Novoeight
UkraineTable of Contents
INSTRUCTIONS FOR MEDICAL USE OF THE MEDICINAL PRODUCT NovoEight® (NovoEight®)
Composition:
Active substance: turoctocog alfa;
One vial of powder contains 1500 IU or 2000 IU or 3000 IU of turoctocog alfa (recombinant human coagulation factor VIII (rDNA));
Excipients: sodium chloride; L-histidine; sucrose; polysorbate 80; L-methionine; calcium chloride, dihydrate; sodium hydroxide; hydrochloric acid.
Solvent: sodium chloride, water for injections.
After reconstitution, 1 ml of NovoEight® solution contains approximately 375 IU, 500 IU or 750 IU of turoctocog alfa (recombinant human coagulation factor VIII (rDNA)).
Pharmaceutical form. Powder and solvent for solution for injection.
Main physicochemical properties: lyophilized powder or loose mass, white or slightly yellow. Solvent: clear, colorless injection solution.
Pharmacotherapeutic group. Hemostatic agents. Coagulation factors. Coagulation factor VIII.
ATC code B02B D02.
Pharmacological properties.
Pharmacodynamics.
Mechanism of action
NovoEight® contains turoctocog alfa, a human coagulation factor VIII (recombinant DNA) with a truncated B-domain. This glycoprotein has the same structure as activated human factor VIII and the same post-translational modifications as the molecule isolated from plasma. It has been established that the tyrosine sulfation site located at Tyr1680 (full-length native factor) – which is important for binding to von Willebrand factor – is fully sulfated in the turoctocog alfa molecule. When administered to patients with hemophilia, factor VIII binds to endogenous von Willebrand factor in the patient's blood. The factor VIII/von Willebrand factor complex consists of two molecules (factor VIII and von Willebrand factor) with different physiological functions. Activated factor VIII acts as a cofactor for activated factor IX, accelerating the conversion of factor X to activated factor X. Activated factor X then converts prothrombin to thrombin. Thrombin, in turn, converts fibrinogen into fibrin, resulting in clot formation. Hemophilia A is an X-linked inherited coagulation disorder caused by reduced levels of factor VIII:C, leading to severe bleeding into joints, muscles, or internal organs, either spontaneously or following trauma or surgical procedures. Replacement therapy increases the plasma level of factor VIII, thereby providing temporary correction of the deficiency and reducing the tendency to bleed.
It should be noted that the annual bleeding rate (ABR) at different factor concentrations and across various clinical studies is not comparable.
Clinical efficacy
Four multicenter, open-label, uncontrolled clinical trials were conducted to evaluate the safety and efficacy of NovoEight® for prophylaxis and treatment of bleeding episodes and during surgical procedures in patients with severe hemophilia A (factor VIII activity ≤ 1%). Three of these studies involved previously treated patients, and one involved previously untreated patients. A total of 298 patients participated in the studies: 175 adults and adolescents aged 12 years and older (≥150 days of exposure to the study drug) without inhibitors detected, 63 pediatric patients under 12 years of age (≥50 days of exposure to the study drug) without inhibitors detected, and 60 patients under 6 years of age who were previously untreated. Of the 238 previously treated patients, 188 continued into an additional safety assessment study. Treatment with NovoEight® was shown to be safe and provided the desired hemostatic and prophylactic effect. A total of 3,293 bleeding episodes were recorded in 298 patients; 2,902 (88.1%) of these bleeding episodes were resolved after 1–2 infusions of NovoEight®.
Table 1. Use of NovoEight® and efficacy outcomes in previously untreated and previously treated patients
| Parameter |
Younger age group children (0 to <6 years), untreated |
Younger age group children (0 to <6 years), treated |
Older age group children (6 to <12 years), treated |
Adolescents (12 to <18 years), treated |
Adults (≥18 years), treated |
Total |
| Number of patients |
60 |
31 |
32 |
24 |
151 |
298 |
| Dose used for prophylaxis per patient (IU/kg BW) |
||||||
| Mean (SD) |
45.2 (14.4) |
41.5 (8.1) |
38.4 (9.4) |
28.5 (9.3) |
28.5 (8.3) |
32.8 (10.9) |
| Minimum; maximum |
4.5; 363.8 |
3.4; 196.3 |
3.2; 62.5 |
17.4; 73.9 |
12.0; 97.4 |
3.2; 363.8 |
| Dose used for treatment of bleeding (IU/kg BW) |
||||||
| Mean (SD) |
43.6 (15.2) |
44.0 (12.6) |
40.4 (10.5) |
29.3 (10.3) |
35.0 (12.3) |
37.5 (13.4) |
| Minimum; maximum |
11.9; 118.9 |
21.4; 193.8 |
24.0; 71.4 |
12.4; 76.8 |
6.4; 104.0 |
6.4; 193.8 |
| Effectiveness ratea, % |
87.0 % |
92.2 % |
88.4 % |
85.1 % |
89.6 % |
88.9 % |
MT − body weight, SD − standard deviation.
