Novoeit®
UkrainaSpis treści
INSTRUKCJA DOTYCZĄCA STOSOWANIA LEKU NOVOEIT® (NovoEight®)
Skład:
substancja czynna: turoctocog alfa;
1 fiolka z proszkiem zawiera 1500 JPM lub 2000 JPM, lub 3000 JPM turoctocogu alfa (czynnika krzepnięcia krwi ludzkiego VIII (rDNA));
substancje pomocnicze: chloro sodu; L-histydyna; sacharoza; polisorbat 80; L-metionina; chlorek wapnia, dwuwodny; wodorotlenek sodu; kwas chlorowodorowy.
Roztwórnik: chloro sodu, woda do wstrzykiwań.
Po odtworzeniu 1 ml leku Novoeit® zawiera około 375 JPM, 500 JPM lub 750 JPM turoctocogu alfa (czynnika krzepnięcia krwi ludzkiego VIII (rDNA)).
Postać leku. Proszek i roztwórnik do sporządzenia roztworu do wstrzykiwań.
Główne właściwości fizykochemiczne: proszek liofilizowany lub luźna masa biała lub lekko żółta. Roztwórnik: przezroczysty, bezbarwny roztwór do wstrzykiwań.
Grupa farmakoterapeutyczna. Środki hemostatyczne. Czynniki krzepnięcia krwi. Czynnik krzepnięcia VIII.
Kod ATX B02B D02.
Właściwości farmakodynamiczne.
Farmakodynamika.
Mechanizm działania
Preparat Novoeit® zawiera turkotokog alfa, ludzki rekombinowany czynnik krzepnięcia krwi VIII (rDNA) z usuniętym domeną B. Ten glikoprotein ma taką samą strukturę jak aktywowany ludzki czynnik VIII oraz takie same modyfikacje potranslacyjne jak cząsteczka izolowana z osocza krwi. Stwierdzono, że reszta tyrozyny poddawana sulfatacji, zlokalizowana na Tyr1680 (pełna długość naturalnego czynnika), ważna dla wiązania się z czynnikiem von Willebranda, jest w pełni zasulfatowana w cząsteczce turkotokogu alfa. Po podaniu pacjentowi z hemofilią czynnik VIII wiąże się z endogennym czynnikiem von Willebranda we krwi chorych. Złożony czynnik VIII/czynnik von Willebranda składa się z dwóch cząsteczek (czynnika VIII i czynnika von Willebranda) o różnych funkcjach fizjologicznych. Aktywowany czynnik VIII działa jako kofaktor aktywowanego czynnika IX, przyspieszając przekształcanie czynnika X w aktywowany czynnik X. Aktywowany czynnik X przekształca protrombinę w trombinę. Następnie trombina przekształca fibrynogen w fibrynę, w wyniku czego powstaje skrzep. Hemofilia A to sprzężone z płcią dziedziczne zaburzenie krzepnięcia krwi spowodowane obniżonym poziomem czynnika VIII:C, prowadzące do obfitych krwawień do stawów, mięśni lub narządów wewnętrznych, zarówno samoistnych, jak i spowodowanych urazem lub zabiegami chirurgicznymi. Dzięki terapii zastępczej poziom czynnika VIII w osoczu wzrasta, co zapewnia tymczasową korekcję niedoboru czynnika oraz zmniejsza skłonność do krwawień.
Należy zaznaczyć, że częstość krwawień w ciągu roku (CKR) przy różnych stężeniach czynnika krzepnięcia i w różnych badaniach klinicznych jest nieporównywalna.
Skuteczność kliniczna
Przeprowadzono cztery wieloośrodkowe, otwarte, niekontrolowane badania kliniczne w celu oceny bezpieczeństwa i skuteczności stosowania preparatu Novoeit® w profilaktyce i leczeniu krwawień oraz podczas zabiegów chirurgicznych u pacjentów z ciężką formą hemofilii A (aktywność czynnika VIII ≤ 1%). Trzy z nich przeprowadzono wśród pacjentów wcześniej leczonych, a czwarte – wśród pacjentów wcześniej nieleczonych. W badaniach wzięło udział 298 pacjentów: 175 dorosłych pacjentów i nastolatków w wieku od 12 lat (≥ 150 dni leczenia badanym preparatem), u których nie wykryto inhibitorów, 63 pacjentów pediatrycznych w wieku poniżej 12 lat (≥ 50 dni leczenia badanym preparatem), u których nie wykryto inhibitorów, oraz 60 pacjentów w wieku do 6 lat, którzy wcześniej nie byli leczeni. 188 z 238 pacjentów wcześniej leczonych kontynuowało udział w dodatkowym badaniu oceniającym bezpieczeństwo. Potwierdzono, że leczenie preparatem Novoeit® jest bezpieczne i zapewnia pożądany efekt hemostatyczny oraz profilaktyczny. Zarejestrowano 3293 przypadki krwawień obserwowane u 298 pacjentów, z których 2902 (88,1%) krwawień ustało po 1–2 infuzjach preparatu Novoeit®.
