Likwestia®
Ukraina
Spis treści
INSTRUKCJA dotyczÄ…ca stosowania leku LIKWESTIA® (LIQUESTIA)
Skład:
substancja czynna: febuksostat;
1 tabletka pokryta powłoką filmową zawiera 80 mg lub 120 mg febuksostatu;
substancje pomocnicze: celuloza mikrokryształyczna, laktoza jednowodna, hydroksypropyloceluloza, sodu laurylosiarczan, sodowa sol kroskarboksymetlocelulozy, laktoza bezwodna, dwutlenek krzemu koloidalny bezwodny, stearynian magnezu;
powłoka filmowa: alkohol poliwinylowy (E 1203), dwutlenek tytanu (E 171), makrogol 3350 (E 1521), talk (E 553b), tlenek żelazka żółty (E 172).
Postać leku. Tabletka pokryta powłoką filmową.
Główne cechy fizykochemiczne:
tabletka pokryta powÅ‚okÄ… filmowÄ… 80 mg: tabletki wydÅ‚użone, dwuwypukÅ‚e, pokryte powÅ‚okÄ… filmowÄ…, od jasnożółtej do żółtej barwy, z oznaczeniem „80” po jednej stronie i gÅ‚adkie po drugiej;
tabletka pokryta powÅ‚okÄ… filmowÄ… 120 mg: tabletki wydÅ‚użone, dwuwypukÅ‚e, pokryte powÅ‚okÄ… filmowÄ…, od jasnożółtej do żółtej barwy, z oznaczeniem „120” po jednej stronie i gÅ‚adkie po drugiej.
Grupa farmakoterapeutyczna.
Leki stosowane w leczeniu padaczki. Leki hamujÄ…ce powstawanie kwasu moczowego. Kod ATC M04A A03.
Właściwości farmakologiczne.
Farmakodynamika.
Mechanizm działania
Kwas moczowy jest końcowym produktem metabolizmu puryn u człowieka i powstaje w wyniku następującej reakcji: hipoksantyna → ksantyna → kwas moczowy. Katalizatorem obu etapów tej reakcji jest ksantynooksydaza. Febukostat jest pochodną 2-arylotiazolu, którego działanie terapeutyczne wiąże się ze zmniejszeniem stężenia kwasu moczowego w surowicy krwi poprzez selektywne hamowanie ksantynooksydazy. Febukostat to silny i selektywny nielikowy inhibitor ksantynooksydazy (NP-SIXO), którego stała hamowania (Ki) in vitro wynosi poniżej 1 nanomola. Wykazano, że febukostat znacząco hamuje aktywność zarówno utlenionej, jak i zredukowanej formy ksantynooksydazy. W stężeniach terapeutycznych febukostat nie wpływa na inne enzymy uczestniczące w metabolizmie puryn lub pirymidyn, takie jak guaninodiezaminaza, hipoksantynoguaninofosforylotransferaza, orotanofosforylotransferaza, orotydynomonofosforan dekarboksylaza lub purynonukleozydofosforylaza.
Skuteczność kliniczna i bezpieczeństwo
Podagra. Skuteczność febukostatu została potwierdzona w trzech głównych badaniach fazy III (dwa główne badania APEX i FACT oraz dodatkowe badanie CONFIRMS, opisane poniżej), w których wzięło udział 4101 pacjentów z hiperurykemią i podagrą. W każdym z tych głównych badań fazy III febukostat skuteczniej niż allopurinol obniżał stężenie kwasu moczowego w surowicy i utrzymywał je na odpowiednim poziomie. Głównym punktem końcowym skuteczności w badaniach APEX i FACT była frakcja pacjentów, u których w ciągu ostatnich trzech miesięcy stężenie kwasu moczowego w surowicy nie przekraczało 6,0 mg/dl (357 µmol/l). W dodatkowym badaniu fazy III CONFIRMS, którego wyniki stały się dostępne po pierwszej rejestracji febukostatu, głównym punktem końcowym skuteczności była frakcja pacjentów, u których stężenie kwasu moczowego w surowicy nie przekraczało 6,0 mg/dl w czasie ostatniej wizyty. Do tych badań nie włączano pacjentów po przeszczepieniu narządów (patrz sekcja „Szczególne ostrzeżenia i środki ostrożności podczas stosowania”).
