NovoEight®
UcraniaContenido
INSTRUCCIONES PARA USO MÉDICO DEL MEDICAMENTO NovoEight®
Composición:
Principio activo: turoctocog alfa;
1 frasco con polvo contiene 1500 UI o 2000 UI o 3000 UI de turoctocog alfa (factor de coagulación VIII humano (ADNr));
Excipientes: cloruro de sodio; L-histidina; sacarosa; polisorbato 80; L-metionina; cloruro de calcio dihidrato; hidróxido de sodio; ácido clorhídrico.
Disolvente: cloruro de sodio, agua para inyección.
Después de la reconstitución, 1 ml del medicamento NovoEight® contiene aproximadamente 375 UI, 500 UI o 750 UI de turoctocog alfa (factor de coagulación VIII humano (ADNr)).
Forma farmacéutica. Polvo y disolvente para solución inyectable.
Principales propiedades físico-químicas: polvo liofilizado o masa granular de color blanco o ligeramente amarillo. Disolvente: solución inyectable transparente e incolora.
Grupo farmacoterapéutico. Agentes hemostáticos. Factores de coagulación sanguínea. Factor de coagulación VIII.
Código ATC B02BD02.
Propiedades farmacológicas.
Farmacodinámica.
Mecanismo de acción
El medicamento NovoEight® contiene turcoctocog alfa, un factor de coagulación VIII humano (ADNr) con dominio B truncado. Esta glicoproteína tiene la misma estructura que el factor VIII humano en estado activado y las mismas modificaciones postraduccionales que la molécula aislada del plasma sanguíneo. Se ha demostrado que el sitio de sulfatación de la tirosina localizado en Tyr1680 (longitud completa del factor nativo), que es importante para la unión al factor de von Willebrand, está completamente sulfatado en la molécula de turcoctocog alfa. Tras la administración al paciente con hemofilia, el factor VIII se une al factor de von Willebrand endógeno en la sangre del paciente. El complejo de factor VIII/factor de von Willebrand está formado por dos moléculas (factor VIII y factor de von Willebrand) con diferentes funciones fisiológicas. El factor VIII activado actúa como cofactor del factor IX activado, acelerando la conversión del factor X en factor X activado. El factor X activado convierte la protrombina en trombina. Posteriormente, la trombina convierte el fibrinógeno en fibrina, formándose así un coágulo. La hemofilia A es un trastorno hereditario ligado al sexo del sistema de coagulación sanguínea debido a la disminución del nivel de factor VIII:C, lo que provoca hemorragias abundantes en articulaciones, músculos u órganos internos, tanto espontáneas como tras traumatismos o intervenciones quirúrgicas. Mediante la terapia sustitutiva, el nivel de factor VIII en plasma aumenta, corrigiendo temporalmente el déficit del factor y reduciendo la predisposición a las hemorragias.
Cabe señalar que la frecuencia anual de hemorragias (FAH) en diferentes concentraciones del factor de coagulación y en diversos estudios clínicos no es comparable.
Eficacia clínica
Se realizaron cuatro estudios clínicos abiertos, multicéntricos y no controlados con el fin de evaluar la seguridad y eficacia del uso del medicamento NovoEight® para la profilaxis y el tratamiento de hemorragias y durante intervenciones quirúrgicas en pacientes con hemofilia A grave (actividad del factor VIII ≤ 1 %). Tres de ellos incluyeron pacientes previamente tratados y uno incluyó pacientes previamente no tratados. En los estudios participaron 298 pacientes: 175 pacientes adultos y adolescentes de 12 años o más (≥ 150 días de tratamiento con el medicamento en estudio), en los que no se detectaron inhibidores; 63 pacientes pediátricos menores de 12 años (≥ 50 días de tratamiento con el medicamento en estudio), en los que no se detectaron inhibidores; y 60 pacientes menores de 6 años que no habían recibido tratamiento previo. 188 de los 238 pacientes previamente tratados continuaron participando en un estudio adicional de evaluación de seguridad. Se ha demostrado que el tratamiento con NovoEight® es seguro y proporciona el efecto hemostático y profiláctico deseado. Se registraron 3293 episodios de hemorragia observados en 298 pacientes, de los cuales 2902 (88,1 %) cesaron tras 1-2 infusiones del medicamento NovoEight®.
Tabla 1. Uso del medicamento NovoEight® y parámetros de eficacia en pacientes previamente no tratados y en aquellos que ya habían recibido tratamiento
| Indicador |
Niños del grupo de edad más joven (de 0 a menos de 6 años), que no habían recibido tratamiento |
Niños del grupo de edad más joven (de 0 a menos de 6 años), que habían recibido tratamiento |
Niños del grupo de edad más avanzada (de 6 a menos de 12 años), que habían recibido tratamiento |
Adolescentes (de 12 a menos de 18 años), que habían recibido tratamiento |
Adultos (≥ 18 años), que habían recibido tratamiento |
Total |
| Número de pacientes |
60 |
31 |
32 |
24 |
151 |
298 |
| Dosis utilizada para profilaxis por paciente (MO/kg de PM) |
||||||
| Valor medio (DE) |
45,2 (14,4) |
41,5 (8,1) |
38,4 (9,4) |
28,5 (9,3) |
28,5 (8,3) |
32,8 (10,9) |
| Mínimo; máximo |
4,5; 363,8 |
3,4; 196,3 |
3,2; 62,5 |
17,4; 73,9 |
12,0; 97,4 |
3,2; 363,8 |
| Dosis utilizada para el tratamiento de hemorragias (MO/kg de PM) |
||||||
| Valor medio (DE) |
43,6 (15,2) |
44,0 (12,6) |
40,4 (10,5) |
29,3 (10,3) |
35,0 (12,3) |
37,5 (13,4) |
| Mínimo; máximo |
11,9; 118,9 |
21,4; 193,8 |
24,0; 71,4 |
12,4; 76,8 |
6,4; 104,0 |
6,4; 193,8 |
| Índice de eficaciaa, % |
87,0 % |
92,2 % |
88,4 % |
85,1 % |
89,6 % |
88,9 % |
MT − masa corporal, DE − desviación estándar.
a La eficacia se definió como excelente o buena.
