Multak®
Ukraina
Spis treści
INSTRUKCJA dotycz¹ca stosowania leku leczniczego MULTAK® (MULTAQ®)
Sk³ad:
substancja czynna: dronedaron;
1 tabletka zawiera dronedaronu chlorowodorek w przeliczeniu na dronedaron 400 mg;
substancje pomocnicze: hipomeloza, skrobiê kukurydzian¹, krosppiwidon (typ A), poloksymer, laktozê monohydrat, dwutlenek krzemu koloidalny bezwodny, stearynian magnezu;
otoczka: hipomeloza, dwutlenek tytanu (E 171), glikol polietylenowy, wosk karneuba.
Postaæ leku. Tabletka powlekana.
G³ówne fizykochemiczne w³aœciwoœci: wyd³u¿one tabletki bia³ego koloru z pow³ok¹ filmow¹, z jednej strony – oznaczenie 4142, z drugiej – podwójna linia falista.
Grupa farmakoterapeutyczna. œrodki stosowane w leczeniu chorób uk³adu sercowo-naczyniowego. œrodki przeciwarytmiczne, klasa III. Dronedaron. Kod ATC C01BD07.
Właściwości farmakologiczne.
Farmakodynamika.
Mechanizm działania. Dronedaron jest blokerem wielokanałowym, który hamuje kanały potasowe (w tym IK(Ach), IKur, IKr, IKs) i w ten sposób wydłuża potencjał czynnościowy mięśnia sercowego oraz okresy refrakcji (klasa III). Hamuje również kanały sodowe (klasa Ib) i kanały wapniowe (klasa IV). Jest niekonkurencyjnym antagonistą receptorów adrenergicznych (klasa II). U zwierząt dronedaron zapobiegał migotaniu przedsionków lub przywracał normalny rytm zatokowy, w zależności od zastosowanego modelu eksperymentalnego. Zapobiegał również komorowemu typowi tachyarytmii oraz migotaniu komór w kilku modelach zwierzęcych. Działanie to najprawdopodobniej wynika z jego właściwości elektrofizjologicznych charakterystycznych dla wszystkich czterech klas w klasyfikacji Vaughan-Williamsa.
Właściwości farmakodynamiczne. W eksperymentalnych modelach na zwierzętach dronedaron spowalnia częstość skurczów serca. Wydłuża długość cyklu Wenckebacha oraz odcinki AH, PQ, QT; jednocześnie nie wykazuje istotnego wpływu lub nieznacznie wydłuża odcinki QTc, HV oraz QRS. Zwiększa efektywne okresy refrakcji (ERP) w przedsionkach, węźle przedsionkowo-komorowym oraz nieco wydłuża ERP w komorach z minimalnym odwrotnym stopniem zależności od częstości pobudzeń.
Dronedaron obniża ciśnienie tętnicze i kurczliwość miokardium (dP/dt max), nie zmieniając frakcji wyrzutowej lewej komory i obniżając jednocześnie zużycie tlenu przez miokard.
Dronedaron wykazuje właściwości naczyniorozkurczowe, które są bardziej wyrażone w odniesieniu do tętnic wieńcowych (związane z aktywacją szlaku sygnałowego tlenku azotu) niż wobec tętnic obwodowych.
Dronedaron wywiera pośrednie działanie antyadrenergiczne; obniża odpowiedź adrenergiczną typu alfa na adrenalinę w zakresie ciśnienia tętniczego oraz odpowiedzi typu beta-1 i beta-2 na izoprotenerol.
Skuteczność i bezpieczeństwo kliniczne.
Redukcja ryzyka hospitalizacji z powodu migotania przedsionków (AF). Skuteczność dronedaronu w redukcji ryzyka hospitalizacji spowodowanej AF została wykazana u pacjentów z AF w momencie włączenia do badania lub w wywiadzie, a także z dodatkowymi czynnikami ryzyka, którzy wzięli udział w wieloośrodkowym międzynarodowym, podwójnie ślepej, randomizowanym, placebo-kontrolowanym badaniu ATHENA. Pacjenci musieli posiadać co najmniej jeden czynnik ryzyka (w tym wiek, nadciśnienie tętnicze, cukrzycę, przebytą incydencję mózgowo-naczyniową, średnicę lewego przedsionka ≥50 mm lub frakcję wyrzutową lewej komory (LVEF) <0,40) w połączeniu z migotaniem/trzepotaniem przedsionków i rytmem zatokowym, oba potwierdzone dokumentacją w ciągu ostatnich 6 miesięcy. Pacjenci, którzy w ciągu 4 tygodni poprzedzających randomizację otrzymywali amiodaron, nie byli włączani do badania. W momencie włączenia do badania pacjenci musieli mieć migotanie/trzepotanie przedsionków lub rytm zatokowy przywrócony po samoistnej konwersji lub po jakiejkolwiek procedurze medycznej skierowanej na konwersję rytmu. W badaniu wzięło udział 4628 pacjentów, którzy zostali zrandomizowani i leczeni badanym lekiem przez okres maksymalnie 30 miesięcy (mediana czasu obserwacji pacjentów wynosiła 22 miesiące), otrzymując albo dronedaron 400 mg dwa razy dziennie (2301 pacjentów), albo placebo (2327 pacjentów), dodatkowo do standardowej terapii, obejmującej beta-blokery (71 %), inhibitory enzymu przekształcającego angiotensynę (ACE) lub blokery receptorów angiotensyny II (ARB) (69 %), glikozydy nasierkowe (14 %), blokery kanałów wapniowych (14 %), statyny (39 %), doustne leki przeciwpłytkowe (60 %), długotrwałą terapię przeciwpłytkową (6 %) oraz/lub diuretyki (54 %).
Pierwszą punktem końcowym badania był czas do pierwszej hospitalizacji z powodu choroby sercowo-naczyniowej lub zgonu z dowolnej przyczyny.
Wiek pacjentów wynosił od 23 do 97 lat, 42 % miało powyżej 75 roku życia. 47 % pacjentów stanowiły kobiety, a większość uczestników badania należała do rasy europejskiej (89 %).
U większości pacjentów występowało nadciśnienie tętnicze (86 %) oraz organiczne schorzenie serca (60 %) (w tym choroba niedokrwiennicza serca – 30 %; przewlekła niewydolność serca (CHF): 30 %; frakcja wyrzutowa lewej komory (LVEF) <45 %: 12 %). U 25 % uczestników występowało AF w momencie włączenia do badania.
Wyniki badania wykazały, że stosowanie dronedaronu prowadziło do zmniejszenia częstości hospitalizacji z powodu chorób sercowo-naczyniowych lub zgonu z dowolnej przyczyny o 24,2 % w porównaniu do placebo (p<0,0001).
Redukcja częstości hospitalizacji z powodu chorób sercowo-naczyniowych lub zgonu z dowolnej przyczyny była podobna we wszystkich podgrupach pacjentów, niezależnie od początkowych cech pacjentów lub leków, które przyjmowali (inhibitory ACE lub ARB II, beta-blokery, glikozydy nasierkowe, statyny, blokery kanałów wapniowych, diuretyki) (patrz rysunek 1).
Rysunek 1. Ocena względnego ryzyka (dronedaron 400 mg dwa razy dziennie w porównaniu do placebo) z 95 % przedziałami ufności według wybranych cech podstawowych – pierwsza hospitalizacja z powodu choroby sercowo-naczyniowej lub zgon z dowolnej przyczyny.