Badanie APEX: badanie skuteczności febukostatu z kontrolą placebo i allopurinolem fazy III (Allopurinol and Placebo-Controlled Efficacy Study of Febuxostat, APEX) było randomizowanym, podwójnie ślepy, wieloośrodkowym badaniem trwającym 28 tygodni. Łącznie zakwalifikowano 1072 pacjentów: placebo (n=134), febukostat 80 mg raz dziennie (n=267), febukostat 120 mg raz dziennie (n=269), febukostat 240 mg raz dziennie (n=134) oraz allopurinol 300 mg raz dziennie (n=258) dla pacjentów z wyjściowym stężeniem kreatyniny w surowicy ≤ 1,5 mg/dl lub 100 mg raz dziennie (n=10) dla pacjentów z wyjściowym stężeniem kreatyniny w surowicy > 1,5 mg/dl i ≤ 2,0 mg/dl). Febukostat w dawce 240 mg (2-krotnie wyższej niż maksymalna zalecana dawka) podawano w celu oceny bezpieczeństwa.
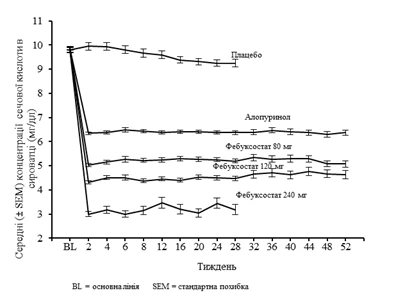
Badanie APEX wykazało istotną statystycznie przewagę obu schematów leczenia – febukostat 80 mg raz dziennie i febukostat 120 mg raz dziennie – w porównaniu do standardowej dawki allopurinolu 300 mg (n = 258)/100 mg (n=10) w obniżeniu stężenia kwasu moczowego w surowicy poniżej 6 mg/dl (357 µmol/l) (patrz tabela 1 poniżej). Na rys. 1 przedstawiono średnie stężenia kwasu moczowego w surowicy w zależności od czasu dla każdej grupy terapeutycznej w obu głównych badaniach fazy III.
Rysunek 1. Średnie stężenia kwasu moczowego w surowicy według danych z połączonych badań kontrolnych (faza III)
**
**
Uwaga: 509 pacjentów otrzymywało allopurinol w dawce 300 mg raz dziennie; 10 pacjentów ze stężeniem kreatyniny w surowicy > 1,5 mg/dl i < 2,0 mg/dl otrzymywało allopurinol w dawce 100 mg raz dziennie (10 z 268 pacjentów w badaniu APEX). Febukostat w dawce 240 mg podawano w celu oceny bezpieczeństwa przy dawce dwukrotnie wyższej niż maksymalna zalecana.
Badanie FACT: badanie skuteczności febukostatu z kontrolą allopurinolu (The Febuxostat Allopurinol Controlled Trial, FACT) fazy III było randomizowanym, podwójnie ślepym, wieloośrodkowym badaniem trwającym 52 tygodnie. Łącznie zakwalifikowano 760 pacjentów: febukostat 80 mg raz dziennie (n = 256), febukostat 120 mg raz dziennie (n = 251) oraz allopurinol 300 mg raz dziennie (n = 253).
Badanie FACT wykazało istotną statystycznie przewagę obu schematów – febukostat 80 mg raz dziennie i febukostat 120 mg raz dziennie – w porównaniu do standardowej dawki allopurinolu 300 mg w zakresie obniżania i utrzymywania stężenia kwasu moczowego w surowicy poniżej 6 mg/dl (357 µmol/l).