Los datos clínicos obtenidos antes de la autorización fueron confirmados mediante un estudio no intervencional de seguridad tras la autorización, realizado con el objetivo de proporcionar información adicional sobre la inmunogenicidad, eficacia y seguridad del medicamento NovoEight® en la práctica clínica habitual. En total, 68 pacientes previamente tratados (> 150 UI [unidades efectivas]), de los cuales 14 pacientes tenían menos de 12 años y 54 pacientes tenían 12 años o más, recibieron tratamiento a demanda (N = 5) o tratamiento profiláctico (N = 63) durante un total de 87,8 paciente-años y 8967 UIE.
Cirugía
Se realizaron un total de 30 intervenciones quirúrgicas en 25 pacientes, de las cuales 26 fueron intervenciones quirúrgicas mayores y 4 fueron menores. La hemostasia fue exitosa en todos los casos de intervenciones quirúrgicas; no se notificaron casos de tratamiento ineficaz.
Se recopilaron datos sobre la inducción de tolerancia inmunitaria (ITI) en pacientes con hemofilia A en los que se detectaron inhibidores del factor VIII. Durante los estudios clínicos con participación de pacientes que no habían recibido tratamiento previo (en inglés: previously untreated patients [PUPs]), 21 pacientes recibieron tratamiento con ITI y 18 pacientes (86 %) finalizaron la terapia ITI con resultados negativos en la prueba de detección de inhibidores.
Farmacocinética.
Todos los estudios farmacocinéticos con turcoctog alfa se realizaron tras la administración intravenosa de una dosis única de 50 UI/kg del medicamento NovoEight® en pacientes con hemofilia A grave (FVIII ≤ 1 %) previamente tratados. El análisis de las muestras de plasma se llevó a cabo mediante un ensayo cromogénico y un ensayo de actividad coagulante en una sola etapa.
La actividad del medicamento NovoEight® en el ensayo FVIII:C se evaluó y comparó con un producto recombinante de FVIII de longitud completa disponible en el mercado. El estudio demostró que los resultados obtenidos son comparables y coherentes para ambos productos y que el medicamento NovoEight® puede determinarse de forma fiable en plasma sin necesidad de un estándar específico para el producto NovoEight®.
Los parámetros farmacocinéticos del medicamento NovoEight® tras la administración de una dosis única, según los resultados del ensayo de actividad coagulante, se muestran en la tabla 2, y según los resultados del ensayo cromogénico en una sola etapa, en la tabla 3.
Tabla 2. Farmacocinética del turcoctog alfa (50 UI/kg) según la edad – Un solo paso, ensayo de actividad coagulante – Media (desviación estándar)
| Indicador |
De 0 a 6 años |
De 6 a 12 años |
≥ 12 años |
| n = 14 |
n = 14 |
n = 33 |
|
| Valor medio (DE) |
Valor medio (DE) |
Valor medio (DE) |
|
| Aumento del nivel (MO/dl)/(MO/kg) |
1,8 (0,7) |
2,0 (0,4) |
2,2 (0,4) |
| AUC ((MO·h)/dl) |
992 (411) |
1109 (374) |
1526 (577) |
| CL (ml/h/kg) |
6,21 (3,66) |
5,02 (1,68) |
3,63 (1,09) |
| t½ (h) |
7,65 (1,84) |
8,02 (1,89) |
11,00 (4,65) |
| Vss (ml/kg) |
56,68 (26,43) |
46,82 (10,63) |
47,40 (9,21) |
| Cmax (MO/dl) |
100 (58) |
107 (35) |
123 (41) |
| Duración media de acción (h) |
9,63 (2,50) |
9,91 (2,57) |
14,19 (5,08) |
AUC – área bajo la curva farmacocinética, que describe la dinámica temporal de la actividad del factor VIII; CL – aclaramiento; t1/2 – período final de semivida; Vss – volumen de distribución en estado de equilibrio; Cmax – actividad máxima del factor VIII.
Tabla 3. Farmacocinética de la turcoctog alfa (50 UOI/kg) según la edad – Análisis cromogénico – Media (desviación estándar)
| Indicador |
De 0 a < 6 años |
De 6 a < 12 años |
≥ 12 años |
| n = 14 |
n = 14 |
n = 33 |
|
| Valor medio (DE) |
Valor medio (DE) |
Valor medio (DE) |
|
| Aumento del nivel (MO/dl)/(MO/kg) |
2,2 (0,6) |
2,5 (0,6) |
2,9 (0,6) |
| AUC ((MO·h)/dl) |
1223 (436) |
1437 (348) |
1963 (773) |
| CL (ml/h/kg) |
4,59 (1,73) |
3,70 (1,00) |
2,86 (0,94) |
| t½ (h) |
9,99 (1,71) |
9,42 (1,52) |
11,22 (6,86) |
| Vss (ml/kg) |
55,46 (23,53) |
41,23 (6,00) |
38,18 (10,24) |
| Cmax (MO/dl) |
112 (31) |
125 (27) |
163 (50) |
| Duración media de acción (h) |
12,06 (1,90) |
11,61 (2,32) |
14,54 (5,77) |
AUC – área bajo la curva farmacocinética, que describe la dinámica temporal de la actividad del factor VIII; CL – aclaramiento; t1/2 – período final de semivida; Vss – volumen de distribución en estado de equilibrio; Cmax – actividad máxima del factor VIII.