| Charakterystyka |
Liczba |
WHR [95 % CI] (a) |
Wartość P (b) |
||
| Wiek (lata) |
|
||||
| < 65 |
873 |
0,89 [0,71; 1,11] |
|||
| [65-75] |
1 830 |
0,71 [0,60; 0,83] |
|||
| ≥ 75 |
1 925 |
0,75 [0,65; 0,87] |
0,27 |
||
| Płeć |
|||||
| Mężczyźni |
2 459 |
0,74 [0,64; 0,85] |
|||
| Kobiety |
2 169 |
0,77 [0,67; 0,89] |
0,65 |
||
| Obecność migotania przedsionków/walnego przedsionkowego |
|||||
| Tak |
1 155 |
0,74 [0,61; 0,91] |
|||
| Nie |
3 473 |
0,76 [0,68; 0,85] |
0,85 |
||
| Choroba serca organiczna |
|||||
| Tak |
2 732 |
0,76 [0,67; 0,85] |
|||
| Nie |
1 853 |
0,77 [0,65; 0,92] |
0,85 |
||
| UDL < 35% lub NYHA ≥ klasa I |
|||||
| Tak |
1 417 |
0,74 [0,63; 0,87] |
|||
| Nie |
3 146 |
0,77 [0,68; 0,87] |
0,71 |
||
| UDL (%) |
|||||
| < 35 |
179 |
0,68 [0,44; 1,03] |
|||
| ≥ 35 |
4 365 |
0,76 [0,69; 0,84] |
0,58 |
||
| Blokery beta |
|||||
| Tak |
3 269 |
0,78 [0,69; 0,87] |
|||
| Nie |
1 359 |
0,71 [0,58; 0,86] |
0,41 |
||
| ACE lub blokery receptorów ATII |
|||||
| Tak |
3 216 |
0,74 [0,66; 0,83] |
|||
| Nie |
1 412 |
0,79 [0,66; 0,95] |
0,59 |
||
| Glikozydy serca |
|||||
| Tak |
629 |
0,76 [0,59; 0,98] |
|||
| Nie |
3 999 |
0,76 [0,68; 0,84] |
0,96 |
||
| Blokery kanałów wapniowych (c) |
|||||
| Tak |
638 |
0,63 [0,48; 0,82] |
|||
| Nie |
3 990 |
0,78 [0,70; 0,87] |
0,15 |
||
| Dronedaron lepiej / placebo lepiej |
|||||
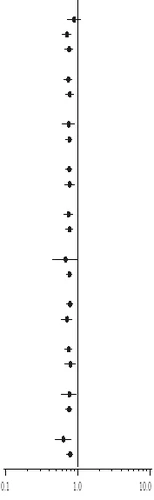
(a) Określone za pomocą modelu regresji Coxa.
(b) Wartość p – interakcja między początkowymi cechami a wskaźnikami leczenia w modelu regresji Coxa.
(c) Blokery kanałów wapniowych o działaniu obniżającym częstość skurczów serca ograniczają się do dyltiazemu, werapamilu i beprydylu.
Podobne wyniki uzyskano w odniesieniu do liczby przypadków hospitalizacji z powodu chorób sercowo-naczyniowych, przy zmniejszeniu ryzyka o 25,5% (p < 0,0001).
W trakcie badania liczba zgonów z dowolnych przyczyn w grupie przyjmującej dronedaron (116/2 301) i w grupie przyjmującej placebo (139/2 327) była podobna.
Utrzymanie rytmu zatokowego. W badaniach EURIDIS i ADONIS wzięło udział łącznie 1237 pacjentów z wcześniejszym epizodem migotania lub trzepotania przedsionków, którzy zostali zakwalifikowani do randomizacji w warunkach ambulatoryjnych w celu otrzymania dronedaronu 400 mg dwa razy dziennie (n = 828) lub placebo (n = 409), dodatkowo do standardowej terapii (w tym doustne leki przeciwwzorące, beta-blokery, inhibitory ACE lub ARB II, leki przeciwpłytkowe długoterminowe, diuretyki, statyny, glikozydy nasercowe oraz blokery kanałów wapniowych). Pacjenci przebyli co najmniej jeden dokumentowany na EKG epizod migotania/trzepotania przedsionków w ciągu ostatnich 3 miesięcy i mieli rytm zatokowy przez co najmniej 1 godzinę; obserwacja pacjentów trwała 12 miesięcy. U pacjentów przyjmujących amiodaron wykonano EKG około 4 godziny po podaniu pierwszej dawki leku w celu potwierdzenia odpowiedniej tolerancji leczenia. Inne leki przeciwarytmiczne odstawiano co najmniej przez okres odpowiadający 5 okresom półtrwania tych leków we krwi, przed podaniem pierwszej dawki badanego leku.
Uczestnicy badania byli w wieku od 20 do 88 lat, większość z nich należała do rasy kaukaskańskiej (97%) i byli mężczyznami (69%). Najczęściej występujące choroby współistniejące to nadciśnienie tętnicze (56,8%) oraz choroby organiczne serca (41,5%), w tym choroba niedokrwienna serca (21,8%).
Wyniki zarówno połączonych danych z badań EURIDIS i ADONIS, jak i danych z tych badań osobno wykazały, że dronedaron systematycznie opóźniał pojawienie się pierwszego nawrotu migotania/trzepotania przedsionków (główny punkt końcowy). W porównaniu z placebo dronedaron zmniejszał ryzyko pierwszego nawrotu migotania/trzepotania przedsionków w ciągu 12-miesięcznego okresu badania o 25% (p = 0,00007). Mediana czasu od momentu randomizacji do pierwszego nawrotu migotania/trzepotania przedsionków w grupie dronedaronu wynosiła 116 dni, co było 2,2 razy dłuższe niż w grupie placebo (53 dni).
W badaniu DIONYSOS porównywano skuteczność i bezpieczeństwo dronedaronu (400 mg dwa razy dziennie) i amiodaronu (600 mg dziennie przez 28 dni, następnie 200 mg dziennie), stosowanych przez okres 6 miesięcy. W badaniu wzięło udział łącznie 504 pacjentów z udokumentowanym migotaniem przedsionków, którzy zostali randomizowani do dwóch grup: 249 pacjentów otrzymywało dronedaron, a 255 pacjentów otrzymywało amiodaron. Częstość występowania zdarzeń głównego punktu końcowego oceny skuteczności leków, którymi były pierwszy nawrót migotania przedsionków lub wcześniejsze odstawienie badanego leku z powodu nietolerancji lub niewystarczającej skuteczności, w ciągu 12 miesięcy wyniosła 75% w grupie dronedaronu i 59% w grupie amiodaronu (stosunek ryzyka (HR) = 1,59, wartość p dla testu logarytmicznego rankingu <0,0001). Ryzyko nawrotu migotania przedsionków wynosiło odpowiednio 63,5% i 42%. Nawroty migotania przedsionków (w tym brak konwersji rytmu) występowały częściej w grupie dronedaronu, natomiast częstotliwość wcześniejszego odstawienia badanego leku z powodu jego nietolerancji była wyższa w grupie amiodaronu. Częstość występowania zdarzeń głównego punktu końcowego oceny bezpieczeństwa leków, którymi były przypadki rozwoju określonych zdarzeń ze strony tarczycy, wątroby, płuc, układu nerwowego, skóry, narządów wzroku lub przewodu pokarmowego oraz wcześniejsze odstawienie badanego leku z powodu dowolnego działania niepożądanego, zmniejszyła się o 20% w grupie dronedaronu w porównaniu z grupą amiodaronu (p=0,129). To zmniejszenie ryzyka wynikało ze statystycznie istotnego obniżenia częstości zdarzeń ze strony tarczycy i układu nerwowego oraz tendencji do zmniejszenia częstości zdarzeń ze strony skóry i narządów wzroku, a także niższej częstości wcześniejszego odstawienia leku w porównaniu z grupą amiodaronu.
W grupie pacjentów przyjmujących dronedaron częściej występowały działania niepożądane ze strony przewodu pokarmowego, głównie z powodu biegunki (12,9% w porównaniu z 5,1%).
Pacjenci z objawami niewydolności serca w spoczynku lub przy minimalnym obciążeniu w ciągu ostatniego miesiąca lub pacjenci hospitalizowani z powodu niewydolności serca w ciągu ostatniego miesiąca. W badaniu ANDROMEDA wzięło udział 627 pacjentów z dysfunkcją lewej komory, którzy zostali hospitalizowani z powodu nowo zdiagnozowanej niewydolności serca lub pogorszenia istniejącej niewydolności serca i mieli co najmniej jeden epizod duszności przy minimalnym obciążeniu fizycznym lub w spoczynku (klasa czynnościowa III lub IV wg NYHA) lub paroksyzmalną duszność nocną w ciągu miesiąca poprzedzającego hospitalizację.
Badanie to zostało zakończone przedwcześnie z powodu zauważonych różnic w liczbie przypadków zgonu podczas przyjmowania dronedaronu (25 pacjentów w grupie dronedaronu w porównaniu z 12 pacjentami w grupie placebo, p = 0,027) (patrz sekcje «Przeciwwskazania» i «Szczególne ostrzeżenia i środki ostrożności dotyczące stosowania»).
Pacjenci z trwałą migotaniem przedsionków.