a Efficacy defined as excellent or good.
The clinical data obtained prior to registration were confirmed by a post-marketing non-interventional safety study conducted to provide additional information on the immunogenicity, efficacy, and safety of NovoEight® under routine clinical practice. Overall, 68 previously treated patients (> 150 ED (effective doses)), including 14 patients under 12 years of age and 54 patients aged 12 years and older, received treatment on-demand (N = 5) or prophylactic treatment (N = 63) for a total of 87.8 patient-years and 8967 ED.
Surgery
A total of 30 surgical procedures were performed in 25 patients, including 26 major surgeries and 4 minor surgeries. Hemostasis was successful in all surgical procedures; no treatment inefficacy was reported.
Data on immune tolerance induction (ITI) were collected in patients with hemophilia A who developed factor VIII inhibitors. During clinical trials involving previously untreated patients (PUPs), 21 patients received ITI treatment, and 18 patients (86%) completed ITI therapy with a negative inhibitor test result.
Pharmacokinetics
All pharmacokinetic studies with turoctocog alfa were conducted after intravenous administration of 50 IU/kg of NovoEight® in patients with severe hemophilia A (FVIII ≤ 1%) who had previously been treated. Plasma samples were analyzed using a one-stage clotting activity assay and a chromogenic assay.
The activity of NovoEight® in the FVIII:C assay was assessed and compared with a marketed full-length recombinant FVIII product. The study demonstrated that the results were comparable and consistent between the two products, and that NovoEight® can be reliably measured in plasma without the need for a product-specific standard.
Pharmacokinetic parameters of NovoEight® after single-dose administration, based on the clotting activity assay, are presented in Table 2, and those based on the one-stage chromogenic assay are presented in Table 3.
Table 2. Pharmacokinetics of turoctocog alfa (50 IU/kg) by age – One-stage clotting activity assay – Mean (standard deviation)
| Parameter |
From 0 to 6 years |
From 6 to 12 years |
≥ 12 years |
| n = 14 |
n = 14 |
n = 33 |
|
| Mean (SD) |
Mean (SD) |
Mean (SD) |
|
| Increase in level (MO/dl)/(MO/kg) |
1.8 (0.7) |
2.0 (0.4) |
2.2 (0.4) |
| AUC ((MO*hr)/dl) |
992 (411) |
1109 (374) |
1526 (577) |
| CL (ml/hr/kg) |
6.21 (3.66) |
5.02 (1.68) |
3.63 (1.09) |
| t½ (hr) |
7.65 (1.84) |
8.02 (1.89) |
11.00 (4.65) |
| Vss (ml/kg) |
56.68 (26.43) |
46.82 (10.63) |
47.40 (9.21) |
| Cmax (MO/dl) |
100 (58) |
107 (35) |
123 (41) |
| Mean duration of action (hr) |
9.63 (2.50) |
9.91 (2.57) |
14.19 (5.08) |
AUC – area under the pharmacokinetic curve describing the time course of factor VIII activity; CL – clearance; t1/2 – terminal half-life; Vss – volume of distribution at steady state; Cmax – maximum factor VIII activity.