Los parámetros farmacocinéticos fueron comparables en pacientes menores de 6 años y en aquellos de 6 a 12 años de edad. Se observaron algunas diferencias en los parámetros farmacocinéticos del medicamento NovoEight® entre pacientes pediátricos y adultos. El aclaramiento más elevado y el t½ más corto en niños en comparación con adultos con hemofilia A podrían explicarse parcialmente por un mayor volumen de plasma por kilogramo de peso corporal en pacientes más jóvenes.
Se realizó un estudio farmacocinético con una dosis única (50 UI/kg) en 35 pacientes con hemofilia (≥ 18 años) de diferentes categorías de IMC. La concentración máxima del fármaco en plasma (Cmax) y la concentración total del fármaco en plasma (AUC) aumentaron con el incremento del IMC, lo que indica que es necesaria una ajuste de la dosis en pacientes con bajo peso (IMC < 18,5 kg/m²) y con sobrepeso (IMC ≥ 30 kg/m²); véase la sección «Posología y forma de administración».
Tabla 4. Parámetros farmacocinéticos del medicamento NovoEight® tras la administración de una dosis única (50 UI/kg) según la categoría de IMCa – Ensayo de coagulación en una sola etapa – Valor medio (desviación estándar)
| Parámetro farmacocinético |
Masa corporal insuficiente, N = 5 |
Masa corporal normal, N = 7 |
Exceso de masa corporal, N = 8 |
Obesidad, clase I, N = 7 |
Obesidad, clase II/III, N = 7 |
| Aumento del nivel (MO/dl)/(MO/kg) |
1,7 (0,2) |
2,0 (0,2) |
2,4 (0,4) |
2,3 (0,3)b |
2,6 (0,3) |
| AUC ((MO*h)/dl) |
1510 (360) |
1920 (610) |
1730 (610) |
2030 (840) |
2350 (590) |
| CL (ml/h/kg) |
3,91 (0,94) |
3,20 (1,00) |
3,63 (1,24) |
3,37 (1,79) |
2,51 (0,63) |
| t½ (h) |
11,3 (2,0) |
11,7 (3,5) |
9,4 (2,9) |
11,2 (3,5) |
11,1 (2,7) |
| Vss (ml/kg) |
56,8 (5,4) |
44,8 (6,5) |
39,6 (6,0) |
42,0 (9,0) |
35,0 (4,6) |
| Cmax (MO/dl) |
100 (11) |
121 (10) |
144 (26) |
140 (21) |
161 (32) |
| Duración media de acción (h) |
15,1 (3,0) |
15,3 (4,8) |
11,9 (3,7) |
14,4 (4,6) |
14,6 (3,7) |
a Grupos de IMC: bajo peso, IMC < 18,5 kg/m²; peso normal, IMC 18,5–24,9 kg/m²; sobrepeso, IMC 25–29,9 kg/m²; obesidad, clase I, IMC 30–34,9 kg/m²; obesidad, clase II/III, IMC ≥ 35 kg/m².
b Los datos se obtuvieron únicamente para 6 pacientes.
Tabla 5. Parámetros farmacocinéticos del medicamento NovoEight® tras la administración de una dosis única (50 UME/kg) en función de la categoría de IMCa – Análisis cromogénico – Media (desviación estándar)
| Parámetro farmacocinético |
Bajo peso corporal, N = 5 |
Peso corporal normal, N = 7 |
Sobrepeso, N = 9 |
Obesidad, clase I, N = 7 |
Obesidad, clase II/III, N = 7 |
| Incremento del nivel (MO/dl)/(MO/kg) |
2,2 (0,4) |
2,9 (0,3) |
3,0 (0,5) |
3,2 (0,5) |
3,5 (0,5) |
| AUC ((MO·h)/dl) |
1860 (700) |
2730 (860) |
2310 (1020) |
2780 (1210) |
3050 (730) |
| CL (ml/h/kg) |
3,28 (0,87) |
2,25 (0,73) |
2,84 (1,09) |
2,58 (1,56) |
1,94 (0,52) |
| t½ (h) |
11,7 (2,4) |
11,5 (3,6) |
9,7 (3,4) |
10,4 (3,2) |
10,5 (2,5) |
| Vss (ml/kg) |
49,1 (10,4) |
31,2 (4,5) |
31,6 (5,8) |
28,9 (5,1) |
25,7 (4,0) |
| Cmax (MO/dl) |
138 (29) |
185 (24) |
194 (31) |
200 (33) |
227 (32) |
| Duración media del efecto (h) |
15,5 (3,2) |
15,2 (4,9) |
12,6 (4,8) |
13,5 (4,6) |
13,9 (3,7) |
a Grupos de IMC: bajo peso, IMC < 18,5 kg/m²; peso normal, IMC 18,5–24,9 kg/m²; sobrepeso, IMC 25–29,9 kg/m²; obesidad, clase I, IMC 30–34,9 kg/m²; obesidad, clase II/III, IMC ≥ 35 kg/m².