Badanie PALLAS było randomizowanym, placebo-kontrolowanym badaniem, w którym oceniano korzyści kliniczne dronedaronu 400 mg podawanego dwa razy dziennie dodatkowo do standardowej terapii u pacjentów z trwałym migotaniem przedsionków i dodatkowymi czynnikami ryzyka (z niewydolnością serca ~ 9%, chorobą wieńcową ~ 41%, przebytym udarem lub TIA ~ 27%; EF LK ≤ 40% ~ 20,7% i pacjenci ≥ 75 lat z nadciśnieniem tętniczym i cukrzycą ~ 18%). Badanie zostało przedwcześnie zakończone po randomizacji 3149 pacjentów (placebo=1577; dronedaron=1572) z powodu istotnego wzrostu przypadków niewydolności serca (placebo=33; dronedaron=80; HR=2,49 (1,66–3,74)]; udaru [placebo=8; dronedaron=17; HR=2,14 (0,92–4,96)] i zgonu z przyczyn sercowo-naczyniowych [placebo=6; dronedaron=15; HR=2,53 (0,98–65,3)] (patrz sekcje «Przeciwwskazania» i «Szczególne ostrzeżenia i środki ostrożności dotyczące stosowania»).
Farmakokinetyka.
Wchłanianie. Dronedaron jest dobrze wchłaniany po doustnym podaniu razem z posiłkiem (co najmniej o 70%). Jednakże z powodu presystemowego metabolizmu „pierwszego przejścia” absolutna biodostępność dronedaronu (podawanego podczas jedzenia) wynosi 15%. Jednoczesne spożycie posiłku zwiększa biodostępność dronedaronu średnio 2–4 razy. Po doustnym podaniu razem z posiłkiem maksymalne stężenia plazmatyczne dronedaronu i jego głównego aktywnego metabolitu obiegowego (metabolit N-debutylowy) osiągane są w ciągu 3–6 godzin. Po wielokrotnym stosowaniu leku w dawce 400 mg dwa razy dziennie stan równowagi osiągany jest w ciągu 4–8 dni leczenia, a średnie stężenie kumulacji dronedaronu mieści się w zakresie od 2,6 do 4,5. Średnie Cmax dronedaronu w stanie równowagi wynosi 84–147 ng/ml, ekspozycja głównego metabolitu N-debutylowego jest podobna do ekspozycji związku pierwotnego. Farmakokinetyka dronedaronu i jego metabolitu N-debutylowego zmienia się umiarkowanie w zależności od wielkości dawki: podwojenie dawki prowadzi do około 2,5–3-krotnego wzrostu wartości Cmax i AUC.
Rozkład. Wiązanie dronedaronu i jego metabolitu N-debutylowego z białkami osocza in vitro wynosi odpowiednio 99,7% i 98,5% i jest nienasycone. Oba związki wiążą się głównie z albuminą. Po wewnątrzżylnym podaniu leku objętość rozkładu w stanie równowagi (Vss) mieści się w zakresie od 1200 do 1400 l.
Biotransformacja. Dronedaron jest intensywnie metabolizowany, głównie za pomocą enzymu CYP 3A4 (patrz sekcja «Interakcje z innymi lekami i inne formy interakcji»). Główną drogą metabolizmu leku jest jego N-debutylowanie z powstawaniem głównego obiegowego aktywnego metabolitu oraz dalszym utlenieniem, utlenieniowym dezaminowaniem z powstawaniem nieaktywnego metabolitu – kwasu propionowego, a następnie utlenieniem i bezpośrednim utlenieniem. W metabolizmie aktywnego metabolitu dronedaronu częściowo uczestniczą monoaminooksydazy (patrz sekcja «Interakcje z innymi lekami i inne formy interakcji»). Metabolit N-debutylowy wykazuje aktywność farmakodynamiczną, ale jest 3–10 razy mniej potężny niż dronedaron. Ten metabolit bierze udział w działaniu farmakologicznym dronedaronu u ludzi.
Wydalanie. Po doustnym podaniu znakowanego radioizotopem leku około 6% podanej dawki wydalone zostało z moczem, głównie w postaci metabolitów (nie wykryto niezmienionego związku pierwotnego w moczu), a 84% – z kałem, głównie w postaci metabolitów. Po wewnątrzżylnym podaniu szybkość wydalenia dronedaronu z osocza wynosi od 130 do 150 l/h. Okres półtrwania terminalnego dronedaronu wynosi około 25–30 godzin, a jego metabolitu N-debutylowego – około 20–25 godzin. Po zakończeniu leczenia dronedaronem w dawce 400 mg dwa razy dziennie pełna eliminacja dronedaronu i jego metabolitu z osocza miała miejsce w ciągu 2 tygodni po ostatnim przyjęciu leku.
Szczególne kategorie pacjentów. Farmakokinetyka dronedaronu u pacjentów z migotaniem przedsionków jest podobna do tej u zdrowych ochotników. Na farmakokinetykę dronedaronu wpływają takie czynniki jak płeć, wiek i masa ciała. Jednak każdy z tych czynników ma ograniczony wpływ na dronedaron.
Płeć. Ekspozycja na dronedaron i jego metabolit N-debutylowy u kobiet była średnio 1,3–1,9 razy wyższa niż u mężczyzn.
Pacjenci w wieku starszym. Wśród wszystkich uczestników badań klinicznych dronedaronu 73% miało co najmniej 65 lat, a 34% – co najmniej 75 lat. U pacjentów w wieku co najmniej 65 lat ekspozycja na dronedaron była o 23% wyższa niż u pacjentów poniżej 65 roku życia.
Pacjenci z zaburzoną funkcją wątroby. U pacjentów z umiarkowaną dysfunkcją wątroby ekspozycja na niezwiązany dronedaron wzrasta dwukrotnie. Ekspozycja na jego aktywny metabolit zmniejsza się o 47% (patrz sekcja «Sposób stosowania i dawki»).
Wpływ ciężkiej dysfunkcji wątroby na farmakokinetykę dronedaronu nie był oceniany (patrz sekcja «Przeciwwskazania»).
Pacjenci z zaburzoną funkcją nerek. Wpływ dysfunkcji nerek na farmakokinetykę dronedaronu nie był oceniany w specjalnie zaplanowanych badaniach. Nie oczekuje się zmian farmakokinetyki dronedaronu na tle dysfunkcji nerek, ponieważ niezmieniony związek nie jest w ogóle wydalany z moczem, a jedynie około 6% dawki podanego leku wydalane jest z moczem w postaci metabolitów (patrz sekcja «Sposób stosowania i dawki»).
Dane przedkliniczne dotyczące bezpieczeństwa. Dronedaron nie wykazywał żadnego efektu genotoksycznego zgodnie z wynikami jednego badania in vivo (test mikrojądrowy u myszy) i czterech badań in vitro.
W wyniku tych badań zaobserwowano wzrost częstości występowania guzów gruczołu mlekowego u samic myszy, sarkom histiocytarnych u myszy oraz hemangiom w węzłach chłonnych mezenterycznych u szczurów, wszystkie wyłącznie po podaniu najwyższej badanej dawki leku (odpowiadającej dawce zapewniającej ekspozycję leku 5–10 razy wyższą niż ta wywołana stosowaniem dawki terapeutycznej u ludzi). Hemangiomy nie są stanami przedrakowymi i nie transformują się w złośliwą hemangiosarkomę ani u zwierząt, ani u ludzi. Żaden z tych wyników nie został uznany za istotny dla człowieka.
W badaniach chronicznej toksyczności leku zaobserwowano niewielki i odwracalny fosfolipidozę (kumulację makrofagów pianistych) w węzłach chłonnych mezenterycznych, głównie u szczurów. Efekt ten uznano za specyficzny dla tego gatunku zwierząt doświadczalnych i nieistotny dla człowieka.
W przypadku stosowania dronedaronu w wysokich dawkach u szczurów lek znacząco wpływał na rozwój embrionalny i płodowy, powodując takie efekty jak zwiększenie strat poimplantacyjnych embrionów, zmniejszenie masy ciała płodu i masy łożyska, a także wady zewnętrzne, wisceralne i szkieletowe u płodów.
Charakterystyki kliniczne.