Table 3. Pharmacokinetics of turoctocog alfa (50 IU/kg) by age – Chromogenic assay – Mean (standard deviation)
| Parameter |
From 0 to < 6 years |
From 6 to < 12 years |
≥ 12 years |
| n = 14 |
n = 14 |
n = 33 |
|
| Mean (SD) |
Mean (SD) |
Mean (SD) |
|
| Peak increase level (MO/dl)/(MO/kg) |
2.2 (0.6) |
2.5 (0.6) |
2.9 (0.6) |
| AUC ((MO*hr)/dl) |
1223 (436) |
1437 (348) |
1963 (773) |
| CL (ml/hr/kg) |
4.59 (1.73) |
3.70 (1.00) |
2.86 (0.94) |
| t½ (hr) |
9.99 (1.71) |
9.42 (1.52) |
11.22 (6.86) |
| Vss (ml/kg) |
55.46 (23.53) |
41.23 (6.00) |
38.18 (10.24) |
| Cmax (MO/dl) |
112 (31) |
125 (27) |
163 (50) |
| Mean duration of action (hr) |
12.06 (1.90) |
11.61 (2.32) |
14.54 (5.77) |
AUC – area under the pharmacokinetic curve describing the time course of factor VIII activity; CL – clearance; t1/2 – terminal half-life; Vss – volume of distribution at steady state; Cmax – maximum factor VIII activity.
Pharmacokinetic parameters were comparable between patients under 6 years of age and those aged 6 to 12 years. Some differences in pharmacokinetic parameters of NovoEight® were observed between pediatric and adult patients. Higher clearance and shorter t½ in children compared to adults with hemophilia A may be partially explained by the larger plasma volume per kilogram of body weight in younger patients.
A pharmacokinetic study with single-dose administration (50 IU/kg) was conducted in 35 hemophilia patients (≥ 18 years) across different BMI categories. Maximum plasma concentration (Cmax) and total plasma concentration (AUC) increased with increasing BMI, indicating that dose adjustment may be required for patients with underweight (BMI < 18.5 kg/m²) and overweight/obesity (BMI ≥ 30 kg/m²); see section "Dosage and administration".
Table 4. Pharmacokinetic parameters of NovoEight® after administration of a single dose (50 IU/kg) according to BMI classa – One-stage clotting assay – Mean (standard deviation)
| Pharmacokinetic parameter |
Underweight, N = 5 |
Normal weight, N = 7 |
Overweight, N = 8 |
Obesity, class I, N = 7 |
Obesity, class II/III, N = 7 |
| Level increase (MO/dl)/(MO/kg) |
1.7 (0.2) |
2.0 (0.2) |
2.4 (0.4) |
2.3 (0.3)b |
2.6 (0.3) |
| AUC ((MO*hr)/dl) |
1510 (360) |
1920 (610) |
1730 (610) |
2030 (840) |
2350 (590) |
| CL (ml/hr/kg) |
3.91 (0.94) |
3.20 (1.00) |
3.63 (1.24) |
3.37 (1.79) |
2.51 (0.63) |
| t½ (hr) |
11.3 (2.0) |
11.7 (3.5) |
9.4 (2.9) |
11.2 (3.5) |
11.1 (2.7) |
| Vss (ml/kg) |
56.8 (5.4) |
44.8 (6.5) |
39.6 (6.0) |
42.0 (9.0) |
35.0 (4.6) |
| Cmax (MO/dl) |
100 (11) |
121 (10) |
144 (26) |
140 (21) |
161 (32) |
| Mean duration of effect (hr) |
15.1 (3.0) |
15.3 (4.8) |
11.9 (3.7) |
14.4 (4.6) |
14.6 (3.7) |
a BMI categories: underweight, BMI < 18.5 kg/m²; normal weight, BMI 18.5–24.9 kg/m²; overweight, BMI 25–29.9 kg/m²; obesity, class I, BMI 30–34.9 kg/m²; obesity, class II/III, BMI ≥ 35 kg/m².
b Data obtained for only 6 patients.
Table 5. Pharmacokinetic parameters of NovoEight® following a single dose (50 IU/kg) according to BMI categorya – Chromogenic assay – Mean (standard deviation)
| Pharmacokinetic parameter |
Underweight, N = 5 |
Normal weight, N = 7 |
Overweight, N = 9 |
Obesity, class I, N = 7 |
Obesity, class II/III, N = 7 |
| Level increase (MO/dl)/(MO/kg) |
2.2 (0.4) |
2.9 (0.3) |
3.0 (0.5) |
3.2 (0.5) |
3.5 (0.5) |
| AUC ((MO*hr)/dl) |
1860 (700) |
2730 (860) |
2310 (1020) |
2780 (1210) |
3050 (730) |
| CL (ml/hr/kg) |
3.28 (0.87) |
2.25 (0.73) |
2.84 (1.09) |
2.58 (1.56) |
1.94 (0.52) |
| t½ (hr) |
11.7 (2.4) |
11.5 (3.6) |
9.7 (3.4) |
10.4 (3.2) |
10.5 (2.5) |
| Vss (ml/kg) |
49.1 (10.4) |
31.2 (4.5) |
31.6 (5.8) |
28.9 (5.1) |
25.7 (4.0) |
| Cmax (MO/dl) |
138 (29) |
185 (24) |
194 (31) |
200 (33) |
227 (32) |
| Mean duration of action (hr) |
15.5 (3.2) |
15.2 (4.9) |
12.6 (4.8) |
13.5 (4.6) |
13.9 (3.7) |
a BMI categories: underweight, BMI < 18.5 kg/m²; normal weight, BMI 18.5–24.9 kg/m²; overweight, BMI 25–29.9 kg/m²; obesity, class I, BMI 30–34.9 kg/m²; obesity, class II/III, BMI ≥ 35 kg/m².