Datos preclínicos de seguridad
Los datos preclínicos indican que no existen riesgos particulares para el ser humano según los resultados de los estudios estándar de farmacología de seguridad y de estudios de toxicidad con dosis repetidas.
Características clínicas.
Indicaciones.
Tratamiento y profilaxis de las hemorragias en pacientes con hemofilia A (déficit congénito del factor VIII).
El medicamento NovoEight® puede utilizarse en pacientes de todas las edades.
Contraindicaciones.
Hipersensibilidad al principio activo o a cualquiera de los excipientes.
Reacción alérgica conocida a proteínas de hámster.
Interacción con otros medicamentos e interacciones de otro tipo.
No se han notificado interacciones entre los factores de coagulación VIII humanos (ADNr) y otros medicamentos.
Características de uso.
Seguimiento
Para mejorar el seguimiento de los medicamentos biológicos, es necesario registrar claramente el nombre y el número de lote del medicamento utilizado.
Hipersensibilidad
Pueden ocurrir reacciones de hipersensibilidad de tipo alérgico con el uso del medicamento NovoEight®. El medicamento contiene trazas de proteínas de hámster que pueden provocar reacciones alérgicas en algunos pacientes. Si aparecen síntomas de hipersensibilidad, se debe recomendar a los pacientes que interrumpan inmediatamente el uso del medicamento y consulten a un médico. Los pacientes deben ser informados sobre los signos tempranos de reacciones de hipersensibilidad, que incluyen erupciones cutáneas, urticaria generalizada, sensación de opresión en el pecho, sibilancias, hipotensión y anafilaxia.
En caso de shock, se debe iniciar el tratamiento farmacológico estándar contra el shock.
Inhibidores
La formación de anticuerpos neutralizantes (inhibidores) contra el factor VIII es una complicación conocida en el tratamiento de personas con hemofilia A. Estos inhibidores suelen ser inmunoglobulinas IgG que interfieren con la actividad procoagulante del factor VIII; cuantitativamente, se determinan en unidades de Bethesda (BU) por mililitro de plasma mediante un ensayo modificado. El riesgo de formación de inhibidores se correlaciona con la exposición al factor VIII y es más alto durante los primeros 50 días de exposición, aunque persiste durante toda la vida del paciente, aunque este riesgo es infrecuente.
La relevancia clínica de la formación de inhibidores dependerá de su título, siendo un título bajo el que conlleva un menor riesgo de respuesta clínica insatisfactoria en comparación con un título alto de inhibidores.
En general, se debe realizar un monitoreo cuidadoso de todos los pacientes que reciben tratamiento con medicamentos que contienen factor de coagulación VIII, en cuanto a la formación de inhibidores, mediante observación clínica adecuada y pruebas de laboratorio. Si no se logra alcanzar el nivel esperado de actividad del factor VIII en plasma o si no se logra controlar la hemorragia con la dosis adecuada, se debe realizar un estudio para detectar la presencia de inhibidores del factor VIII. En pacientes con niveles altos de inhibidores, la terapia con factor VIII puede ser ineficaz, por lo que se deben considerar otros métodos de tratamiento. El tratamiento de estos pacientes debe realizarse bajo la supervisión de médicos con experiencia en el manejo de pacientes con hemofilia y presencia de inhibidores del factor VIII.
Precauciones relacionadas con los excipientes
El medicamento contiene 30,5 mg de sodio por vial con el medicamento reconstituido, lo que equivale al 1,5 % del consumo diario máximo recomendado de sodio para un adulto según la OMS, que es de 2 g.
Complicaciones cardiovasculares
En pacientes con factores de riesgo cardiovasculares preexistentes, la terapia sustitutiva con factor VIII puede aumentar el riesgo de complicaciones cardiovasculares.
Complicaciones relacionadas con el catéter
Si es necesario colocar un catéter venoso central, se deben considerar los riesgos de complicaciones relacionadas con el catéter, incluyendo infecciones locales, bacteriemia y trombosis en el sitio de cateterización.
Con el fin de mantener el vínculo entre el estado del paciente y el lote del medicamento, se recomienda encarecidamente registrar el nombre y el número de lote del medicamento cada vez que se administre NovoEight® a un paciente.
Niños
Las advertencias y precauciones indicadas se aplican tanto a adultos como a niños.
Uso durante el embarazo o la lactancia.
No se han realizado estudios sobre el efecto del medicamento NovoEight® en la función reproductiva de animales. Debido a que la hemofilia A en mujeres es rara, no existe experiencia en el uso del factor VIII durante el embarazo ni la lactancia. Por lo tanto, el factor VIII debe usarse durante el embarazo y la lactancia solo cuando existan indicaciones claramente definidas.
Capacidad para afectar la velocidad de reacción al conducir vehículos o manejar maquinaria.
El medicamento NovoEight® no afecta la capacidad para conducir vehículos ni para trabajar con otras máquinas.
Vía de administración y dosis.
El tratamiento debe iniciarse bajo la supervisión de un médico con experiencia en el tratamiento de la hemofilia.