Tabela 1. Stosowanie preparatu Novoeit® i wskaźniki skuteczności u pacjentów wcześniej nieleczonych i u tych, którzy wcześniej leczono
| Wskaźnik |
Dzieci w młodszym przedziale wiekowym (od 0 do mniej niż 6 lat), które nie poddawano leczeniu |
Dzieci w młodszym przedziale wiekowym (od 0 do mniej niż 6 lat), które poddawano leczeniu |
Dzieci w starszym przedziale wiekowym (od 6 do mniej niż 12 lat), które poddawano leczeniu |
Młodzież (od 12 do mniej niż 18 lat), która poddawano leczeniu |
Dorośli (≥ 18 lat), którzy poddawano leczeniu |
Łącznie |
| Liczba pacjentów |
60 |
31 |
32 |
24 |
151 |
298 |
| Dawka stosowana w profilaktyce na jednego pacjenta (JM/kg MT) |
||||||
| Średnia (SD) |
45,2 (14,4) |
41,5 (8,1) |
38,4 (9,4) |
28,5 (9,3) |
28,5 (8,3) |
32,8 (10,9) |
| Minimum; maksimum |
4,5; 363,8 |
3,4; 196,3 |
3,2; 62,5 |
17,4; 73,9 |
12,0; 97,4 |
3,2; 363,8 |
| Dawka stosowana w leczeniu krwawień (JM/kg MT) |
||||||
| Średnia (SD) |
43,6 (15,2) |
44,0 (12,6) |
40,4 (10,5) |
29,3 (10,3) |
35,0 (12,3) |
37,5 (13,4) |
| Minimum; maksimum |
11,9; 118,9 |
21,4; 193,8 |
24,0; 71,4 |
12,4; 76,8 |
6,4; 104,0 |
6,4; 193,8 |
| Wskaźnik skutecznościa, % |
87,0 % |
92,2 % |
88,4 % |
85,1 % |
89,6 % |
88,9 % |
MT – masa ciała, SW – standardowe odchylenie.
a Skuteczność określono jako doskonałą lub dobrą.
Dane kliniczne uzyskane przed rejestracją zostały potwierdzone nieinterwencyjnym badaniem bezpieczeństwa po rejestracji, przeprowadzonym w celu dostarczenia dodatkowych informacji dotyczących immunogenności, skuteczności i bezpieczeństwa leku Novoeit® w warunkach standardowej praktyki klinicznej. Ogółem 68 wcześniej leczonych pacjentów (> 150 ED (skutecznych dawek)), w tym 14 pacjentów w wieku do 12 lat i 54 pacjentów w wieku od 12 lat, otrzymywało leczenie w przypadku potrzeby (N = 5) lub leczenie profilaktyczne (N = 63) przez łącznie 87,8 pacjentolata i 8967 ED.
Zabiegi chirurgiczne
Przeprowadzono łącznie 30 zabiegów chirurgicznych u 25 pacjentów, w tym 26 dużych zabiegów chirurgicznych i 4 małe. Hemostaza była skuteczna we wszystkich przypadkach zabiegów chirurgicznych; nie zgłoszono braku skuteczności leczenia.
Zebrano dane dotyczące indukcji tolerancji immunologicznej (ITI) u pacjentów z hemofilą A, u których wykryto inhibitory czynnika VIII. W trakcie badań klinicznych z udziałem pacjentów, którzy wcześniej nie byli leczeni (ang. previously untreated patients (PUPs)), 21 pacjentów otrzymywało leczenie ITI, a 18 pacjentów (86 %) ukończyło terapię ITI z negatywnym wynikiem testu na obecność inhibitorów.
Farmakokinetyka.
Wszystkie badania farmakokinetyczne z zastosowaniem turkotokogu alfa przeprowadzono po dożylnej podaniu 50 J/ kg leku Novoeit® u pacjentów z ciężką hemofilią A (FVIII ≤ 1 %), którzy wcześniej otrzymywali leczenie. Analizę próbek osocza przeprowadzono przy użyciu jednostopniowego testu aktywności koagulacyjnej oraz testu chromogennego.
Aktywność leku Novoeit® w analizie FVIII:C oceniano i porównywano z dostępnym na rynku rekombinowanym lekiem FVIII pełnej długości. Badanie wykazało, że uzyskane wyniki są porównywalne i spójne dla obu leków oraz że lek Novoeit® można wiarygodnie oznaczać w osoczu bez konieczności stosowania oddzielnego standardu dla leku Novoeit®.
Pokazania farmakokinetyczne leku Novoeit® po podaniu dawki pojedynczej, wynikające z analizy aktywności koagulacyjnej krwi, przedstawiono w tabeli 2, a wynikające z jednostopniowego testu chromogennego – w tabeli 3.
Tabela 2. Farmakokinetyka turkotokogu alfa (50 J/kg) w zależności od wieku – jedna faza, analiza aktywności koagulacyjnej krwi – wartość średnia (standardowe odchylenie)
| Wskaźnik |
Od 0 do 6 lat |
Od 6 do 12 lat |
≥ 12 lat |
| n = 14 |
n = 14 |
n = 33 |
|
| Wartość średnia (SD) |
Wartość średnia (SD) |
Wartość średnia (SD) |
|
| Przyrost poziomu (JM/dl)/(JM/kg) |
1,8 (0,7) |
2,0 (0,4) |
2,2 (0,4) |
| AUC ((JM*godz)/dl) |
992 (411) |
1109 (374) |
1526 (577) |
| CL (ml/godz/kg) |
6,21 (3,66) |
5,02 (1,68) |
3,63 (1,09) |
| t½ (godz) |
7,65 (1,84) |
8,02 (1,89) |
11,00 (4,65) |
| Vss (ml/kg) |
56,68 (26,43) |
46,82 (10,63) |
47,40 (9,21) |
| Cmax (JM/dl) |
100 (58) |
107 (35) |
123 (41) |
| Średni czas działania (godz) |
9,63 (2,50) |
9,91 (2,57) |
14,19 (5,08) |
AUC – pole pod krzywą farmakokinetyczną opisującą dynamikę czasową aktywności czynnika VIII; CL – klirens; t1/2 – końcowy okres półtrwania; Vss – objętość rozkładu w stanie stacjonarnym; Cmax – maksymalna aktywność czynnika VIII.