Wskazania. Multak® wskazany jest w celu utrzymania rytmu zatokowego po udanej kardiowersji u dorosłych pacjentów klinicznie stabilnych z migotaniem przedsionków (AF) przerywowym lub utrzymującym się. Ze względu na jego profil bezpieczeństwa (patrz sekcje „Przeciwwskazania” i „Szczególne ostrzeżenia i środki ostrożności podczas stosowania”) Multak® należy stosować wyłącznie po rozważeniu alternatywnych metod leczenia. Multak® nie powinien być stosowany u pacjentów z dysfunkcją skurczową lewej komory lub u pacjentów z niewydolnością serca lub z wywiadem epizodów niewydolności serca.
Przeciwwskazania.
Podwyższona wrażliwość na substancję czynną lub którykolwiek z substancji pomocniczych.
Blokada przedsionkowo-komorowa II lub III stopnia, całkowita blokada nogi pęczka Hisa, blokada obwodowa, dysfunkcja węzła zatokowego, zaburzenia przewodnictwa w przedsionkach lub zespół słabości węzła zatokowego (z wyjątkiem przypadków jednoczesnego stosowania z działającym stymulatorem serca).
Bradykardia <50 uderzeń na minutę.
Trwałe migotanie przedsionków (AF) o trwaniu AF ≥ 6 miesięcy (lub o nieznanym czasie trwania), jeśli lekarz podjął decyzję o zaprzestaniu prób przywrócenia rytmu zatokowego.
Niestabilność hemodynamiczna.
Niewydolność serca lub dysfunkcja skurczowa lewej komory w wywiadzie lub obecnie.
Toksyczne uszkodzenie wątroby lub płuc związane z wcześniejszym stosowaniem amiodaronu.
Jednoczesne stosowanie silnych inhibitorów cytochromu 450 (CYP) 3A4, takich jak ketokonazol, itrakonazol, worykonazol, pozakonazol, telitromycyna, klaritromycyna, nefazodon i rytonawir (patrz sekcja „Interakcje z innymi lekami i inne formy interakcji”).
Leki powodujące tachykardię typu torsades de pointes, np. fenotiazyne, cizapryd, beprydył, trójcykliczne leki przeciwdrgawkowe, terfenadyna i niektóre doustne makrolidy (np. erytromycyna), leki przeciwarytmiczne klasy I i III (patrz sekcja „Interakcje z innymi lekami i inne formy interakcji”).
Interwał QTc, określony według wzoru Bazetta: ≥ 500 ms.
Ciężka niewydolność wątroby.
Ciężka niewydolność nerek (klirens kreatyniny < 30 ml/min).
Jednoczesne stosowanie z dabigatranem.
Interakcje z innymi lekami i inne formy interakcji.
Dronedarone jest metabolizowany głównie przez CYP 3A4 (patrz sekcja „Farmakokinetyka”). Dlatego inhibitory i induktory CYP 3A4 mogą oddziaływać z dronedaronem.
Dronedarone jest umiarkowanym inhibitorem CYP 3A4, słabym inhibitorem CYP 2D6 i silnym inhibitorem białka P-glikoproteiny (P-gP). Z tego powodu dronedarone może oddziaływać z lekami, które są substratami białka P-glikoproteiny, CYP 3A4 lub CYP 2D6. Wykazano również, że dronedarone i/lub jego metabolity hamują białka transportowe z rodziny organicznych transporterów anionów (OAT), organicznych transporterów polipeptydowych anionów (OATP) i organicznych transporterów kationów (OCT) w warunkach in vitro. Dronedarone nie wykazuje istotnego potencjału hamowania CYP 1A2, CYP 2C9, CYP 2C19, CYP 2C8 i CYP 2B6.
Możliwe są interakcje farmakodynamiczne leku z beta-blokerami, blokerami kanałów wapniowych i glikozydami naparstnicy.
Leki powodujące rozwój torsades de pointes. Leki sprzyjające rozwojowi torsades de pointes, takie jak fenotiazyne, cizapryd, beprydył, trójcykliczne leki przeciwdrgawkowe, niektóre doustne makrolidy (takie jak erytromycyna), terfenadyna i leki przeciwarytmiczne klasy I i III, są przeciwwskazane ze względu na możliwy ryzyko działania proarytymicznego (patrz sekcja „Przeciwwskazania”). Należy zachować ostrożność również przy jednoczesnym stosowaniu leku z beta-blokerami lub cyfogryksyną.
Wpływ innych leków na Multak®.
Silne inhibitory enzymu CYP 3A4. Wielokrotne przyjmowanie ketokonazolu w dawce 200 mg dziennie prowadziło do 17-krotnego wzrostu ekspozycji na dronedarone. W związku z tym jednoczesne stosowanie ketokonazolu oraz innych silnych inhibitorów enzymu CYP 3A4, takich jak itrakonazol, worykonazol, pozakonazol, rytonawir, telitromycyna, klaritromycyna lub nefazodon, jest przeciwwskazane (patrz sekcja „Przeciwwskazania”).
Umiarkowane/słabe inhibitory enzymu CYP 3A4.
Erytromycyna. Erytromycyna, doustny makrolid, może wywoływać rozwój torsades de pointes i z tego powodu jest przeciwwskazana (patrz sekcja „Przeciwwskazania”). Stosowanie powtarzanych dawek erytromycyny (500 mg trzy razy dziennie przez 10 dni) prowadziło do 3,8-krotnego wzrostu stężenia równowagowego dronedaronu.
Blokery kanałów wapniowych. Blokery kanałów wapniowych, dyltiazen i werapamil, są substratami i/lub umiarkowanymi inhibitorami enzymu CYP 3A4. Ponadto ze względu na ich właściwości obniżające częstość skurczów serca werapamil i dyltiazen mogą oddziaływać z dronedaronem z punktu widzenia farmakodynamiki.
Stosowanie powtarzanych dawek dyltiazemu (240 mg dwa razy dziennie), werapamilu (240 mg raz dziennie) i nifedypiny (20 mg dwa razy dziennie) prowadziło do wzrostu ekspozycji na dronedarone odpowiednio o 1,7, 1,4 i 1,2 raza. Dronedarone (400 mg dwa razy dziennie) prowadziło również do wzrostu ekspozycji na blokery kanałów wapniowych (werapamilu o 1,4 raza i nizoldypiny o 1,5 raza). W trakcie badań klinicznych 13 % pacjentów przyjmowało blokery kanałów wapniowych jednocześnie z dronedaronem. Nie zaobserwowano wzrostu ryzyka hipotensji tętniczej, bradykardii i niewydolności serca.
Ogólnie ze względu na interakcję farmakokinetyczną i możliwą interakcję farmakodynamiczną blokery kanałów wapniowych hamujące funkcje węzła zatokowego i przedsionkowo-komorowego, takie jak werapamil i dyltiazen, należy stosować z ostrożnością razem z dronedaronem. Należy rozpoczynać ich stosowanie w niskich dawkach, które można stopniowo zwiększać dopiero po ocenie EKG. Pacjentom, którzy na początku stosowania dronedaronu już przyjmują blokery kanałów wapniowych, należy wykonać EKG i w razie potrzeby skorygować dawkę blokera kanałów wapniowych (patrz sekcja „Szczególne ostrzeżenia i środki ostrożności podczas stosowania”).
Inne umiarkowane/słabe inhibitory enzymu CYP 3A4. Inne umiarkowane inhibitory enzymu CYP 3A4 mogą również zwiększać ekspozycję na dronedarone.
Induktory enzymu CYP 3A4. Ryfampicyna (600 mg raz dziennie) zmniejszała ekspozycję na dronedarone o 80 %, nie wpływając istotnie na ekspozycję jego aktywnego metabolitu. W związku z tym jednoczesne stosowanie ryfampicyny oraz innych silnych induktorów enzymu CYP 3A4, takich jak fenylobarbital, karbamazepina, fenytoina lub dziurawiec zwyczajny, nie jest zalecane, ponieważ zmniejszają one ekspozycję na dronedarone.
Inhibitory monoaminooksydazy (MAO). W badaniu in vitro inhibitory MAO wpływały na metabolizm aktywnego metabolitu dronedaronu. Kliniczne znaczenie tych danych jest nieznane (patrz sekcje „Szczególne ostrzeżenia i środki ostrożności podczas stosowania” i „Farmakokinetyka”).
Wpływ leku Multak® na inne leki.
Interakcje z lekami metabolizowanymi przez enzym CYP 3A4.