Monitorización del tratamiento
Durante el curso del tratamiento se recomienda la determinación adecuada de los niveles del factor VIII para ajustar la dosis y la frecuencia de las inyecciones repetidas. Los pacientes pueden presentar respuestas variables al factor VIII, así como diferentes periodos de semivida y recuperación. La dosis calculada según el peso corporal podría requerir ajustes en pacientes con bajo o exceso de peso. Los estudios de farmacocinética tras la administración de una dosis única en adultos mostraron que la concentración máxima del fármaco en plasma (Cmáx) y la concentración total del fármaco en plasma (AUC) aumentan con el incremento del IMC. Esto sugiere que podría ser necesaria una ajuste de la dosis. Los pacientes con bajo peso corporal (IMC < 18,5 kg/m²) podrían requerir un aumento de la dosis, mientras que los pacientes con obesidad (IMC ≥ 30 kg/m²) podrían necesitar una reducción. Sin embargo, actualmente no hay suficientes datos para recomendar ajustes específicos de dosis; véase la sección «Farmacocinética».
En particular, durante intervenciones quirúrgicas importantes, es obligatorio realizar una monitorización cuidadosa de la terapia sustitutiva mediante análisis de coagulación (actividad del factor VIII en plasma sanguíneo).
Al realizar un análisis uniestadio de coagulación basado en el tiempo de tromboplastina parcial in vitro (TTPa) para determinar la actividad del factor VIII en sangre, tanto el tipo de reactivo TTPa como el preparado de referencia utilizado en el ensayo pueden influir significativamente en los resultados de la determinación de la actividad del factor VIII en plasma. Asimismo, pueden existir diferencias importantes entre los resultados obtenidos mediante el análisis uniestadio basado en TTPa y los obtenidos mediante análisis cromogénico según la Farmacopea Europea. Esto es especialmente importante cuando se cambia de laboratorio y/o de reactivos utilizados en el análisis.
Dosificación
La dosificación y la duración de la terapia sustitutiva dependen del grado de deficiencia del factor VIII, la localización y gravedad de la hemorragia, así como del estado clínico del paciente.
La cantidad de unidades del factor VIII se expresa en unidades internacionales (UI), referidas al estándar vigente de la OMS para los productos del factor VIII. La actividad del factor VIII en plasma sanguíneo se expresa en porcentaje (en relación con el nivel normal en plasma humano) o en unidades internacionales (en relación con el estándar internacional para el factor VIII en plasma sanguíneo).
Una unidad internacional (UI) de actividad del factor VIII equivale a la cantidad de factor VIII presente en 1 ml de plasma sanguíneo de una persona normal.
Tratamiento a demanda
El cálculo de la dosis necesaria de factor VIII se basa en la observación empírica de que 1 unidad internacional (UI) de factor VIII por kg de peso corporal aumenta la actividad del factor VIII en plasma en 2 UI/dl. La dosis necesaria se determina mediante la siguiente fórmula:
Cantidad necesaria de unidades = peso corporal (kg) × incremento deseado del nivel de factor VIII (%) (UI/dl) × 0,5 (UI/kg por 1 UI/dl).
La cantidad a administrar y la frecuencia de uso siempre deben determinarse individualmente en cada caso, basándose en la eficacia clínica.
En caso de presentarse los fenómenos hemorrágicos indicados a continuación, la actividad del factor VIII no debe disminuir por debajo del nivel indicado (en % respecto al valor normal o en UI/dl) durante el periodo correspondiente. La tabla 6 puede utilizarse como guía para la dosificación en casos de hemorragia y cirugía.
Tabla 6. Guía de dosificación para hemorragias e intervenciones quirúrgicas
| Gravedad del sangrado/tipo de procedimiento quirúrgico |
Nivel necesario del factor VIII (%) (UI/dl) |
Frecuencia de administración (horas)/duración del tratamiento (días) |
| Sangrado |
||
| Signos iniciales de hemartrosis, hemorragia muscular o hemorragias en la cavidad oral |
20−40 |
Repetir cada 12−24 horas al menos 1 día hasta la detención del sangrado, determinada por la ausencia de dolor o hasta la cicatrización |
| Hemartrosis más marcada, hemorragia muscular o hematoma |
30−60 |
Repetir la infusión cada 12−24 horas durante 3−4 días o más, hasta la desaparición del dolor y la recuperación de la función |
| Hemorragias que ponen en peligro la vida |
60−100 |
Repetir la infusión cada 8−24 horas hasta la desaparición del riesgo vital |
| Intervenciones quirúrgicas |
||
| Pequeñas intervenciones quirúrgicas, incluyendo extracción dental |
30−60 |
Cada 24 horas al menos 1 día, hasta la cicatrización |
| Grandes intervenciones quirúrgicas |
80−100 (antes y después de la cirugía) |
Repetir la infusión cada 8−24 horas hasta una adecuada cicatrización de la herida; después continuar el tratamiento durante al menos 7 días más con el fin de mantener la actividad del factor VIII entre 30 y 60 % (UI/dl) |
Prevención
La dosis recomendada habitual para la profilaxis prolongada de hemorragias en pacientes con hemofilia A grave es de 20-40 UI del factor VIII por kg de peso corporal cada día o de 20-50 UI del factor VIII por kg de peso corporal tres veces por semana. En adultos y adolescentes (de más de 12 años de edad), el régimen de administración puede prever una frecuencia menor de aplicación del medicamento (40-60 UI/kg cada tercer día o dos veces por semana). A veces, especialmente en pacientes más jóvenes, pueden ser necesarios intervalos más cortos entre las administraciones o dosis más altas.
Intervención quirúrgica
La experiencia con el uso del medicamento en niños durante intervenciones quirúrgicas es limitada.
Pacientes de edad avanzada
No existe experiencia con el uso del medicamento en pacientes mayores de 65 años.