Tabela 3. Farmakokinetyka turkotokogu alfa (50 JМ/кг) w zależności od wieku – Analiza chromogenowa – Średnia wartość (odchylenie standardowe)
| Wskaźnik |
Od 0 do < 6 lat |
Od 6 do < 12 lat |
≥ 12 lat |
| n = 14 |
n = 14 |
n = 33 |
|
| Średnia wartość (SD) |
Średnia wartość (SD) |
Średnia wartość (SD) |
|
| Przyrost poziomu (MO/dl)/(MO/kg) |
2,2 (0,6) |
2,5 (0,6) |
2,9 (0,6) |
| AUC ((MO·godz)/dl) |
1223 (436) |
1437 (348) |
1963 (773) |
| CL (ml/godz/kg) |
4,59 (1,73) |
3,70 (1,00) |
2,86 (0,94) |
| t½ (godz.) |
9,99 (1,71) |
9,42 (1,52) |
11,22 (6,86) |
| Vss (ml/kg) |
55,46 (23,53) |
41,23 (6,00) |
38,18 (10,24) |
| Cmax (MO/dl) |
112 (31) |
125 (27) |
163 (50) |
| Średni czas działania (godz.) |
12,06 (1,90) |
11,61 (2,32) |
14,54 (5,77) |
AUC – pole pod krzywą farmakokinetyczną opisującą dynamikę czasową aktywności czynnika VIII; CL – klirens; t1/2 – końcowy okres półwyprowadzenia; Vss – objętość rozłożenia w stanie równowagi; Cmax – maksymalna aktywność czynnika VIII.
Pokaźniki farmakokinetyczne były porównywalne u pacjentów w wieku do 6 lat oraz u pacjentów w wieku od 6 do 12 lat. Obserwowano pewne różnice w parametrach farmakokinetycznych leku Novoeit® u dzieci i dorosłych. Wyższy klirens oraz krótszy t½ u dzieci w porównaniu z dorosłymi pacjentami z hemofilią A mogą być częściowo wyjaśnione większą objętością osocza krwi na kilogram masy ciała u młodszych pacjentów.
Przeprowadzono badanie farmakokinetyki po pojedynczej dawce (50 JM/kg) z udziałem 35 pacjentów z hemofilią (≥ 18 lat) w różnych kategoriach BMI. Maksymalne stężenie leku w osoczu (Cmax) oraz całkowite stężenie leku w osoczu (AUC) wzrastały wraz ze zwiększającym się BMI, co wskazuje na potrzebę korekty dawki u pacjentów z niedowagą (BMI < 18,5 kg/m²) i nadwagą (BMI ≥ 30 kg/m²), patrz punkt „Sposób założenia i dawki”.
Tabela 4. Parametry farmakokinetyczne leku Novoeit® po podaniu pojedynczej dawki (50 JM/kg) w zależności od klasy BMIa – jednoetapowy test krzepnięcia krwi – Średnia wartość (odchylenie standardowe)
| Wskaźnik farmakokinetyczny |
Niedowaga, N = 5 |
Masa ciała w normie, N = 7 |
Nadwaga, N = 8 |
Otyłość, klasa I, N = 7 |
Otyłość, klasa II/III, N = 7 |
| Wzrost stężenia (J m/decy)/ (J m/kg) |
1,7 (0,2) |
2,0 (0,2) |
2,4 (0,4) |
2,3 (0,3)b |
2,6 (0,3) |
| AUC ((J m*godz)/decy) |
1510 (360) |
1920 (610) |
1730 (610) |
2030 (840) |
2350 (590) |
| CL (ml/godz/kg) |
3,91 (0,94) |
3,20 (1,00) |
3,63 (1,24) |
3,37 (1,79) |
2,51 (0,63) |
| t½ (godz) |
11,3 (2,0) |
11,7 (3,5) |
9,4 (2,9) |
11,2 (3,5) |
11,1 (2,7) |
| Vss (ml/kg) |
56,8 (5,4) |
44,8 (6,5) |
39,6 (6,0) |
42,0 (9,0) |
35,0 (4,6) |
| Cmax (J m/decy) |
100 (11) |
121 (10) |
144 (26) |
140 (21) |
161 (32) |
| Średni czas działania (godz) |
15,1 (3,0) |
15,3 (4,8) |
11,9 (3,7) |
14,4 (4,6) |
14,6 (3,7) |
a Grupy BMI: niedobór masy ciała, BMI < 18,5 kg/m²; prawidłowa masa ciała, BMI 18,5–24,9 kg/m²; nadmiar masy ciała, BMI 25–29,9 kg/m²; otyłość, klasa I, BMI 30–34,9 kg/m²; otyłość, klasa II/III, BMI ≥ 35 kg/m².
b Dane uzyskano jedynie dla 6 pacjentów.
Tabela 5. Wskaźniki farmakokinetyczne leku Novoeit® po podaniu dawki jednorazowej (50 JЕ/kg) w zależności od klasy BMIa – analiza chromogenna – wartość średnia (odchylenie standardowe)
| Farmakokinetyczny parametr |
Niedostateczna masa ciała, N = 5 |
Masa ciała w normie, N = 7 |
Nadmierna masa ciała, N = 9 |
Otyłość, klasa I, N = 7 |
Otyłość, klasa II/III, N = 7 |
| Wzrost stężenia (MO/dl)/(MO/kg) |
2,2 (0,4) |
2,9 (0,3) |
3,0 (0,5) |
3,2 (0,5) |
3,5 (0,5) |
| AUC ((MO·godz)/dl) |
1860 (700) |
2730 (860) |
2310 (1020) |
2780 (1210) |
3050 (730) |
| CL (ml/godz/kg) |
3,28 (0,87) |
2,25 (0,73) |
2,84 (1,09) |
2,58 (1,56) |
1,94 (0,52) |
| t½ (godz) |
11,7 (2,4) |
11,5 (3,6) |
9,7 (3,4) |
10,4 (3,2) |
10,5 (2,5) |
| Vss (ml/kg) |
49,1 (10,4) |
31,2 (4,5) |
31,6 (5,8) |
28,9 (5,1) |
25,7 (4,0) |
| Cmax (MO/dl) |
138 (29) |
185 (24) |
194 (31) |
200 (33) |
227 (32) |
| Średni czas działania (godz) |
15,5 (3,2) |
15,2 (4,9) |
12,6 (4,8) |
13,5 (4,6) |
13,9 (3,7) |
a Grupy BMI: niedobór masy ciała, BMI < 18,5 kg/m2; prawidłowa masa ciała, BMI 18,5–24,9 kg/m2; nadmiar masy ciała, BMI 25–29,9 kg/m2; otyłość, klasa I, BMI 30–34,9 kg/m2; otyłość, klasa II/III, BMI ≥ 35 kg/m2.