Dabigatran. Gdy dabigatran etyksylat w dawce 150 mg raz dziennie był stosowany jednoczesnie z dronedaronem w dawce 400 mg dwa razy dziennie, stężenia AUC0-24 i Cmax dabigatranu wzrastały odpowiednio o 100 % i 70 %. Brak danych klinicznych dotyczących jednoczesnego stosowania tych leków pacjentom z AF. Jednoczesne stosowanie tych leków jest przeciwwskazane (patrz sekcja „Przeciwwskazania”).
Statyny. Dronedarone może zwiększać ekspozycję na statyny, które są substratami enzymu CYP 3A4 i/lub substratami P-gP. Dronedarone (400 mg dwa razy dziennie) powodowało 4-krotne i 2-krotne zwiększenie ekspozycji na simwastatynę i kwas simwastatynowy. Przewiduje się, że dronedarone może również zwiększać ekspozycję na lawastatynę w podobnym stopniu jak ekspozycję na kwas simwastatynowy. Obserwowano słabą interakcję dronedaronu z atorwastatyną (prowadzącą do średniego 1,7-krotnego wzrostu ekspozycji na atorwastatynę). Obserwowano słabą interakcję dronedaronu ze statynami transportowanymi przez OATP, takimi jak rosuwastatyna (prowadzącą do średniego 1,4-krotnego wzrostu ekspozycji na rosuwastatynę).
W badaniach klinicznych nie uzyskano danych dowodowych dotyczących niebezpieczeństwa jednoczesnego stosowania dronedaronu i statyn metabolizowanych przez enzym CYP 3A4. Jednakże zgłaszano spontaniczne doniesienia o zarejestrowanych przypadkach rabdomiolizy przy stosowaniu dronedaronu w połączeniu ze statynami (w szczególności z simwastatyną), dlatego należy ostrożnie przepisywać statyny jednoczesnie z lekiem.
Zaleca się niższe dawki początkowe i utrzymaniowe statyn zgodnie z zaleceniami podanymi w instrukcjach do ich stosowania, pacjenci powinni być monitorowani pod kątem objawów toksyczności mięśniowej (patrz sekcja „Szczególne ostrzeżenia i środki ostrożności podczas stosowania”).
Blokery kanałów wapniowych. Interakcja dronedaronu z blokerami kanałów wapniowych została opisana powyżej (patrz sekcja „Szczególne ostrzeżenia i środki ostrożności podczas stosowania”).
Immunosupresanty. Stosowanie dronedaronu może prowadzić do zwiększenia stężenia immunosupresantów we krwi (takrolimus, sirolimus, ewerolimus i cyklosporyna). W przypadku jednoczesnego stosowania z dronedaronem zaleca się monitorowanie ich stężenia we krwi i odpowiednie dostosowanie dawek.
Środki antykoncepcyjne doustne. U zdrowych ochotników przyjmujących dronedarone (800 mg dwa razy dziennie) jednoczesnie ze środkami antykoncepcyjnymi doustnymi nie zaobserwowano zmniejszenia stężenia etyniloestradiolu i lewonorgestrelu.
Interakcje z lekami metabolizowanymi przez enzym CYP 2D6.
Beta-blokery. Sotalol należy odstawić przed rozpoczęciem stosowania leku Multak® (patrz sekcje „Przeciwwskazania” i „Sposób stosowania i dawki”). Dronedarone może zwiększać ekspozycję na beta-blokery metabolizowane przez enzym CYP 2D6. Ponadto beta-blokery mogą oddziaływać z dronedaronem z punktu widzenia farmakodynamiki. Dronedarone w dawce 800 mg dziennie zwiększał ekspozycję na metoprolol o 1,6 raza, a na propranolol – o 1,3 raza (czyli znacznie mniej niż 6-krotne różnice obserwowane u wolnych i szybkich metabolizatorów enzymu CYP 2D6). W badaniach klinicznych przy stosowaniu dronedaronu razem z beta-blokerami częściej obserwowano bradykardię.
Ogólnie ze względu na interakcję farmakokinetyczną i możliwą interakcję farmakodynamiczną beta-blokery należy stosować z ostrożnością razem z dronedaronem. Stosowanie tych leków należy rozpoczynać od niskich dawek, które należy zwiększać stopniowo dopiero po ocenie EKG. Pacjentom, którzy na początku stosowania dronedaronu już przyjmują beta-blokery, należy wykonać EKG i w razie potrzeby skorygować dawkę beta-blokera (patrz sekcja „Szczególne ostrzeżenia i środki ostrożności podczas stosowania”).
Antydepresanty. Ponieważ u człowieka dronedarone jest słabym inhibitorem enzymu CYP 2D6, przewiduje się, że jego interakcja z lekami antydepresyjnymi metabolizowanymi przez enzym CYP 2D6 jest ograniczona.
Interakcje z substratami P-gP.
Digoksyna. Dronedarone (400 mg dwa razy dziennie) zwiększało ekspozycję na digoksynę 2,5 raza poprzez hamowanie transportera P-gP. Ponadto leki naparstnicy mogą oddziaływać z dronedaronem z punktu widzenia farmakodynamiki. Możliwe jest działanie synergistyczne na częstość skurczów serca i przewodnictwo przedsionkowo-komorowe. W badaniach klinicznych podczas stosowania dronedaronu razem z lekami naparstnicy obserwowano wzrost stężenia leków naparstnicy we krwi i/lub częstsze zaburzenia przewodu pokarmowego, wskazujące na toksyczność naparstnicy.
Dawkę digoksyny należy zmniejszyć o około 50 %, należy dokładnie monitorować stężenie digoksyny w surowicy krwi, a także zaleca się monitorowanie kliniczne i EKG.
Interakcje z lekami metabolizowanymi przez enzym CYP 3A4 i P-gP.
Rywaroksaban. Dronedarone prawdopodobnie zwiększa działanie rywaroksabanu (substratu CYP 3A4 i P-gp), zatem jednoczesne stosowanie może zwiększyć ryzyko krwawień. Jednoczesne stosowanie rywaroksabanu i dronedaronu nie jest zalecane.
Apiksaban. Dronedarone może zwiększyć działanie apiksabanu (substratu CYP 3A4 i P-gp). Jednak przy jednoczesnym stosowaniu z lekami, które nie są silnymi inhibitorami zarówno CYP 3A4, jak i P-gp, takimi jak dronedarone, nie jest wymagana korekta dawki apiksabanu.
Edoksaban. W badaniach in vivo działanie edoksabanu (substratu CYP 3A4 i P-gp) zwiększało się przy stosowaniu z dronedaronem. Dawkę edoksabanu należy zmniejszyć zgodnie z zaleceniami podanymi w instrukcji do stosowania edoksabanu.
Interakcje z warfaryną i lozartanem (substratami enzymu CYP 2C9)
Warfaryna i inne antagoniści witaminy K. Dronedarone (600 mg dwa razy dziennie) zwiększało stężenie S-warfaryny 1,2 raza, nie wpływając na stężenie R-warfaryny, a międzynarodowe znormalizowane stosunki (INR) zwiększały się jedynie 1,07 raza.
Jednak zgłaszano przypadki klinicznie istotnego wzrostu INR (≥ 5 razy), które obserwowano zazwyczaj w ciągu pierwszego tygodnia od rozpoczęcia stosowania dronedaronu u pacjentów przyjmujących doustne leki przeciwkrzepliwe. W związku z tym wartość INR należy dokładnie kontrolować po rozpoczęciu stosowania dronedaronu u pacjentów przyjmujących antagoniści witaminy K.
Lozartan i inne BRI II (blokery receptorów angiotensyny II). Nie zaobserwowano interakcji między dronedaronem a lozartanem, a także nie przewiduje się interakcji między dronedaronem a innymi BRI II.
Interakcje z teofiliną (substratem enzymu CYP 1A2). Dronedarone w dawce 400 mg dwa razy dziennie nie zwiększa równowagowej ekspozycji na teofilinę.
Interakcje z metforminą (substratem OCT1 i OCT2). Nie zaobserwowano interakcji między dronedaronem a metforminą, substratem OCT1 i OCT2.
Interakcje z omeprazolem (substratem enzymu CYP 2C19). Dronedarone nie wpływa na farmakokinetykę omeprazolu, substratu enzymu CYP 2C19.
Interakcje z klopidogrelem. Dronedarone nie wpływa na farmakokinetykę klopidogrelu i jego aktywnego metabolitu.
Inne informacje.