Niños
Para la profilaxis prolongada de hemorragias en pacientes menores de 12 años, se recomiendan dosis de 25-50 UI del factor VIII por kg de peso corporal cada día o de 25-60 UI del factor VIII por kg de peso corporal tres veces por semana. Las recomendaciones de dosificación para niños a partir de 12 años son las mismas que para pacientes adultos.
Vía de administración
Administración intravenosa.
La velocidad de infusión recomendada del medicamento NovoEight® es de 1-2 ml/min. La velocidad debe determinarse según el nivel de comodidad del paciente.
Las instrucciones para la reconstitución del medicamento antes de la administración se indican en las Instrucciones de uso del medicamento NovoEight®.
Conservación tras la reconstitución
Se ha demostrado la estabilidad química y física del medicamento reconstituido durante 24 horas a una temperatura de 2-8 °C; 4 horas a una temperatura no superior a 30 °C, siempre que el medicamento se haya conservado no más de 9 meses a temperatura ambiente no superior a 30 °C desde el momento de fabricación hasta el inicio de la reconstitución. Conservar 4 horas a una temperatura no superior a 40 °C, siempre que el medicamento se haya conservado no más de 3 meses a temperatura ambiente de 30-40 °C desde el momento de fabricación hasta el inicio de la reconstitución. Desde el punto de vista microbiológico, el medicamento reconstituido debe utilizarse inmediatamente. Si el medicamento no se utiliza inmediatamente, el usuario será responsable del período y condiciones de conservación. En caso de reconstitución en condiciones asépticas controladas y validadas, el período de conservación del medicamento reconstituido no debe exceder el período indicado anteriormente.
Cualquier medicamento reconstituido no utilizado que se haya conservado más de 4 horas a temperatura ambiente hasta 40 °C debe eliminarse.
Instrucciones de uso del medicamento NovoEight®
LEA ATENTAMENTE LAS INSTRUCCIONES ANTES DE UTILIZAR EL MEDICAMENTO NOVOEIGHT®.
El medicamento NovoEight® se suministra en forma de polvo. Antes de la inyección (administración), debe reconstituirse con el diluyente suministrado en la jeringa. El diluyente es solución de cloruro de sodio al 0,9 % (9 mg/ml). El medicamento NovoEight® se administra por vía intravenosa (inyección intravenosa). El contenido del envase (véase más abajo) está destinado a la reconstitución y administración del medicamento NovoEight®.
Para la administración del medicamento también se necesita un conjunto para infusión (sistema de tubos y aguja tipo "mariposa"), torundas estériles impregnadas con alcohol, gasas y esparadrapo. Estos dispositivos y materiales no están incluidos en el envase del medicamento NovoEight®.
No utilice el equipo sin haber recibido la debida formación del médico o enfermero.
Lávese siempre las manos antes de usarlo y asegúrese de que el área a su alrededor esté limpia.
Durante la preparación y administración del medicamento directamente en la vena, es muy importante cumplir estrictamente las normas de asepsia y antisepsia. Si la técnica de inyección es incorrecta, existe el riesgo de infección sanguínea.
No abra el equipo hasta que esté listo para usarlo.
No utilice el equipo si ha caído o está dañado. En su lugar, use un nuevo envase.
No utilice el equipo si ha expirado su fecha de caducidad. En su lugar, use un nuevo envase. La fecha de caducidad está impresa tras las palabras «Cad.» en la caja de cartón, en el frasco, en el adaptador para frasco y en la jeringa precargada.
No utilice el equipo si sospecha que está contaminado. En su lugar, use un nuevo envase.
No deseche ninguna parte del kit hasta que haya administrado la solución preparada.
El equipo está destinado para uso único.
Contenido del envase:
1 frasco con polvo NovoEight®
1 adaptador para frasco
1 jeringa precargada con diluyente
1 émbolo (se encuentra debajo de la jeringa)
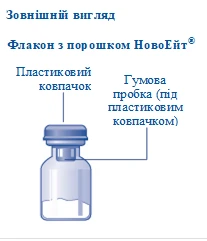
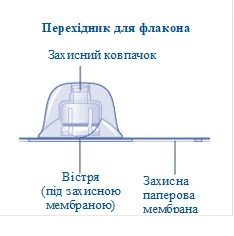
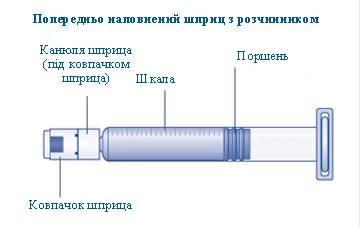

Fig. A
|
|
||
| Fig. B
|
|
||
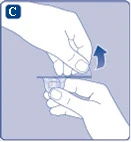
Fig. C
Si la membrana protectora de papel está dañada o rota, no utilice el conector para frasco. No saque el conector para frasco del envase con los dedos. Si toca la punta del conector para frasco, podría transmitir microorganismos desde sus dedos. |
|
||
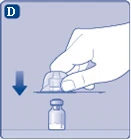
| Fig. D
Una vez conectado, no desmonte el conector del frasco. |
|
||
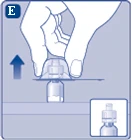
| Fig. E
Retire la tapa protectora del conector para frasco. No desmonte el conector para frasco del frasco al retirar la tapa protectora. |
|
||
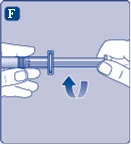
Fig. F
|
|
||
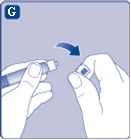
| Fig. G
Si la tapa de la jeringa no está bien ajustada o falta, no utilice esta jeringa precargada. |
|
||
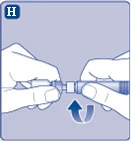
| Fig. H
|
|
||
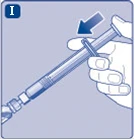
Fig. I
|
|
||
| Fig. J
No agite el frasco, ya que podría formarse espuma.