Dane przedkliniczne dotyczące bezpieczeństwa
Dane przedkliniczne wskazują na brak szczególnych ryzyk dla człowieka na podstawie wyników standardowych badań farmakologicznych dotyczących bezpieczeństwa oraz badań toksyczności przy powtarzanych dawkach.
Dane kliniczne.
Wskazania.
Leczenie i profilaktyka krwawień u pacjentów z hemofilią typu A (wrodzonym niedoborem czynnika VIII).
Preparat Novoeit® może być stosowany u pacjentów z wszystkich grup wiekowych.
Przeciwwskazania.
Nadwrażliwość na substancję czynną lub na którykolwiek z substancji pomocniczych.
Znana reakcja alergiczna na białka chomika.
Interakcje z innymi lekami i inne rodzaje interakcji.
Nie odnotowano interakcji ludzkich czynników krzepnięcia krwi VIII (rDNA) z innymi lekami.
Szczególne wytyczne dotyczące stosowania.
Śledzenie
W celu poprawy śledzenia leków biologicznych należy dokładnie zapisywać nazwę i numer serii zastosowanego leku.
Nadwrażliwość
Podczas stosowania leku Novoeit® możliwe są reakcje nadwrażliwościowe o charakterze alergicznym. Lek zawiera śladowe ilości białek chomika, które u niektórych pacjentów mogą wywoływać reakcje alergiczne. W przypadku wystąpienia objawów nadwrażliwości pacjentom należy zalecić natychmiastowe zaprzestanie stosowania leku i skontaktowanie się z lekarzem. Pacjentów należy poinformować o wczesnych objawach reakcji nadwrażliwościowych, takich jak wysypka, uogólnione pokrzywienie, uczucie ucisku w klatce piersiowej, świsty podczas oddychania, obniżenie ciśnienia krwi oraz anafilaksja.
W przypadku szoku należy rozpocząć standardowe leczenie przeciwszokowe.
Inhibitory
Powstawanie przeciwciał neutralizujących (inhibitorów) wobec czynnika VIII jest znanym powikłaniem leczenia osób z hemofilią typu A. Inhibitory te są zazwyczaj immunoglobulinami klasy IgG, które hamują działanie prokoagulacyjne czynnika VIII; ilościowo wyraża się je w jednostkach Bethesda (J.B.) na 1 ml osocza, stosując zmodyfikowany test. Ryzyko powstawania inhibitorów koreluje z ekspozycją na czynnik VIII i jest najwyższe w ciągu pierwszych 50 dni ekspozycji, jednak utrzymuje się przez całe życie pacjenta, choć jest rzadkie.
Kliniczne znaczenie powstawania inhibitorów zależy od ich miana; niższe miano wiąże się z mniejszym ryzykiem niewystarczającej odpowiedzi klinicznej niż wysokie miano inhibitorów.
Ogólnie należy dokładnie monitorować wszystkich pacjentów otrzymujących leczenie lekami zawierającymi czynnik krzepnięcia VIII pod kątem powstawania inhibitorów, poprzez odpowiednią obserwację kliniczną i badania laboratoryjne. Jeśli nie uda się osiągnąć oczekiwanej aktywności czynnika VIII we krwi lub jeśli nie uda się kontrolować krwawienia odpowiednią dawką, należy przeprowadzić badanie na obecność inhibitorów czynnika VIII. U pacjentów z wysokim poziomem inhibitorów terapia czynnikiem VIII może być nieskuteczna, należy wtedy rozważyć inne metody leczenia. Leczenie takich pacjentów należy prowadzić pod nadzorem lekarzy posiadających doświadczenie w leczeniu pacjentów z hemofilią i obecnością inhibitorów czynnika VIII.
Ostrzeżenia związane ze składnikami pomocniczymi
Lek zawiera 30,5 mg sodu w fiolce z odtworzonym lekiem, co odpowiada 1,5% maksymalnego dziennej dawki spożycia sodu dla dorosłego, zalecanego przez WHO, wynoszącego 2 g.
Powikłania ze strony układu sercowo-naczyniowego
U pacjentów z istniejącymi czynnikami ryzyka sercowo-naczyniowego terapia zastępcza z zastosowaniem czynnika VIII może zwiększać ryzyko powikłań sercowo-naczyniowych.
Powikłania związane z cewnikiem
W przypadku konieczności założenia cewnika dożylnego centralnego należy wziąć pod uwagę ryzyko powikłań związanych z cewnikiem, w tym infekcji miejscowych, bakteriemii oraz zakrzepicy w miejscu wprowadzenia cewnika.
W celu zapewnienia śledzenia związku pomiędzy stanem pacjenta a serią leku, zaleca się konieczność zapisywania nazwy i numeru serii leku przy każdym podaniu leku Novoeit® pacjentowi.
Dzieci
Podane ostrzeżenia i środki ostrożności dotyczą zarówno dorosłych, jak i dzieci.
Stosowanie w czasie ciąży lub karmienia piersią.