Pantoprazol (40 mg raz dziennie), lek zwiększający kwasowość soku żołądkowego bez wpływu na cytochrom P450, nie wywierał istotnego wpływu na farmakokinetykę dronedaronu.
Sok grejpfrutowy (inhibitor enzymu CYP 3A4). Wielokrotne przyjmowanie soku grejpfrutowego po 300 ml trzy razy dziennie prowadziło do 3-krotnego wzrostu ekspozycji na dronedarone. W związku z tym pacjentów należy ostrzec przed spożywaniem napojów zawierających sok grejpfrutowy podczas przyjmowania dronedaronu (patrz sekcja „Szczególne ostrzeżenia i środki ostrożności podczas stosowania”).
Szczególności stosowania.
Podczas stosowania dronedaronu zaleca się dokładne monitorowanie poprzez regularną ocenę funkcji serca, wątroby i płuc (patrz niżej). W przypadku nawrotu migotania przedsionków należy rozważyć możliwość odstawienia dronedaronu. Leczenie dronedaronem należy przerwać, jeśli u pacjenta rozwinie się którykolwiek z zaburzeń, które mogą prowadzić do przeciwwskazań (patrz sekcja „Przeciwwskazania”). Należy monitorować stosowanie równoczesne z takimi środkami, jak digoksyna i leki przeciwkrzepliwe.
Pacjenci, u których podczas leczenia rozwija się trwałe migotanie przedsionków. Badanie kliniczne z udziałem pacjentów z trwałym migotaniem przedsionków (przy trwaniu migotania przedsionków co najmniej 6 miesięcy) i czynnikami ryzyka wystąpienia chorób układu sercowo-naczyniowego zostało przerwane przedwcześnie z powodu nadmiernej liczby przypadków zgonów z przyczyn sercowo-naczyniowych, udarów mózgu i niewydolności serca u pacjentów otrzymujących Multak® (patrz sekcja „Farmakodynamika”). Zaleca się regularne, co najmniej raz na 6 miesięcy, wykonywanie EKG. Jeśli u pacjentów leczonych lekiem Multak® rozwija się trwałe migotanie przedsionków, Multak® należy odstawić.
Pacjenci z niewydolnością serca lub przewlekłą dysfunkcją lewej komory w wywiadzie lub obecnie. Multak® jest przeciwwskazany pacjentom z niestabilną hemodynamiką, z niewydolnością serca lub przewlekłą dysfunkcją lewej komory w wywiadzie lub obecnie (patrz sekcja „Przeciwwskazania”).
U pacjentów należy dokładnie ocenić występowanie objawów zastoinowej niewydolności serca. Napływają spontaniczne doniesienia o przypadkach pierwszego wystąpienia lub pogorszenia istniejącej niewydolności serca podczas leczenia lekiem Multak®. Pacjentom należy zalecić zgłaszanie się do lekarza w przypadku rozwoju lub obecności u nich objawów lub objawów niewydolności serca, takich jak przyrost masy ciała, obrzęki zastoinowe lub nasilająca się duszność. W przypadku rozwoju niewydolności serca leczenie lekiem Multak® należy przerwać.
Pacjenci powinni być monitorowani pod kątem rozwoju przewlekłej dysfunkcji lewej komory podczas leczenia. W przypadku rozwoju przewlekłej dysfunkcji lewej komory leczenie lekiem Multak® należy przerwać.
Pacjenci z chorobą wieńcową. Lek należy stosować z ostrożnością u pacjentów z chorobą wieńcową.
Pacjenci w podeszłym wieku. Lek należy stosować z ostrożnością u pacjentów w podeszłym wieku (w wieku powyżej 75 lat) z wieloma współistniejącymi chorobami (patrz sekcja „Sposób stosowania i dawki” oraz „Właściwości farmakodynamiczne”).
Uszkodzenia wątroby. W okresie posrejestrowym zgłaszano przypadki uszkodzeń hepatocytów, w tym zagrożenie życia ostrej niewydolnością wątroby, u pacjentów przyjmujących Multak®. Przed rozpoczęciem leczenia dronedaronem ocena wskaźników funkcji wątroby powinna być przeprowadzona po 1 tygodniu i po 1 miesiącu od rozpoczęcia leczenia, a następnie powtarzana co miesiąc przez sześć miesięcy, w 9. i 12. miesiącu i okresowo po tym czasie. Jeśli poziomy alaninotransaminazy (ALT) są trzy lub więcej razy wyższe od górnej granicy normy (GGN), poziomy ALT należy ponownie określić w ciągu 48–72 godzin. Jeśli ponowne oznaczenie potwierdza, że poziomy ALT są trzy lub więcej razy wyższe od GGN, leczenie dronedaronem należy przerwać. Tym pacjentom należy kontynuować badania odpowiednimi metodami i przeprowadzać dokładne monitorowanie do czasu normalizacji poziomów ALT.
Pacjenci powinni natychmiast powiadomić swojego lekarza o wszelkich objawach możliwego uszkodzenia wątroby (trwający, po raz pierwszy stwierdzony ból brzucha; utrata apetytu, nudności, wymioty, podwyższenie temperatury ciała, osłabienie, zwiększone zmęczenie, żółtaczka, ciemne zabarwienie moczu lub świąd).
Zwiększenie stężenia kreatyniny w osoczu. W przypadku przyjmowania 400 mg dronedaronu dwa razy dziennie u zdrowych ochotników i u chorych obserwowano zwiększenie stężenia kreatyniny w osoczu (średnie zwiększenie – o 10 µmol/l). U większości pacjentów zwiększenie to pojawia się wkrótce po rozpoczęciu leczenia, a poziom kreatyniny osiąga plateau po 7 dniach. Zaleca się oznaczenie stężenia kreatyniny w osoczu przed przepisaniem dronedaronu i po 7 dniach od rozpoczęcia jego przyjmowania. Jeśli obserwuje się zwiększenie kreatyninemii, poziom kreatyniny w surowicy należy ponownie ocenić po 7 dniach. Jeśli nie obserwuje się wzrostu stężenia kreatyniny w surowicy, ten zwiększony wskaźnik powinien być nadal używany jako nowy początkowy poziom kreatyniny u danego pacjenta, biorąc pod uwagę, że jest to oczekiwane dla dronedaronu. Jeśli poziom kreatyniny w surowicy nadal rośnie, należy rozważyć konieczność przeprowadzenia bardziej szczegółowego badania i odstawienia leku.
Zwiększenie stężenia kreatyniny nie powinno koniecznie prowadzić do przerwania leczenia inhibitorami ACE lub blokerami receptora angiotensyny II (ARB II).
W okresie posrejestrowym zgłaszano bardziej wyraźne zwiększenie stężenia kreatyniny po rozpoczęciu stosowania dronedaronu. W niektórych przypadkach zgłaszano również zwiększenie stężenia azotu mocznika we krwi, prawdopodobnie z powodu hipoperfuzji wtórnej w wyniku rozwoju niewydolności serca (azotemia przednerkowa). W takich przypadkach leczenie dronedaronem należy przerwać (patrz sekcje „Przeciwwskazania” i „Szczególności stosowania”). Zaleca się okresowe monitorowanie funkcji nerek i rozważenie konieczności przeprowadzenia dalszych badań w razie potrzeby.
Zaburzenia równowagi elektrolitowej. Ponieważ leki przeciwarytmiczne mogą być nieskuteczne lub mogą wywoływać działanie arytmogenne u pacjentów z hipokaliemią, przed rozpoczęciem stosowania i podczas leczenia dronedaronem należy skorygować wszelki niedobór potasu lub magnezu.
Wydłużenie odcinka QT. Działanie farmakologiczne dronedaronu może powodować umiarkowane wydłużenie odcinka QTc według wzoru Bazetta (około 10 ms), co wiąże się ze zwiększeniem czasu repolaryzacji. Te zmiany są związane z efektem terapeutycznym dronedaronu i nie odzwierciedlają jego toksyczności. Podczas leczenia zaleca się monitorowanie pacjenta, w tym wykonywanie EKG (elektrokardiografii). Jeśli odcinek QTc według wzoru Bazetta ≥500 ms, należy przerwać przyjmowanie dronedaronu (patrz sekcja „Przeciwwskazania”).
Z uwagi na doświadczenie kliniczne, dronedaron wykazuje niewielkie działanie proarytmogenne, a w ramach badania ATHENA obserwowano zmniejszenie liczby zgonów z powodu arytmii podczas przyjmowania tego leku (patrz sekcja „Właściwości farmakodynamiczne”).