|
Si la dosis necesaria es mayor que la contenida en un solo frasco, repita los pasos de la A a la J con frascos adicionales, conectores para frascos y jeringas precargadas hasta obtener la dosis necesaria. |
||
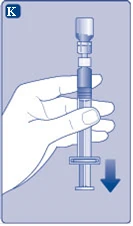
| Fig. K
Si accidentalmente entra aire en la jeringa, expúlselo de nuevo al frasco.
|
|
||
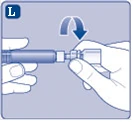
| Fig. L
|
|
||
El medicamento NovoEight® ya está listo para su administración intravenosa.
Inyección del medicamento NovoEight® mediante adaptadores intravenosos sin aguja (vía IV) ¡Atención! La jeringa precargada está hecha de vidrio y está diseñada para usarse con el tipo estándar de conexión Luer-Lok. Algunos adaptadores sin aguja con espiga interna no son compatibles con esta jeringa precargada. Esta incompatibilidad puede impedir la administración del medicamento y/o dañar el adaptador sin aguja. Inyección de la solución mediante un dispositivo de acceso venoso central (DAVC), como un catéter venoso central (CVC) o un puerto subcutáneo:
|
|||
| Desecho Fig. M
No deseche estos materiales con la basura doméstica. |
|
||
| No desmonte el equipo antes de desecharlo. No reutilice el equipo. |
|||

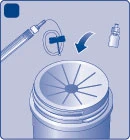
Niños.
El medicamento se puede administrar a niños según las instrucciones indicadas en la sección «Instrucciones de uso y dosis».
Sobredosificación.
No se han notificado síntomas de sobredosificación con el factor de coagulación VIII recombinante.
Reacciones adversas.
Resumen del perfil de seguridad
Se han observado reacciones de hipersensibilidad o alérgicas (que pueden incluir angioedema, sensación de ardor y picor en el lugar de la infusión, escalofríos, sofocos, urticaria generalizada, cefalea, erupción cutánea, hipotensión arterial, letargo, náuseas, inquietud, taquicardia, opresión en el pecho, hormigueo, vómitos, sibilancias) raramente, y en algunos casos pueden progresar hasta una anafilaxia grave (incluyendo shock).
Muy raramente se han observado la formación de anticuerpos contra proteínas de hámster y reacciones de hipersensibilidad relacionadas.
En pacientes con hemofilia A pueden desarrollarse anticuerpos neutralizantes (inhibidores) contra el factor VIII. Si se forman estos inhibidores, la respuesta clínica será insuficiente. En tales casos, se recomienda consultar con un centro especializado en el tratamiento de la hemofilia.
Lista de reacciones adversas
En la Tabla 7, las reacciones adversas se presentan según la clasificación por órganos y sistemas corporales (SOC), indicando los términos de MedDRA (Diccionario Médico para Actividades Regulatorias).
La frecuencia se define de la siguiente manera: muy frecuentes (≥ 1/10), frecuentes (≥ 1/100 hasta < 1/10), poco frecuentes (≥ 1/1.000 hasta < 1/100), raras (≥ 1/10.000 hasta < 1/1.000), muy raras (< 1/10.000); frecuencia desconocida (no puede estimarse a partir de los datos disponibles).
Dentro de cada grupo por frecuencia, las reacciones adversas se presentan en orden decreciente de gravedad.
Tabla 7. Frecuencia de reacciones adversas al medicamento en estudios clínicos
| Clases de sistemas de órganos |
Frecuenciaa en pacientes previamente tratados (PTPs) |
Frecuenciaa en pacientes previamente no tratados (PUPs) |
Reacción adversa |
| Alteraciones del sistema sanguíneo y del sistema linfático |
No frecuenteb |
Muy frecuenteb |
Inhibición del factor VIII |
| Trastornos psiquiátricos |
No frecuente |
Insomnio |
|
| Alteraciones del sistema nervioso |
No frecuente |
Dolor de cabeza, mareo, sensación de ardor |
|
| Trastornos cardiacos |
No frecuente |
Taquicardia sinusal, infarto agudo de miocardio |
|
| Alteraciones del sistema vascular |
No frecuente |
Hipertensión arterial, linfedema, hiperemia |
|
| Frecuente |
Alegrías, tromboflebitis de venas superficiales |
||
| Alteraciones de la piel y del tejido subcutáneo |
Frecuente |
Erupción cutánea, erupción eritematosa |
|
| No frecuente |
Erupción cutánea, queratosis liquenoide, sensación de ardor en la piel |
||
| Alteraciones del sistema osteomuscular y del tejido conjuntivo |
No frecuente |
Rigidez muscular y articular, artropatía, dolor en las extremidades, dolor muscular y óseo |
|
| Frecuente |
Hemartrosis, hemorragia muscular |
||
| Alteraciones del sistema respiratorio, órganos del tórax y mediastino |
Frecuente |
Tos |
|
| Trastornos generales y condiciones en el sitio de administración |
Frecuente |
Reacciones en el sitio de inyecciónv |
|
| Frecuente |
Hipertemia, eritema en el sitio de cateterización |
||
| No frecuente |
Cansancio, sensación de calor, edema periférico, hipertemia |
||
| Alteraciones en los resultados de pruebas de laboratorio |
Frecuente |
Niveles elevados de enzimas hepáticash |
|
| Frecuente |
Resultado positivo en la prueba de anticuerpos contra el factor VIII |
||
| No frecuente |
Frecuencia cardíaca elevada |
||
| Alteraciones del tracto gastrointestinal |
Frecuente |
Vómitos |
|
| Lesiones, envenenamientos y complicaciones de procedimientos |
Frecuente |
Dosis administrada incorrecta |
|
| Frecuente |
Reacción relacionada con la realización del procedimiento de infusión |
||
| No frecuente |
Contusión |
||
| Disfunción técnica |
Frecuente |
Oclusión del dispositivo |
a Calculado sobre la base del número total de pacientes individuales en todos los estudios clínicos (301), de los cuales 242 habían recibido tratamiento previo (en inglés: previously treated patients (PTPs)) y 60 no habían recibido tratamiento previo (en inglés: previously untreated patients (PUPs)).