Badania wpływu leku Novoeit® na funkcję rozrodczą u zwierząt nie były prowadzone. Ponieważ hemofilia typu A u kobiet występuje rzadko, brak jest doświadczenia w stosowaniu czynnika VIII w czasie ciąży i karmienia piersią. Dlatego czynnik VIII należy stosować u kobiet w ciąży i w okresie laktacji wyłącznie w przypadku wyraźnych wskazań.
Wpływ na zdolność prowadzenia pojazdów lub obsługiwania maszyn.
Lek Novoeit® nie wpływa na zdolność prowadzenia pojazdów i obsługiwanie innych maszyn.
Sposób stosowania i dawki
Leczenie należy rozpoczynać pod nadzorem lekarza posiadającego doświadczenie w leczeniu hemofilii.
Monitorowanie leczenia
Podczas leczenia zaleca się odpowiednie oznaczanie poziomów czynnika VIII w celu dostosowania dawki i częstotliwości powtarzanych wstrzyknięć. U pacjentów może występować różna odpowiedź na czynnik VIII, różne okresy półtrwania i odnowienie. Dawkę obliczaną na podstawie masy ciała może być konieczne dostosować u pacjentów z niedowagą lub nadwagą. Badania farmakokinetyki leku po podaniu dawki pojedynczej dorosłym pacjentom wykazały, że maksymalne stężenie leku we krwi (Cmax) oraz całkowite stężenie leku we krwi (AUC) wzrasta wraz ze zwiększonym wskaźnikiem masy ciała (BMI). Oznacza to, że może być konieczna korekta dawki. Pacjenci z niedowagą (BMI < 18,5 kg/m²) mogą wymagać zwiększenia dawki, a pacjenci z otyłością (BMI ≥ 30 kg/m²) – jej zmniejszenia. Obecnie jednak brakuje wystarczających danych do sformułowania konkretnych zaleceń dotyczących korekty dawki, patrz sekcja „Farmakokinetyka”.
W szczególności, w przypadku poważnych zabiegów operacyjnych konieczne jest staranne monitorowanie terapii zastępczej poprzez analizę krzepnięcia (aktywność czynnika VIII we krwi).
Podczas wykonywania jednostopniowego testu krzepnięcia in vitro opartego na czasie czynniku aktywującym tromboplastynę (aPTT) do oznaczania aktywności czynnika VIII we krwi pacjentów, zarówno rodzaj odczynnika aPTT, jak i preparat odniesienia stosowany w teście mogą znacząco wpływać na wyniki oznaczania aktywności czynnika VIII we krwi. Mogą również występować istotne różnice między wynikami uzyskanymi w jednostopniowym teście krzepnięcia opartym na aPTT a wynikami analizy chromogenowej zgodnie z Europejską Farmakopeą. Jest to szczególnie istotne przy zmianie laboratorium i/lub odczynników stosowanych w analizie.
Dawkowanie
Dawkowanie i długość trwania terapii zastępczej zależą od stopnia niedoboru czynnika VIII, lokalizacji i nasilenia krwawienia oraz stanu klinicznego pacjenta.
Ilość jednostek czynnika VIII wyrażana jest w jednostkach międzynarodowych (j.m.); odpowiada to obowiązującemu standardowi WHO dla preparatów czynnika VIII. Aktywność czynnika VIII we krwi wyrażana jest w procentach (w stosunku do normalnego poziomu we krwi człowieka) lub w jednostkach międzynarodowych (w stosunku do międzynarodowego standardu dla czynnika VIII we krwi).
Jedna jednostka międzynarodowa (j.m.) aktywności czynnika VIII odpowiada ilości czynnika VIII zawartej w 1 ml osocza krwi osoby zdrowej.
Leczenie w razie potrzeby
Obliczenie wymaganej dawki czynnika VIII opiera się na wynikach empirycznych, zgodnie z którymi 1 jednostka międzynarodowa (j.m.) czynnika VIII na 1 kg masy ciała zwiększa aktywność czynnika VIII we krwi o 2 j.m./dl. Wymaganą dawkę oblicza się według następującego wzoru:
Wymagana liczba jednostek = masa ciała (kg) × pożądane zwiększenie poziomu czynnika VIII (%) (j.m./dl) × 0,5 (j.m./kg na 1 j.m./dl).
Ilość, którą należy podać, oraz częstotliwość stosowania należy zawsze określać indywidualnie w każdym przypadku, kierując się skutecznością kliniczną.
W przypadku wystąpienia poniżej wymienionych objawów krwawienia aktywność czynnika VIII nie powinna spadać poniżej podanego poziomu aktywności we krwi (w % normy lub j.m./dl) przez odpowiedni okres czasu. Tabelę 6 można wykorzystać jako wytyczne dotyczące dawkowania w przypadku krwawień i zabiegów chirurgicznych.
Tabela 6. Wytyczne dotyczące dawkowania w przypadku krwawień i zabiegów chirurgicznych
| Stopień krwawienia/rodzaj zabiegu chirurgicznego |
Wymagany poziom czynnika VIII (%) (j.m./ml) |
Częstotliwość podawania (godziny)/czas trwania terapii (dni) |
| Krwawienie |
||
| Początkowe objawy hemartrozy, krwawienia do mięśni lub krwawienia z jamy ustnej |
20−40 |
Podawać powtórnie co 12−24 godziny przez co najmniej 1 dzień do ustania krwawienia, co stwierdza się po braku bólu lub pełnym gojeniu |
| Bardziej nasilona hemartroza, krwawienie do mięśni lub hematoma |
30−60 |
Podawać powtórnie infuzję co 12−24 godziny przez 3−4 dni lub dłużej, aż do ustąpienia bólu i przywrócenia sprawności |
| Krwawienia stanowiące zagrożenie dla życia |
60−100 |
Podawać powtórnie infuzję co 8−24 godziny do ustąpienia zagrożenia dla życia |
| Zabiegi chirurgiczne |
||
| Małe zabiegi chirurgiczne, w tym ekstrakcja zęba |
30−60 |
Co 24 godziny przez co najmniej 1 dzień do pełnego gojenia |
| Duże zabiegi chirurgiczne |
80−100 (przed i po operacji) |
Podawać powtórnie infuzję co 8−24 godziny do wystarczającego gojenia rany, a następnie kontynuować terapię przez co najmniej 7 dni w celu utrzymania aktywności czynnika VIII na poziomie 30–60% (j.m./ml) |
Profilaktyka
W celu długoterminowej profilaktyki krwawień u pacjentów z ciężką hemofilią A zalecana dawka wynosi 20–40 JМ czynnika VIII na 1 kg masy ciała co drugi dzień lub 20–50 JМ czynnika VIII na 1 kg masy ciała 3 razy w tygodniu. U dorosłych i u nastolatków (powyżej 12 roku życia) możliwe jest rzadsze stosowanie leku (40–60 JМ/kg co trzeci dzień lub dwa razy w tygodniu). Czasami, szczególnie u młodszych pacjentów, mogą być konieczne krótsze odstępy między dawkami lub wyższe dawki.