Jednak efekty proarytmogenne mogą wystąpić w niektórych sytuacjach, np. przy jednoczesnym stosowaniu leku z lekami sprzyjającymi wystąpieniu arytmii i/lub zaburzeniom równowagi elektrolitowej (patrz sekcje „Szczególności stosowania” i „Interakcje z innymi lekami i inne rodzaje interakcji”).
Zaburzenia ze strony układu oddechowego, narządów klatki piersiowej i śródpiersia. W okresie posrejestrowym zgłaszano przypadki chorób śródmiąższowych płuc, w tym zapalenia płuc i włóknienia płuc. Pojawienie się duszności i nieproduktywnego kaszlu może być związane z toksycznym działaniem leku na płuca, a tym pacjentom wskazana jest dokładna ocena stanu klinicznego. W przypadku potwierdzenia toksycznego działania leku na płuca należy go odstawić.
Interakcje (patrz sekcja „Interakcje z innymi lekami i inne rodzaje interakcji”).
Digoksyna. Przepisanie dronedaronu pacjentom przyjmującym digoksynę prowadzi do zwiększenia stężenia digoksyny w osoczu krwi i tym samym nasila objawy i oznaki związane z toksycznym działaniem digoksyny. Zaleca się kliniczne, EKG i biologiczne monitorowanie, a także zmniejszenie dawki digoksyny o połowę. Możliwe jest również działanie synergistyczne tych leków na częstość skurczów serca i przewodnictwo przedsionkowo-komorowe.
Jednoczesne stosowanie beta-blokerów lub blokerów kanałów wapniowych, które hamują funkcje węzła zatokowego i przedsionkowo-komorowego, należy prowadzić z ostrożnością. Stosowanie tych leków należy rozpocząć od niskich dawek, które można zwiększać drogą dozowania tylko po ocenie EKG. Pacjentom, którzy na początku stosowania dronedaronu już przyjmują blokery kanałów wapniowych lub beta-blokery, należy wykonać EKG i w razie potrzeby skorygować dawkę.
Leki przeciwkrzepliwe. Zgodnie z klinicznymi zaleceniami dotyczącymi leczenia migotania przedsionków pacjentom wymagana jest odpowiednia terapia przeciwkrzepliwa. Po rozpoczęciu stosowania dronedaronu pacjentom przyjmującym antagoniści witaminy K należy dokładnie monitorować międzynarodowe znormalizowane stosunki (INR), zgodnie z zaleceniami zawartymi w instrukcjach do tych leków.
Nie zaleca się stosowania takich mocnych induktorów CYP3A4, jak ryfampicyna, fenylobarbital, karbamazepina, fenytoina lub ziele świętojańskie.
Inhibitory monoaminooksydazy mogą zmniejszać klirens aktywnego metabolitu dronedaronu i dlatego należy je stosować z ostrożnością.
Z ostrożnością należy stosować statyny. Zaleca się niskie dawki początkowe i utrzymujące statyn, a pacjentom należy monitorować objawy toksyczności mięśni.
Pacjentów należy ostrzec przed spożywaniem napojów z sokiem grejpfrutowym podczas przyjmowania dronedaronu.
Pacjenci z nietolerancją galaktozy. Ze względu na obecność laktozy w tym leku nie należy go przyjmować pacjentom z takimi rzadkimi, dziedzicznymi zaburzeniami, jak nietolerancja galaktozy, deficyt laktazy saamskiej lub zespół złego wchłaniania glukozy-galaktozy.
Stosowanie w okresie ciąży lub karmienia piersią.
Ciąża. Do tej pory nie ma wystarczających danych dowodowych dotyczących stosowania dronedaronu u kobiet w ciąży. Badania doświadczalne na zwierzętach wykazały toksyczność leku w odniesieniu do funkcji rozrodczych (teratogenność u zwierząt). Dlatego dronedaron jest przeciwwskazany w ciąży.
Test na ciążę
Przed rozpoczęciem stosowania leku Multak® u kobiety w wieku rozrodczym należy upewnić się, że nie jest w ciąży.
Kontracepcja
Kobietom w wieku rozrodczym podczas leczenia dronedaronem oraz przez 7 dni po przyjęciu ostatniej dawki należy stosować skuteczne metody antykoncepcji.
Okres karmienia piersią. Nie wiadomo, czy dronedaron i jego metabolity wydzielają się w mleku matki. Dostępne dane farmakodynamiczne/toksykologiczne z badań na zwierzętach wykazały obecność dronedaronu i jego metabolitów w mleku matki. Ryzyko dla noworodków/dzieci nie można wykluczyć. Należy zalecać kobietom powstrzymywanie się od karmienia piersią podczas terapii lekiem Multak® oraz przez 7 dni (około 5 okresów półtrwania leku) po przyjęciu ostatniej dawki. Decyzję o tym, czy przerwać karmienie piersią czy przerwać/odstawić leczenie lekiem Multak®, należy podjąć z uwzględnieniem korzyści z karmienia piersią dla dziecka i konieczności leczenia dla kobiety.
Plodność. Dronedaron nie wpływał na płodność w badaniach na zwierzętach.
Wpływ na zdolność prowadzenia pojazdów lub obsługi innych mechanizmów. Wpływ leku Multak® na zdolność prowadzenia pojazdów lub pracy z innymi mechanizmami jest nieobecny lub nieznaczny. Jednak na zdolność prowadzenia pojazdów lub pracy z innymi mechanizmami mogą wpływać niepożądane reakcje, takie jak zwiększona zmęczalność.
Sposób stosowania i dawki.
Leczenie należy rozpoczynać i prowadzić wyłącznie pod nadzorem specjalisty (patrz rozdział „Szczególne wskazania dotyczące stosowania”).
Leczenie lekiem Multak® można rozpoczynać w warunkach ambulatoryjnych.
Przed rozpoczęciem stosowania leku Multak® należy przerwać leczenie lekami przeciwnagrzmiotowymi klasy I lub III (flekainid, propafenon, chinidyna, dysopirydynamid, dofetylid, sotalol, amiodaron).
Dane dotyczące optymalnego czasu przejścia z amiodaronu na Multak® są obecnie ograniczone. Należy pamiętać, że działanie amiodaronu może utrzymywać się przez dłuższy czas po przerwaniu jego stosowania ze względu na długi okres półwylu. Jeśli planowane jest przejście na Multak®, należy je przeprowadzić pod nadzorem specjalisty (patrz rozdziały „Przeciwwskazania” oraz „Farmakodynamika”).
Dorosłym zalecana dawka wynosi 400 mg 2 razy na dobę. Należy przyjmować po 1 tabletce rano i wieczorem podczas jedzenia.
Zaleca się połykanie tabletu w całości, popijając wodą. Nie wolno dzielić tabletki na dwie równe dawki.
Nie należy popijać tabletek Multak® sokiem grejpfrutowym (patrz rozdział „Interakcje z innymi lekami oraz inne rodzaje oddziaływań”).
W przypadku pominięcia dawki pacjenci powinni przyjąć następną dawkę zgodnie z ustalonym schematem – nie należy podwajać dawki.
Osoby w wieku starszym. Skuteczność i bezpieczeństwo stosowania leku u osób starszych, które nie cierpią na inne choroby układu sercowo-naczyniowego, były porównywalne do tych u młodszych pacjentów.
Lek należy przepisywać z ostrożnością pacjentom w wieku powyżej 75 lat, u których występują choroby współistniejące (patrz rozdziały „Przeciwwskazania”, „Szczególne wskazania dotyczące stosowania” oraz „Farmakodynamika”). Choć w ramach badania farmakokinetyki z udziałem zdrowych ochotników stwierdzono zwiększone stężenie leku w osoczu u kobiet w wieku starszym, nie uznaje się za konieczne dostosowywanie dawki (patrz rozdziały „Farmakodynamika” oraz „Farmakokinetyka”).
Niewydolność wątroby. Multak® jest przeciwwskazany u pacjentów z ciężką niewydolnością wątroby ze względu na brak odpowiednich danych. U pacjentów z łagodną lub umiarkowaną niewydolnością wątroby nie jest wymagana korekta dawki (patrz rozdział „Farmakokinetyka”).