W tabeli 1 przedstawiono wyniki oceny głównego punktu końcowego skuteczności.
Tabela 1
Frakcja pacjentów ze stężeniem kwasu moczowego w surowicy < 6,0 mg/dl (357 µmol/l) podczas trzech ostatnich miesięcznych wizyt
| Badanie |
Febuksostat 80 mg 1 raz na dobę |
Febuksostat 120 mg 1 raz na dobę |
Allopurinol 300/100 mg 1 raz na dobę 1 |
| APEX (28 tygodni) |
48 % * (n=262) |
65 % *, # (n=269) |
22 % (n=268) |
| FACT (52 tygodnie) |
53 %* (n=255) |
62 %* (n=250) |
21 % (n=251) |
| Wyniki połączone |
51 %* (n=517) |
63 %*, # (n=519) |
22 % (n=519) |
| 1 Wyniki u pacjentów otrzymujących 100 mg 1 raz na dobę (n = 10: pacjenci z wyjściowym stężeniem kreatyniny w surowicy > 1,5 mg/dl i ≤ 2,0 mg/dl) lub 300 mg 1 raz na dobę (n=509), w trakcie analizy zostały połączone. * p < 0,001 w porównaniu z allopurinolem. # p < 0,001 w porównaniu z dawką 80 mg. |
|||
Możliwość szybkiego obniżania stężenia kwasu moczowego we krwi przez febuksostat była szybka i trwała. Obniżenie stężenia kwasu moczowego we krwi < 6,0 mg/dl (357 µmol/l) zaobserwowano już w drugim tygodniu badania i utrzymywało się dalej w trakcie leczenia.
Badanie CONFIRMS: badanie CONFIRMS było randomizowanym, kontrolowanym badaniem fazy III trwającym 26 tygodni, przeprowadzonym w celu oceny bezpieczeństwa i skuteczności febuksostatu w dawkach 40 mg i 80 mg w porównaniu z alopurynolem w dawkach 300 mg i 200 mg u pacjentów z podagrą i hiperurykemią. Ogółem zrandomizowano 2269 pacjentów: febuksostat 40 mg 1 raz dziennie (n=757), febuksostat 80 mg 1 raz dziennie (n=756) i alopurynol 300/200 mg 1 raz dziennie (n=756). Co najmniej 65% pacjentów miało zaburzenia funkcji nerek od lekkiego do umiarkowanego stopnia (z klirem kreatyniny 30–89 ml/min). Profilaktyka napadów podagry była obowiązkowa przez 26 tygodni.
Odsetek pacjentów z poziomem kwasu moczowego we krwi < 6,0 mg/dl (357 µmol/l) w ostatniej wizycie wyniósł 45% w grupie febuksostatu 40 mg, 67% w grupie febuksostatu 80 mg i 42% w grupie alopurynolu 300/200 mg.
Pierwotny punkt końcowy w podgrupie pacjentów z zaburzeniami funkcji nerek. W badaniu APEX oceniano skuteczność leku u 40 pacjentów z zaburzeniami funkcji nerek (tj. z wyjściowym stężeniem kreatyniny we krwi > 1,5 mg/dl i ≤ 2,0 mg/dl). Pacjentom zrandomizowanym do grupy alopurynolu dawkę leku zmniejszono do 100 mg 1 raz dziennie. Pierwotny punkt końcowy skuteczności osiągnięto w grupach przyjmujących febuksostat u 44% pacjentów (80 mg 1 raz dziennie), u 45% (120 mg 1 raz dziennie) i u 60% (240 mg 1 raz dziennie) w porównaniu z 0% w grupach alopurynolu 100 mg 1 raz dziennie i placebo.