Zabiegi chirurgiczne
Doświadczenie dotyczące stosowania leku u dzieci w trakcie zabiegów chirurgicznych jest ograniczone.
Pacjenci starsi
Brak doświadczenia w stosowaniu leku u pacjentów w wieku powyżej 65 lat.
Dzieci
W celu długoterminowej profilaktyki krwawień u pacjentów w wieku do 12 roku życia zalecane są dawki 25–50 JМ czynnika VIII na 1 kg masy ciała co drugi dzień lub 25–60 JМ czynnika VIII na 1 kg masy ciała 3 razy w tygodniu. Zalecenia dotyczące dawkowania leku dzieciom w wieku od 12 lat są takie same, jak u dorosłych pacjentów.
Sposób stosowania
Podanie dożylnie.
Zalecana szybkość infuzji leku Novoeit® wynosi 1–2 ml/min. Szybkość należy dostosować do poziomu komfortu pacjenta.
Instrukcja odtwarzania leku przed podaniem znajduje się w Instrukcji stosowania leku Novoeit®.
Przechowywanie po odtworzeniu
Stwierdzono chemiczną i fizyczną stabilność odtworzonego leku przez 24 godziny w temperaturze 2–8 °C; 4 godziny w temperaturze nie wyższej niż 30 °C, pod warunkiem, że lek był przechowywany nie dłużej niż 9 miesięcy w temperaturze pokojowej nie wyższej niż 30 °C od momentu produkcji do rozpoczęcia odtwarzania. Przechowywanie przez 4 godziny w temperaturze nie wyższej niż 40 °C jest dopuszczalne pod warunkiem, że lek był przechowywany nie dłużej niż 3 miesiące w temperaturze pokojowej 30–40 °C od momentu produkcji do rozpoczęcia odtwarzania. Z mikrobiologicznego punktu widzenia odtworzony lek należy stosować natychmiast. Jeśli lek nie został użyty od razu, użytkownik ponosi odpowiedzialność za warunki i czas przechowywania. W przypadku odtworzenia w kontrolowanych i zwalidowanych warunkach jałowych czas przechowywania odtworzonego leku nie powinien przekraczać okresu wskazanego powyżej.
Każdy niewykorzystany odtworzony lek, który był przechowywany dłużej niż 4 godziny w temperaturze pokojowej do 40 °C, należy zutylizować.
Instrukcja stosowania leku Novoeit®
UWAŻNIE PRZECZYTAJ INSTRUKCJĘ PRZED ZASTOSOWANIEM LECZUJĄCEGO NOVOEIT®.
Lek Novoeit® dostarczany jest w postaci proszku. Przed wstrzyknięciem (podaniem) należy go rozpuścić za pomocą rozpuszczalnika dołączonego w strzykawce. Rozpuszczalnikiem jest 0,9 % roztwór chlorku sodu (9 mg/ml). Lek Novoeit® podaje się dożylnie (wstrzyknięcie dożylne). Zawartość opakowania (patrz poniżej) przeznaczona jest do odtworzenia i podania leku Novoeit®.
Do podania leku potrzebny jest również zestaw do infuzji (układ rurek i igła „motyl”), jałowe waciki nasączone alkoholem, gaziki i plaster. Urządzenia i materiały te nie są zawarte w opakowaniu leku Novoeit®.
Nie należy używać zestawu bez wcześniejszego szkolenia przeprowadzonego przez lekarza lub pielęgniarkę.
Przed użyciem należy zawsze umyć ręce i upewnić się, że otoczenie jest czyste.
Podczas przygotowywania i podawania leku bezpośrednio do żyły niezwykle ważne jest przestrzeganie reguł aseptyki i antyseptyki. Nieprawidłowa technika wstrzykiwania może prowadzić do zakażenia krwi.
Nie należy otwierać zestawu, dopóki nie będzie się gotowym do jego użycia.
Nie należy używać zestawu, jeśli upadł lub został uszkodzony. Należy wtedy użyć nowego opakowania.
Nie należy używać zestawu po upływie daty ważności. Należy wtedy użyć nowego opakowania. Data ważności jest wydrukowana po słowach „Przydatny do” na tekturowej puszce, na fiolce, na łączniku fiolki oraz na wstępnie napełnionej strzykawce.
Nie należy używać zestawu, jeśli podejrzewa się jego zanieczyszczenie (kontaminację). Należy wtedy użyć nowego opakowania.
Nie należy wyrzucać żadnej części zestawu, dopóki nie poda się przygotowanego roztworu.
Zestaw przeznaczony jest do jednorazowego użycia.