Preclinical safety data
Preclinical data indicate no special risk to humans based on the results of standard safety pharmacology and repeated-dose toxicity studies.
Clinical characteristics.
Indications.
Treatment and prevention of bleeding episodes in patients with hemophilia A (congenital factor VIII deficiency).
NovoEight® can be used in patients of all age groups.
Contraindications.
Hypersensitivity to the active substance or to any of the excipients.
Known allergic reaction to hamster proteins.
Interaction with other medicinal products and other forms of interaction.
No interactions of human coagulation factor VIII (rDNA) with other medicinal products have been reported.
Special precautions for use
Traceability
To improve traceability of biological medicinal products, it is essential to clearly record the name and batch number of the administered product.
Hypersensitivity
Allergic-type hypersensitivity reactions may occur during administration of NovoEight®. The product contains trace amounts of hamster proteins, which may cause allergic reactions in some patients. If symptoms of hypersensitivity occur, patients should be advised to immediately discontinue use of the medicinal product and contact their physician. Patients should be informed about early signs of hypersensitivity reactions, including rash, generalized urticaria, chest tightness, wheezing, hypotension, and anaphylaxis.
In case of shock, standard anti-shock medical treatment should be initiated.
Inhibitors
Development of neutralizing antibodies (inhibitors) to factor VIII is a known complication in the treatment of patients with haemophilia A. These inhibitors are typically immunoglobulins IgG that interfere with the procoagulant activity of factor VIII; quantitatively, they are measured in Bethesda Units (BU) per 1 ml of plasma using a modified assay. The risk of inhibitor development correlates with factor VIII exposure and is highest during the first 50 exposure days, but persists throughout the patient's life, although this risk is uncommon.
The clinical significance of inhibitor formation depends on their titer, with a low titer posing a lower risk of inadequate clinical response compared to a high inhibitor titer.
Overall, all patients receiving treatment with factor VIII coagulation factor products should be carefully monitored for the development of inhibitors through appropriate clinical observation and laboratory testing. If the expected factor VIII plasma activity level cannot be achieved, or if bleeding cannot be controlled with an appropriate dose, testing for the presence of factor VIII inhibitors should be performed. In patients with high inhibitor titers, factor VIII therapy may be ineffective, and alternative treatment approaches should be considered. Management of such patients should be conducted under the supervision of physicians experienced in treating patients with haemophilia and factor VIII inhibitors.
Precautions related to excipients
The medicinal product contains 30.5 mg of sodium per vial of reconstituted product, which is equivalent to 1.5% of the maximum daily sodium intake for an adult recommended by WHO, set at 2 g.
Cardiovascular complications
In patients with existing cardiovascular risk factors, replacement therapy with factor VIII may increase the risk of cardiovascular complications.
Catheter-related complications
If a central venous catheter is required, the risk of catheter-related complications, including local infections, bacteremia, and thrombosis at the catheter site, should be considered.
To maintain the link between the patient's condition and the product batch, it is strongly recommended to record the name and batch number of the product each time NovoEight® is administered to a patient.
Paediatric population
The aforementioned warnings and precautions apply to both adults and children.
Use during pregnancy or breastfeeding
Studies on the effect of NovoEight® on reproductive function in animals have not been conducted. Since haemophilia A in females is rare, experience with the use of factor VIII during pregnancy and breastfeeding is lacking. Therefore, factor VIII should be used in pregnant women and women during lactation only when clearly indicated.