Nie zaobserwowano istotnych różnic klinicznych w obniżeniu stężenia kwasu moczowego we krwi w procentach u zdrowych ochotników niezależnie od stanu czynności nerek (58% w grupie z normalną czynnością nerek i 55% w grupie z ciężkim zaburzeniem czynności nerek).
Przeprowadzony prospektywny analiza z udziałem pacjentów z podagrą i zaburzeniami funkcji nerek w ramach badania CONFIRMS wykazała, że febuksostat był istotnie bardziej skuteczny: poziom kwasu moczowego we krwi obniżał się < 6,0 mg/dl w porównaniu z alopurynolem 300 mg / 200 mg u pacjentów z podagrą i zaburzeniem czynności nerek od lekkiego do umiarkowanego stopnia (65% badanych).
Pierwotny punkt końcowy w podgrupie pacjentów ze stężeniem kwasu moczowego we krwi ≥ 10 mg/dl. Wyjściowe stężenie kwasu moczowego we krwi ≥ 10 mg/dl zaobserwowano u około 40% pacjentów (połączone badania APEX i FACT). Wśród tych pacjentów pierwotny punkt końcowy skuteczności (stężenie kwasu moczowego we krwi poniżej 6 mg/dl w ostatnich 3 wizytach) osiągnięto w podgrupach febuksostatu: u 41% pacjentów (80 mg 1 raz dziennie), u 48% pacjentów (120 mg 1 raz dziennie) i u 66% pacjentów (240 mg 1 raz dziennie) w porównaniu z 9% w grupie alopurynolu 300 mg/100 mg 1 raz dziennie i 0% w grupie placebo.
Zgodnie z danymi badania CONFIRMS, odsetek pacjentów, którzy osiągnęli pierwotny punkt końcowy skuteczności (ze stężeniem kwasu moczowego we krwi < 6,0 mg/dl w ostatniej wizycie), w grupie pacjentów z wyjściowym stężeniem kwasu moczowego we krwi ≥ 10 mg/dl, którzy otrzymywali febuksostat 40 mg 1 raz dziennie, wyniósł 27% (66/249), febuksostat 80 mg 1 raz dziennie – 49% (125/254), alopurynol 300 mg/200 mg 1 raz dziennie – 31% (72/230).
Wyniki kliniczne: odsetek pacjentów wymagających leczenia napadów podagry
Badanie APEX: w trakcie 8-tygodniowego okresu profilaktycznego odsetek pacjentów z grupy terapeutycznej febuksostatu 120 mg (36%), którzy wymagali leczenia napadów podagry, porównywano z pacjentami przyjmującymi febuksostat 80 mg (28%), alopurynol 300 mg (23%) lub placebo (20%). Częstotliwość napadów była wyższa po okresie profilaktycznym i stopniowo malała z czasem. Od 46% do 55% pacjentów leczono z powodu napadów podagry od 8 do 28 tygodnia. Napady podagry występujące w ostatnich 4 tygodniach badań (24–28 tydzień) obserwowano u 15% pacjentów (febuksostat 80, 120 mg), 14% pacjentów (alopurynol 300 mg) i 20% pacjentów (placebo).
Badanie FACT: w trakcie 8-tygodniowego okresu profilaktycznego odsetek pacjentów z grupy terapeutycznej febuksostatu 120 mg (36%), którzy wymagali leczenia napadów podagry, porównywano z obiema grupami terapeutycznymi przyjmującymi febuksostat 80 mg (22%) lub alopurynol 300 mg (21%). W trakcie 8-tygodniowego okresu profilaktycznego częstotliwość napadów wzrosła i stopniowo malała z czasem (64% i 70% pacjentów leczonych z powodu napadów podagry od 8 do 52 tygodnia). Napady podagry w ostatnich 4 tygodniach badań (49–52 tydzień) obserwowano u 6–8% pacjentów (febuksostat 80 mg, 120 mg) i u 11% pacjentów (alopurynol 300 mg).