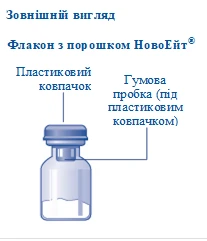
Zawartość opakowania:
1 fiolka z proszkiem Novoeit®
1 łącznik fiolki
1 wstępnie napełniona strzykawka z rozpuszczalnikiem
1 tłok strzykawki (znajduje się pod strzykawką)
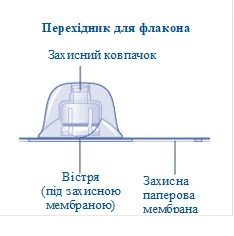
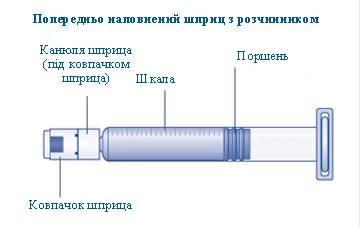

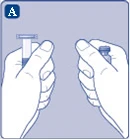
Rys. A
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Rys. B
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
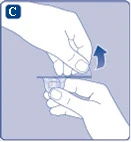
Rys. C
Jeśli uszczelnienie ochronnej papierowej membrany jest naruszone lub membrana jest uszkodzona, nie używaj przejściówki do fiolki. Nie wyjmuj przejściówki do fiolki z opakowania palcami. Dotknięcie igły przejściówki może prowadzić do przeniesienia mikroorganizmów z Twoich palców. |
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
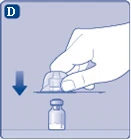
| Rys. D
Po przyłączeniu nie odłączaj przejściówki od fiolki. |
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
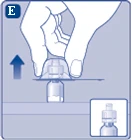
| Rys. E
Ściągnij ochronny kapturzek z przejściówki do fiolki. Nie odłączaj przejściówki do fiolki od fiolki, gdy ściągasz ochronny kapturzek. |
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
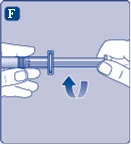
Rys. F
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
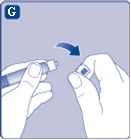
| Rys. G
Jeśli kapturzek strzykawki nie trzyma się dobrze lub jest uszkodzony, nie używaj tej wstępnie napełnionej strzykawki. |
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
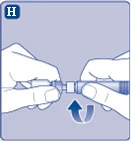
| Rys. H
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
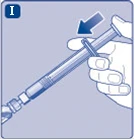
Rys. I
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Rys. J
Nie wstrząsaj fiolką, ponieważ może to prowadzić do powstawania piany.
|
Jeśli wymagana dawka jest większa niż zawarta w jednej fiolce, powtórz kroki od A do J z dodatkowymi fiolkami, przejściówkami do fiolki i wstępnie napełnionymi strzykawkami, aż uzyskasz wymaganą dawkę. |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
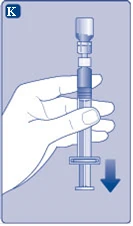
| Rys. K
Jeśli przypadkowo nabrało się powietrze do strzykawki, wypuść je z powrotem do fiolki.
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
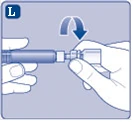
| Rys. L
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Lek Novoeit® jest teraz gotowy do podania dożylnego.
Wstrzykiwanie leku Novoeit® przez bezigłowe adaptery do cewników dożylnych (doż.) Uwaga! Wstępnie napełniona strzykawka wykonana jest ze szkła i przeznaczona jest do użytku ze standardowym połączeniem Luer-Lock. Niektóre bezigłowe adaptery z wewnętrznym kolcem są niekompatybilne z tą wstępnie napełnioną strzykawką. Taka niekompatybilność może utrudnić podanie leku i/lub spowodować uszkodzenie bezigłowego adaptera. Wstrzykiwanie roztworu przez urządzenie do dostępu do żył centralnych (UDŻC), takie jak cewnik żył centralnych (CŻC) lub port podskórny:
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Utylizacja Rys. M
Nie wyrzucaj tych materiałów do zwykłych odpadów komunalnych. |
Dzieci. Lek stosuje się u dzieci zgodnie z instrukcjami podanymi w sekcji „Sposób i dawki stosowania”. Przedawkowanie. Nie zgłaszano objawów przedawkowania rekombinowanym czynnikiem krzepnięcia krwi VIII. Reakcje niepożądane.Podsumowanie profilu bezpieczeństwa Reakcje nadwrażliwości lub alergiczne (mogące obejmować obrzęk naczynioruchowy, uczucie pieczenia i mrowienia w miejscu wlewu, dreszcze, zawroty głowy, uogólnione pokrzywienie, ból głowy, wysypkę, hipotensję tętniczą, letargię, nudności, niepokój, tachykardię, uczucie ucisku w klatce piersiowej, mrowienie, wymioty, świsty oddechowe) obserwowano rzadko, a w niektórych przypadkach mogły one postępować do ciężkiej reakcji anafilaktycznej (w tym szoku). Bardzo rzadko obserwowano powstawanie przeciwciał przeciwko białkom chomika oraz związane z tym reakcje nadwrażliwości. U pacjentów z hemofilią typu A mogą powstawać przeciwciała neutralizujące (inhibitory) przeciwko czynnika VIII. W przypadku powstania takich inhibitorów odpowiedź kliniczna będzie niewystarczająca. W takich przypadkach zaleca się skonsultowanie się ze specjalistycznym ośrodkiem leczenia hemofilii. Wykaz reakcji niepożądanych W tabeli 7 działania niepożądane są przedstawione zgodnie z klasyfikacją wg układów narządów (SOC) z podaniem terminów preferencyjnych słownika medycznego działalności regulacyjnej (MedDRA). Częstotliwość określono następująco: bardzo często (≥ 1/10), często (≥ 1/100 do < 1/10), rzadko (≥ 1/1000 do < 1/100), rzadko (≥ 1/10000 do < 1/1000), bardzo rzadko (< 1/10000); częstotliwość nieznana (niemożliwe oszacowanie na podstawie dostępnych danych). W ramach każdej grupy wg częstości działania niepożądane są uporządkowane według malejącego stopnia nasilenia. Tabela 7. Częstotliwość reakcji niepożądanych na lek w badaniach klinicznych