Ability to affect reaction rate while driving or operating machinery
NovoEight® does not affect the ability to drive or operate machinery.
Administration and Dosage
Treatment should be initiated under the supervision of a physician experienced in the management of hemophilia.
Monitoring of Treatment
During treatment, appropriate monitoring of factor VIII levels is recommended to adjust the dose and frequency of repeat injections. Patients may exhibit varying responses to factor VIII, as well as differences in half-life and recovery. The dose calculated based on body weight may require adjustment in patients with underweight or overweight. Pharmacokinetic studies following single-dose administration in adult patients have shown that maximum plasma concentration (Cmax) and total plasma concentration (AUC) of the drug increase with increasing BMI. This suggests that dose adjustments may be necessary. Patients with underweight (BMI < 18.5 kg/m²) may require higher doses, while obese patients (BMI ≥ 30 kg/m²) may require lower doses. However, currently there is insufficient data to provide specific recommendations for dose adjustment; see section "Pharmacokinetics".
In particular, careful monitoring of replacement therapy through coagulation assays (plasma factor VIII activity) is mandatory during major surgical procedures.
When performing a one-stage coagulation assay based on activated partial thromboplastin time (in vitro aPTT) to determine factor VIII activity in patient plasma, both the type of aPTT reagent used and the reference standard applied in the assay may significantly influence the results of factor VIII activity measurement. Moreover, substantial discrepancies may occur between results obtained using the one-stage aPTT-based coagulation assay and those obtained using a chromogenic assay according to the European Pharmacopoeia. This is particularly important when changing laboratories and/or reagents used for testing.
Dosage
The dosage and duration of replacement therapy depend on the severity of factor VIII deficiency, the location and severity of bleeding, and the patient's clinical condition.
Factor VIII quantities are expressed in International Units (IU), corresponding to the current WHO standard for factor VIII products. Factor VIII activity in plasma is expressed as a percentage (relative to the normal level in human plasma) or in International Units (relative to the international standard for factor VIII in plasma).
One International Unit (IU) of factor VIII activity is equivalent to the amount of factor VIII present in 1 mL of plasma from a normal human.
On-demand Treatment
The calculation of the required factor VIII dose is based on the empirical finding that 1 International Unit (IU) of factor VIII per kg of body weight raises factor VIII activity in plasma by 2 IU/dL. The required dose can be determined using the following formula:
Required number of units = body weight (kg) × desired increase in factor VIII level (%) (IU/dL) × 0.5 (IU/kg per 1 IU/dL).
The amount to be administered and the frequency of administration must always be individually determined based on clinical efficacy.
In the event of the hemorrhagic manifestations listed below, factor VIII activity should not be allowed to fall below the specified plasma activity levels (expressed as % of normal or IU/dL) for the appropriate period. Table 6 can be used as a guideline for dosing in bleeding episodes and surgical interventions.
Table 6. Dosing Guidelines for Bleeding Episodes and Surgical Interventions
| Severity of bleeding/surgical procedure type |
Required factor VIII level (%) (IU/dL) |
Infusion frequency (hours)/duration of therapy (days) |
| Bleeding |
||
| Early signs of hemarthrosis, muscle hematoma, or oral bleeding |
20−40 |
Repeat every 12−24 hours for at least 1 day until cessation of bleeding, determined by absence of pain, or until healing |
| More pronounced hemarthrosis, muscle hematoma, or hematoma |
30−60 |
Repeat infusion every 12−24 hours for 3−4 days or longer until pain resolves and function is restored |
| Life-threatening hemorrhage |
60−100 |
Repeat infusion every 8−24 hours until the life-threatening condition has resolved |
| Surgical procedures |
||
| Minor surgical procedures, including tooth extraction |
30−60 |
Every 24 hours for at least 1 day until healing |
| Major surgical procedures |
80−100 (before and after surgery) |
Repeat infusion every 8−24 hours until adequate wound healing is achieved, followed by continued therapy for at least 7 additional days to maintain factor VIII activity between 30 and 60% (IU/dL) |
Prophylaxis
For long-term prevention of bleeding episodes in patients with severe haemophilia A, the usual recommended dose is 20–40 IU of factor VIII per kg body weight every other day or 20–50 IU of factor VIII per kg body weight three times a week. In adults and adolescents (aged over 12 years), a less frequent dosing regimen may be considered (40–60 IU/kg every third day or twice weekly). Sometimes, especially in younger patients, shorter intervals between administrations or higher doses may be required.
