1 scatola di cartone contiene 1 flacone di vetro da 2 ml o 12 ml con polvere bianca per soluzione iniettabile, chiuso con tappo di gomma clorobutile e sigillato con capsula di alluminio con capsulotto protettivo rimovibile in polipropilene, e 1 siringa pre-riempita in vetro tipo 1 da 3 ml o 10 ml contenente 2 ml o 5 ml di solvente (istidina, acqua per preparazioni iniettabili).
La siringa è chiusa da un lato con un capsulotto in gomma bromobutile e polipropilene e dall’altro con un pistone in gomma bromobutile e un fermo in polipropilene. Nell’insieme è incluso anche un mandrino del pistone in polipropilene. Viene fornito anche un adattatore per flacone con filtro di purificazione fine da 25 µm in confezionamento individuale.
Categoria di dispensazione. Su prescrizione medica.
Produttore/detentore dell’autorizzazione.
A/T Novo Nordisk / Novo Nordisk A/S.
Indirizzo del produttore e sede operativa.
Novo Alle, Bagsvaerd, 2880, Danimarca / Novo Alle, Bagsvaerd, 2880, Denmark.