a Obliczono na podstawie całkowitej liczby poszczególnych pacjentów we wszystkich badaniach klinicznych (301), w tym 242 wcześniej leczonych pacjentów (ang. previously treated patients (PTPs)) oraz 60 nieleczonych wcześniej pacjentów (ang. previously untreated patients (PUPs)). b Dane dotyczące częstości oparte są na wynikach badań z wykorzystaniem wszystkich leków zawierających czynnik VIII, przeprowadzonych u pacjentów z ciężką hemofilią typu A. c Reakcje w miejscu iniekcji obejmują rumień w miejscu iniekcji, ekstrawazację w miejscu iniekcji oraz świąd w miejscu iniekcji. d Wzrost stężenia enzymów wątrobowych obejmuje alaninotransaminazę, asparginianotransaminazę, gamma-glutamylotransferazę i bilirubinę. Opis poszczególnych działań niepożądanych W trakcie badań klinicznych leku Novoeit® z udziałem pacjentów wcześniej leczonych zarejestrowano łącznie 35 działań niepożądanych u 23 z 242 pacjentów. Najczęstsze działania niepożądane to reakcje w miejscu iniekcji, komplikacje związane z podaniem niewłaściwej dawki oraz podwyższone stężenie enzymów wątrobowych. Dwa z 35 działań niepożądanych zaobserwowano u jednego z 31 pacjentów w wieku do 6 lat, żadnych nie odnotowano u pacjentów w wieku od 6 do 12 lat, jedno zaobserwowano u jednego z 24 pacjentów w wieku od 12 do 18 lat oraz 32 działania u 21 z 155 pacjentów dorosłych (w wieku od 18 lat). Dzieci W badaniach klinicznych z udziałem 63 dzieci w wieku od 0 do 12 lat oraz 24 nastolatków w wieku od 12 do 18 lat z ciężką hemofilią typu A nie zaobserwowano różnic w profilu bezpieczeństwa leku Novoeit® pomiędzy dziećmi a dorosłymi pacjentami. W trakcie badania z udziałem pacjentów w wieku od 0 do 6 lat, którzy wcześniej nie byli leczeni, zarejestrowano łącznie 46 działań niepożądanych u 33 z 60 pacjentów otrzymujących Novoeit®. Najczęstsze działanie niepożądane to inhibicja czynnika VIII (patrz sekcja „Szczególne ostrzeżenia i środki ostrożności”). U 92,3% wszystkich pacjentów oraz u 93,8% pacjentów z potwierdzonym wysokim mianem inhibitorów wykryto wysokie ryzyko mutacji genetycznych. Żadne inne czynniki nie były istotnie związane z powstawaniem inhibitorów. Zgłaszanie działań niepożądanych oraz braku skuteczności leku Zgłaszanie działań niepożądanych po rejestracji leku ma istotne znaczenie. Umożliwia to monitorowanie stosunku korzyści do ryzyka w trakcie stosowania danego leku. Osoby medyczne i farmaceutyczne, a także pacjenci lub ich ustawieni reprezentanci, powinni zgłaszać wszystkie przypadki podejrzewanych działań niepożądanych oraz braku skuteczności leku poprzez Automatyczny System Informacyjny do Farmakonadzoru pod adresem: https://aisf.dec.gov.ua. Okres ważności. Okres ważności gotowego leku wynosi 30 miesięcy. Nie należy stosować leku po upływie okresu ważności podanego na opakowaniu, fiolce oraz na etykiecie strzykawki. Ostateczną datą stosowania leku jest ostatni dzień bieżącego miesiąca. Warunki przechowywania. Lek należy przechowywać w temperaturze 2–8 °C. Nie zamarzać. Przechowywać w miejscu niedostępnym dla dzieci. W okresie ważności lek może być przechowywany w temperaturze pokojowej (nie wyższej niż 30 °C) przez okres nie dłuższy niż 9 miesięcy, licząc od daty produkcji do momentu rozpoczęcia rekonstytucji; lek może być przechowywany w temperaturze pokojowej (30–40 °C) przez okres nie dłuższy niż 3 miesiące, licząc od daty produkcji do momentu rozpoczęcia rekonstytucji. Nie należy ponownie umieszczać leku w lodówce. Na opakowaniu należy wskazać datę i temperaturę wyjęcia leku z lodówki. Przechowywać w opakowaniu zewnętrznym w celu ochrony przed światłem. Niezgodność. Ponieważ nie przeprowadzono badań zgodności, niniejszego leku nie wolno mieszać z innymi lekami. Opakowanie. Jedna fiolka szklana (typ I) o pojemności 5 ml zawierająca proszek, zamknięta korkiem gumowym chlorobutylowym i uszczelniona aluminiową pokrywką z plastikowym kapturkiem, w zestawie z roztworem do rozcieńczenia (0,9% roztwór chlorku sodu) w ilości 4 ml w strzykawce wstępnie napełnionej o pojemności 5 ml z ogranicznikiem przesuwu tłoka z polipropylenu, tłokiem z gumy bromobutylowej, nakrywką końcówki z korkiem gumowym bromobutylowym, tłokiem strzykawki z polipropylenu, oraz sterylnym łącznikiem do fiolki w opakowaniu indywidualnym, w pudełku tekturowym. Kategoria wydania. Na receptę. Wnioskodawca/Wytwórca. A/T Novo Nordisk. Miejsce pochodzenia producenta oraz adres miejsca prowadzenia działalności. Novo Allé, Bagsværd, 2880, Dania. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||