Surgery
Experience with the use of the medicinal product in children undergoing surgery is limited.
Elderly
There is no experience with the use of the medicinal product in patients aged > 65 years.
Paediatric population
For long-term prevention of bleeding episodes in patients under 12 years of age, recommended doses are 25–50 IU of factor VIII per kg body weight every other day or 25–60 IU of factor VIII per kg body weight three times a week. Dosing recommendations for children aged 12 years and older are the same as for adult patients.
Method of administration
Intravenous administration.
The recommended infusion rate of NovoEight® is 1–2 ml/min. The rate should be adjusted according to the patient's comfort level.
Instructions for reconstituting the medicinal product prior to administration are provided in the Instructions for use of NovoEight®.
Storage after reconstitution
Chemical and physical in-use stability of the reconstituted product has been demonstrated for 24 hours at 2–8 °C; 4 hours at temperatures not exceeding 30 °C, provided the product has been stored for no more than 9 months at room temperature not exceeding 30 °C from the date of manufacture until reconstitution. Storage for 4 hours at temperatures not exceeding 40 °C is acceptable, provided the product has been stored for no more than 3 months at room temperature of 30–40 °C from the date of manufacture until reconstitution. From a microbiological point of view, the reconstituted product should be used immediately. If not used immediately, the user is responsible for the storage duration and conditions. When reconstitution is performed under controlled and validated aseptic conditions, the storage period of the reconstituted product should not exceed the time limits stated above.
Any unused reconstituted medicinal product stored for longer than 4 hours at room temperature up to 40 °C should be discarded.
Instructions for use of NovoEight®
READ THESE INSTRUCTIONS CAREFULLY BEFORE USING NOVOEIGHT®.
NovoEight® is supplied as a powder. Before injection (administration), it must be reconstituted with the solvent provided in the pre-filled syringe. The solvent is 0.9 % sodium chloride solution (9 mg/ml). NovoEight® is administered intravenously (intravenous injection). The contents of the pack (see below) are intended for reconstitution and administration of NovoEight®.
An infusion set (tubing and butterfly needle), sterile alcohol swabs, gauze pads, and adhesive bandages are also required for administration. These devices and materials are not included in the NovoEight® pack.
Do not use the equipment without proper training from your doctor or nurse.
Always wash your hands before use and ensure that your surroundings are clean.
During preparation and administration of the medicinal product directly into the vein, it is very important to follow aseptic and antiseptic procedures. Improper injection technique may lead to blood infection.
Do not open the equipment until you are ready to use it.
Do not use the equipment if it has been dropped or damaged. Instead, use a new pack.
Do not use the equipment if the expiry date has passed. Instead, use a new pack. The expiry date is printed after "Exp" on the carton, vial, vial adapter, and pre-filled syringe.
Do not use the equipment if you suspect it is contaminated. Instead, use a new pack.
Do not discard any part of the kit until you have administered the prepared solution.
The equipment is intended for single use only.
Contents of the pack:
1 vial of NovoEight® powder
1 vial adapter
1 pre-filled syringe with solvent
1 syringe plunger rod (located under the syringe)
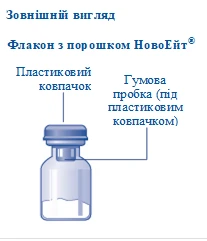
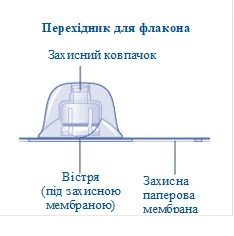
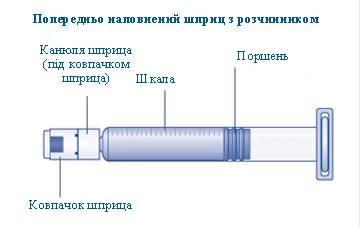

Fig. A
|
|
||
| Fig. B
|
|
||
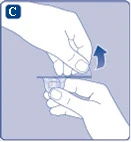
Fig. C
If the seal of the protective paper membrane is broken or damaged, do not use the vial adapter. Do not remove the vial adapter from the packaging with your fingers. If you touch the tip of the vial adapter, this may transfer microorganisms from your fingers. |
|
||
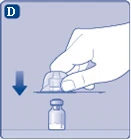
| Fig. D
After attachment, do not disconnect the vial adapter from the vial. |
|
||
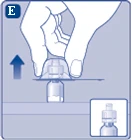
| Fig. E
Remove the protective cap from the vial adapter. Do not disconnect the vial adapter from the vial when removing the protective cap. |
|
||
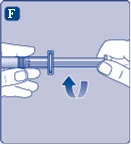
Fig. F
|
|
||
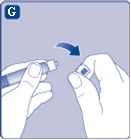
| Fig. G
If the cap on the syringe is loose or missing, do not use this pre-filled syringe. |
|
||
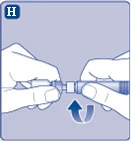
| Fig. H
|
|
||
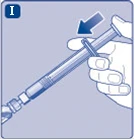
Fig. I
|
|
||
| Fig. J
Do not shake the vial, as this may cause foaming.