Niewydolność nerek. Multak® jest przeciwwskazany u pacjentów z ciężką niewydolnością nerek (klirens kreatyniny (CrCl) <30 ml/min) (patrz rozdział „Przeciwwskazania”). U pacjentów z innymi formami niewydolności nerek nie jest wymagana korekta dawki (patrz rozdziały „Szczególne wskazania dotyczące stosowania” oraz „Farmakokinetyka”).
Dzieci. Brak doświadczenia w stosowaniu u dzieci i młodzieży (do 18 roku życia). Odpowiednie dane są niedostępne.
Przedawkowanie.
Nie wiadomo, czy dronedarone i/lub jego metabolity mogą być usuwane metodą dializy (hemodializy, dializy otrzewnowej lub hemofiltraции).
Nie istnieje specyficzny antydotum. W przypadku przedawkowania leczenie powinno mieć charakter wspierający i skierowane na złagodzenie objawów.
Niepożądane działania.
Profil bezpieczeństwa dronedaronu w dawce 400 mg dwa razy dziennie u pacjentów z migotaniem przedsionków (AF) lub trzepotaniem przedsionków (AT) został ustalony w pięciu badaniach kontrolowanych placebo. W tych badaniach zrandomizowano i leczono łącznie 6285 pacjentów, z których 3282 otrzymywało dronedaron 400 mg dwa razy dziennie, a 2875 − placebo. Średnia długość leczenia w tych badaniach wynosiła 13 miesięcy. W badaniu ATHENA maksymalna długość okresu dalszej obserwacji wynosiła 30 miesięcy. Niektóre niepożądane reakcje zostały również zidentyfykowane w okresie nadzoru pozarejestracyjnego.
Ocena wpływu takich cech pacjentów, jak płeć lub wiek, na częstość występowania niepożądanych zjawisk podczas leczenia wykazała wpływ płci (zwiększone ryzyko u kobiet) na częstość występowania dowolnych niepożądanych zjawisk oraz na częstość poważnych niepożądanych zjawisk.
Podczas badań klinicznych leczenie zostało wcześnie przerwane z powodu wystąpienia niepożądanych reakcji u 11,8 % pacjentów otrzymujących dronedaron i u 7,7 % pacjentów z grupy placebo. Najczęstszą przyczyną przerwania terapii lekiem Multak® były zaburzenia przewodu pokarmowego (3,2 % pacjentów w porównaniu do 1,8 % w grupie placebo). We wszystkich pięciu badaniach podczas stosowania dronedaronu w dawce 400 mg dwa razy dziennie najczęściej obserwowane działania niepożądane to biegunka, nudności i wymioty, zwiększona zmęczoność oraz osłabienie.
W poniższej tabeli przedstawiono niepożądane reakcje sklasyfikowane według klas układów narządów. Częstość występowania określono jako: bardzo często (≥ 1/10), często (≥ 1/100, < 1/10); rzadko (≥ 1/1000, < 1/100); bardzo rzadko (≥ 1/10000, < 1/1000); nieznane (nie można oszacować ze względu na brak danych). W ramach każdej grupy według częstości występowania działania niepożądane są uporządkowane według malejącego stopnia ich nasilenia.
| Klasa układu narządów |
Bardzo często (≥ 1/10) |
Często (od ≥ 1/100 do < 1/10) |
Nieczęsto (od ≥ 1/1000 do < 1/100) |
Rzadko (od ≥ 1/10000 do < 1/1000) |
| Zaburzenia układu odpornościowego |
Reakcje anafilaktyczne, w tym obrzęk naczynioruchowy |
|||
| Zaburzenia ze strony układu nerwowego |
Dysgezja |
Agezja |
||
| Zaburzenia serca |
Niewydolność serca (patrz niżej) |
Bradykardia (patrz sekcje „Przeciwwskazania” i „Szczególne ostrzeżenia i środki ostrożności”) |
||
| Zaburzenia naczyniowe |
Waskułopatia, w tym waskułopatia leukocyto-klastyczna |
|||
| Zaburzenia ze strony układu oddechowego, narządów klatki piersiowej i jamy opłucnowej |
Choroba płuc międzybłoniowych, w tym zapalenie płuc i włóknienie płuc (patrz niżej) |
|||
| Zaburzenia żołądkowo-jelitowe |
Diareia Wymioty Światłowstręt Ból brzucha Trudności trawienne |
|||
| Zaburzenia wątrobowo-żółciowe |
Odchylenia od normy w badaniach funkcji wątroby |
Uszkodzenie wątroby typu hepatocytarnego, w tym zagrożenie życia ostrą niewydolnością wątroby (patrz sekcja „Szczególne ostrzeżenia i środki ostrożności”) |
||
| Choroby skóry i tkanki podskórnej |
Wysypka (w tym uogólniona, makularna, makulo- grudkowa) Zwędzenie |
Erytema (w tym rumień i wysypka rumieniowa) Egzema Odbicia światłoczułości Aleryczne zapalenie skóry Zapalenie skóry |
||
| Zaburzenia ogólne i reakcje w miejscu podania |
Zwiększona zmęczliwość Astenia |
|||
| Zaburzenia wykryte w badaniach |
Zwiększenie stężenia kreatyniny we krwi* Wydłużenie przedziału QTc według wzoru Bazetta# |
*Podwyższony poziom kreatyniny we krwi ≥ 10 % po 5 dniach od rozpoczęcia leczenia (patrz punkt «Szczególne ostrzeżenia i środki ostrożności stosowania»).
#Wydłużenie interwału QTc obliczonego według wzoru Bazetta (> 450 ms u mężczyzn i > 470 ms u kobiet) (patrz punkt «Szczególne ostrzeżenia i środki ostrożności stosowania»).
Opis wybranych działań niepożądanych. W pięciu badaniach klinicznych kontrolowanych placebo, niewydolność serca występowała z częstością podobną w grupie pacjentów przyjmujących dронedarone i w grupie placebo (bardzo często; odpowiednio 11,2 % i 10,9 %). Należy brać pod uwagę ogólnie charakterystyczny dla pacjentów z migotaniem przedsionków wzrost częstości występowania niewydolności serca. O przypadkach niewydolności serca zgłaszano również w oparciu o dane z nadzoru popo rejestracyjnego (częstość nieznana) (patrz punkt «Szczególne ostrzeżenia i środki ostrożności stosowania»).
W trakcie pięciu badań kontrolowanych placebo, działania niepożądane ze strony płuc stwierdzono u 0,6 % pacjentów przyjmujących dronedarone, w porównaniu do 0,8 % pacjentów w grupie placebo. O przypadkach choroby śródmiąższowej płuc, w tym zapalenia płuc i włóknienia płuc, zgłaszano na podstawie danych z nadzoru popo rejestracyjnego (częstość nieznana). Część z tych pacjentów wcześniej przyjmowała amiodaron (patrz punkt «Szczególne ostrzeżenia i środki ostrożności stosowania»).
Zgłaszanie podejrzewanych działań niepożądanych.
Zgłaszanie działań niepożądanych po dopuszczeniu leku do obrotu ma istotne znaczenie. Umożliwia to ciągłe monitorowanie stosunku korzyści do ryzyka związanego z zastosowaniem leku. Osoby pracujące w zawodach medycznych i farmaceutycznych, a także pacjenci lub ich prawni opiekunowie powinni zgłaszać wszystkie przypadki podejrzewanych działań niepożądanych oraz braku działania leku za pośrednictwem zautomatyzowanego systemu informacyjnego nadzoru farmakologicznego pod adresem: https://aisf.dec.gov.ua.
Okres ważności. 3 lata.
Warunki przechowywania. Przechowywać w miejscu niedostępnym dla dzieci. Przechowywać w oryginalnym opakowaniu w temperaturze poniżej 30 ºC.
Opakowanie. № 60 (10×6): po 10 tabletek w blistrze; po 6 blisterów w tekturowym pudełku.
Kategoria receptury. Na receptę.
Producent.
SANOFI WINTHROP INDUSTRIE.
Adres producenta i miejsce prowadzenia działalności.
1 rue de la Vierge Ambard et Lagrave 33565 CARBON BLANC CEDEX, Francja.
Wniosek składający.
SPÓŁKA Z OGRANICZONĄ ODPOWIEDZIALNOŚCIĄ «Sanofi-Aventis Ukraina», Ukraina.
Adres wniosku składającego. Ukraina, 01033, miasto Kijów, ul. Żyliańska 48-50A