b Las tasas de frecuencia se basan en datos de estudios con el uso de todos los medicamentos del factor VIII en pacientes con hemofilia A grave.
c Las reacciones en el sitio de inyección incluyen eritema en el sitio de inyección, extravasación en el sitio de inyección y prurito en el sitio de inyección.
d Las enzimas hepáticas con niveles elevados incluyen alanina aminotransferasa, aspartato aminotransferasa, gamma-glutamil transferasa y bilirrubina.
Descripción de reacciones adversas individuales
Durante los estudios clínicos con NovoEight® en pacientes previamente tratados, se registraron en total 35 reacciones adversas en 23 de 242 pacientes. Las reacciones adversas más frecuentes fueron reacciones en el sitio de inyección, complicaciones relacionadas con la administración de una dosis incorrecta y aumento de los niveles de enzimas hepáticas. Dos de las 35 reacciones adversas se observaron en uno de 31 pacientes menores de 6 años, ninguna se observó en pacientes de 6 a 12 años, una en un paciente de 12 a 18 años (1 de 24) y 32 en 21 de 155 pacientes adultos (de 18 años o más).
niños
En estudios clínicos con 63 niños de 0 a 12 años y 24 adolescentes de 12 a 18 años con hemofilia A grave, no se observaron diferencias en el perfil de seguridad de NovoEight® entre niños y adultos.
En un estudio con pacientes de 0 a 6 años que no habían recibido tratamiento previo, se registraron en total 46 reacciones adversas en 33 de 60 pacientes tratados con NovoEight®. La reacción adversa más frecuente fue la aparición de inhibidores del factor VIII (véase la sección «Propiedades farmacodinámicas»). En el 92,3 % del total de pacientes y en el 93,8 % de los pacientes con título confirmado alto de inhibidores se identificó un alto riesgo de mutaciones genéticas. No se encontraron otros factores significativamente asociados con la formación de inhibidores.
Notificación de reacciones adversas y falta de eficacia del medicamento
La notificación de reacciones adversas tras la autorización del medicamento es importante. Permite realizar un seguimiento continuo de la relación beneficio-riesgo del medicamento. Los profesionales médicos y farmacéuticos, así como los pacientes o sus representantes legales, deben informar sobre todos los casos sospechosos de reacciones adversas y falta de eficacia del medicamento a través del Sistema de Información Automatizado de Farmacovigilancia en el enlace: https://aisf.dec.gov.ua.
Período de validez.
El período de validez del medicamento terminado es de 30 meses.
No utilizar el medicamento después de la fecha de caducidad indicada en la caja, el frasco y la etiqueta de la jeringa. La fecha límite de uso del medicamento es el último día del mes indicado.
Condiciones de almacenamiento.
Conservar el medicamento a una temperatura de 2-8 °C. No congelar. Mantener fuera del alcance de los niños. Durante el período de validez, el medicamento puede conservarse a temperatura ambiente (no superior a 30 °C) desde el momento de fabricación hasta el inicio de la reconstitución durante un máximo de 9 meses; el medicamento puede conservarse a temperatura ambiente (30-40 °C) desde el momento de fabricación hasta el inicio de la reconstitución durante un máximo de 3 meses. No se debe volver a colocar el medicamento en el refrigerador. Debe anotarse en la caja la fecha y la temperatura en que se retiró el medicamento del refrigerador.
Conservar en el embalaje exterior para protegerlo de la luz.
Incompatibilidad.
Dado que no se han realizado estudios de compatibilidad, este medicamento no debe mezclarse con otros medicamentos.
Envase.
Un frasco de vidrio (tipo I) de 5 ml con polvo, cerrado con tapón de goma de clorobutilo y sellado con tapa de aluminio con tapa plástica desmontable, acompañado de un solvente (solución de cloruro de sodio al 0,9 %) en jeringa precargada de 4 ml, jeringa de 5 ml con tope de émbolo de polipropileno, émbolo de goma de bromobutilo, capuchón para la boquilla con tapón de goma de bromobutilo, vástago del émbolo de polipropileno, así como un conector estéril para frasco, todo en un embalaje individual dentro de una caja de cartón.
Categoría de dispensación.
Medicamento sujeto a prescripción médica.
Titular del registro / Fabricante.
A/T Novo Nordisk.
Dirección del fabricante y lugar de actividad.
Novo Allé, Bagsværd, 2880, Dinamarca.