|
If your required dose is greater than that contained in one vial, repeat steps from A to J with additional vials, vial adapters, and pre-filled syringes until you obtain your required dose. |
||
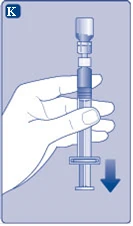
| Fig. K
If air has accidentally entered the syringe, expel it back into the vial.
|
|
||
| Fig. L
|
|
||
NovoEight® is now ready for intravenous administration.
Injecting NovoEight® through needle-free intravenous (IV) catheter adapters Caution! The pre-filled syringe is made of glass and is intended for use with a standard Luer-Lock connection. Some needle-free adapters with internal spikes are incompatible with this pre-filled syringe. Such incompatibility may prevent drug administration and/or damage the needle-free adapter. Injecting the solution through a central venous access device (CVAD), such as a central venous catheter (CVC) or implanted port:
|
|||
| Disposal Fig. M
Do not dispose of these materials with household waste. |
|
||
| Do not disassemble the device prior to disposal. Do not reuse the device. |
|||

Children.
The medicinal product can be used in children according to the instructions provided in the section "Dosage and method of administration".
Overdose.
There have been no reports of symptoms related to overdosage with recombinant coagulation factor VIII.
Adverse Reactions
Summary of Safety Profile
Hypersensitivity or allergic reactions (which may include angioneurotic edema, burning and pruritus at the infusion site, chills, flushing, generalized urticaria, headache, rash, hypotension, lethargy, nausea, restlessness, tachycardia, chest tightness, paresthesia, vomiting, wheezing) have been observed rarely, and in some cases may progress to severe anaphylaxis (including shock).
Formation of antibodies to hamster proteins and associated hypersensitivity reactions has been observed very rarely.
In patients with hemophilia A, neutralizing antibodies (inhibitors) to factor VIII may develop. If such inhibitors are formed, clinical response will be inadequate. In such cases, it is recommended to consult a specialized hemophilia treatment center.
List of Adverse Reactions
In Table 7, adverse events are listed by MedDRA System Organ Classes (SOC), with preferred terms from the Medical Dictionary for Regulatory Activities (MedDRA).
Frequency is defined as follows: very common (≥ 1/10), common (≥ 1/100 to < 1/10), uncommon (≥ 1/1000 to < 1/100), rare (≥ 1/10000 to < 1/1000), very rare (< 1/10000); frequency not known (cannot be estimated based on available data).
Within each frequency group, adverse reactions are listed in order of decreasing severity.
Table 7. Frequency of adverse reactions to the drug in clinical studies
| System organ classes |
Frequencya in previously treated patients (PTPs) |
Frequencya in previously untreated patients (PUPs) |
Adverse reaction |
| Blood and lymphatic system disorders |
Uncommon |
Very common |
Inhibitor to factor VIII |
| Psychiatric disorders |
Uncommon |
Insomnia |
|
| Nervous system disorders |
Uncommon |
Headache, dizziness, burning sensation |
|
| Cardiac disorders |
Uncommon |
Sinus tachycardia, acute myocardial infarction |
|
| Vascular disorders |
Uncommon |
Arterial hypertension, lymphedema, hyperemia |
|
| Common |
Flushing, thrombophlebitis of superficial veins |
||
| Skin and subcutaneous tissue disorders |
Common |
Rash, erythematous rash |
|
| Uncommon |
Rash, lichenoid keratosis, burning sensation of the skin |
||
| Musculoskeletal and connective tissue disorders |
Uncommon |
Muscle and joint stiffness, arthropathy, limb pain, muscle and bone pain |
|
| Common |
Hemarthrosis, muscle hemorrhage |
||
| Respiratory, thoracic and mediastinal disorders |
Common |
Cough |
|
| General disorders and administration site conditions |
Common |
Injection site reactions |
|
| Common |
Hyperthermia, erythema at catheterization site |
||
| Uncommon |
Fatigue, feeling of warmth, peripheral edema, hyperthermia |
||
| Investigations |
Common |
Elevated liver enzymesg |
|
| Common |
Positive test for factor VIII antibodies |
||
| Uncommon |
Increased heart rate |
||
| Gastrointestinal disorders |
Common |
Vomiting |
|
| Injury, poisoning and procedural complications |
Common |
Incorrect dose administered |
|
| Common |
Reaction related to infusion procedure |
||
| Uncommon |
Contusion |
||
| Device malfunction |
Common |
Device occlusion |
a Based on the total number of individual patients in all clinical studies (301), of which 242 were previously treated patients (PTPs) and 60 were previously untreated patients (PUPs).
b Frequency rates are based on data from studies involving all factor VIII products in patients with severe haemophilia A.
c Injection site reactions include injection site erythema, injection site extravasation, and injection site pruritus.
d Elevated liver enzymes include alanine aminotransferase, aspartate aminotransferase, gamma-glutamyl transferase, and bilirubin.
Description of selected adverse reactions
In clinical studies of NovoEight® involving previously treated patients, a total of 35 adverse reactions were reported in 23 out of 242 patients. The most common adverse reactions were injection site reactions, complications associated with incorrect dosing, and elevated liver enzyme levels. Two of the 35 adverse reactions occurred in one out of 31 patients under 6 years of age, no events occurred in patients aged 6 to 12 years, one event occurred in one out of 24 patients aged 12 to 18 years, and 32 events occurred in 21 out of 155 adult patients (aged 18 years and older).
Paediatric population
In clinical studies involving 63 children aged 0 to 12 years and 24 adolescents aged 12 to 18 years with severe haemophilia A, no differences in the safety profile of NovoEight® were observed between paediatric and adult patients.
In a study involving previously untreated patients aged 0 to 6 years, a total of 46 adverse reactions were reported in 33 out of 60 patients treated with NovoEight®. The most common adverse reaction was factor VIII inhibition (see section "Special warnings and precautions for use"). High-risk genetic mutations were identified in 92.3% of all patients and in 93.8% of patients with confirmed high inhibitor titres. No other factors were significantly associated with inhibitor development.
Reporting of adverse reactions and lack of drug efficacy
Reporting of adverse reactions after drug authorization is important. It enables continuous monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals, pharmacists, patients, or their legal representatives should report all suspected adverse reactions and lack of drug efficacy through the Automated Information System for Pharmacovigilance at: https://aisf.dec.gov.ua.
Shelf life.
The shelf life of the finished medicinal product is 30 months.
Do not use the medicinal product after the expiry date stated on the carton, vial, and syringe label. The expiry date refers to the last day of the stated month.
Storage conditions.
Store at 2–8 °C. Do not freeze. Keep out of the reach of children. During the shelf life, the product may be stored at room temperature (not exceeding 30 °C) for up to 9 months from the date of manufacture until reconstitution begins; the product may be stored at room temperature (30–40 °C) for up to 3 months from the date of manufacture until reconstitution begins. The product must not be returned to the refrigerator. The date and temperature of removal from the refrigerator must be indicated on the carton.
Store in the outer packaging to protect from light.
Incompatibilities.
Since compatibility studies have not been conducted, this medicinal product must not be mixed with other medicinal products.
Packaging.
One 5 ml glass vial (Type I) containing powder, closed with a chlorobutyl rubber stopper and sealed with an aluminium cap with a removable plastic cap, supplied with solvent (0.9% sodium chloride solution) in a 4 ml pre-filled syringe (5 ml syringe) with a polypropylene plunger stop, bromobutyl rubber plunger, bromobutyl rubber tip cap, polypropylene plunger rod, and a sterile vial adapter in an individual package, all contained in a cardboard box.
Prescription status.
Prescription only.
Marketing Authorisation Holder/Manufacturer.
Novo Nordisk A/S.
Manufacturer's address and location of operations.
Novo Allé, Bagsværd, 2880, Denmark.