Odsetek pacjentów wymagających leczenia zaostrzeń podagry (badania APEX i FACT) był niższy w grupach, w których średnie stężenie kwasu moczowego we krwi po leczeniu obniżało się do < 6,0 mg/dl, < 5,0 mg/dl lub < 4,0 mg/dl w porównaniu z grupami, w których średnie stężenie kwasu moczowego wynosiło ≥ 6,0 mg/dl w ostatnich 32 tygodniach leczenia (od 20–24 tygodnia do 49–52 tygodnia).
W trakcie badania CONFIRMS odsetek pacjentów wymagających leczenia napadów podagry (1 raz na 6 miesięcy) wyniósł 31% i 25% odpowiednio w grupach przyjmujących febuksostat 80 mg i alopurynol. Nie zaobserwowano różnic w stosunku pacjentów wymagających leczenia napadów podagry między grupami przyjmującymi febuksostat 80 mg i 40 mg.
Długoterminowe badania otwarte rozszerzone. Badanie EXCEL (C02-021): EXCEL było trzyletnim, otwartym, wieloośrodkowym, randomizowanym, rozszerzonym, kontrolowanym alopurynolem badaniem bezpieczeństwa fazy III, przeprowadzonym w celu oceny bezpieczeństwa u pacjentów, którzy ukończyli podstawowe badania fazy III (APEX lub FACT). Ogółem do badania włączono 1086 pacjentów przyjmujących: febuksostat 80 mg 1 raz dziennie (n = 649), febuksostat 120 mg 1 raz dziennie (n = 292) lub alopurynol 300/100 mg 1 raz dziennie (n = 145). U około 69% pacjentów nie wymagano korekty terapii w celu osiągnięcia ostatecznej stabilnej terapii. Pacjentów, u których poziom stężenia kwasu moczowego we krwi przy trzykrotnym kolejnym pomiarze wynosił > 6,0 mg/dl, wyłączono z badania.
Poziomy stężenia kwasu moczowego we krwi w czasie nie uległy zmianie (np. u 91% i 93% pacjentów, którzy początkowo przyjmowali febuksostat w dawkach odpowiednio 80 mg i 120 mg, poziomy stężenia kwasu moczowego we krwi były poniżej 6,0 mg/dl w 36. miesiącu).
Zgodnie z danymi trzyletniej obserwacji u mniej niż 4% pacjentów wymagających leczenia napadów zaobserwowano zmniejszenie częstotliwości napadów podagry w okresie 16–24 miesięcy i 30–36 miesięcy (tj. u ponad 96% pacjentów nie było potrzeby leczenia napadów).
U 46% i 38% pacjentów, którzy otrzymywali ostateczną stabilną terapię febuksostatem w dawce odpowiednio 80 lub 120 mg 1 raz dziennie, zaobserwowano całkowite zniknięcie pierwotnego wyczuwalnego tofusu od początku do ostatniej wizyty.
Badanie FOCUS (TMX-01-005) było pięcioletnim, otwartym, wieloośrodkowym, rozszerzonym badaniem bezpieczeństwa fazy II, przeprowadzonym u pacjentów, którzy zakończyli 4-tygodniowe podwójne ślepe dawkowanie febuksostatu w badaniu TMX-00-004. Badanie obejmowało 116 pacjentów, którzy początkowo przyjmowali febuksostat 80 mg 1 raz dziennie. U 62% pacjentów nie wymagano korekty dawki w celu utrzymania poziomu stężenia kwasu moczowego we krwi poniżej 6,0 mg/dl, a 38% pacjentów wymagało korekty dawki w celu osiągnięcia ostatecznej stabilnej koncentracji.
Odsetek pacjentów ze stężeniem kwasu moczowego we krwi poniżej 6,0 mg/dl (357 µmol/l) w ostatniej wizycie wyniósł więcej niż 80% (81–100%) w każdej z grup dawkowych febuksostatu.
W badaniach klinicznych fazy III u pacjentów przyjmujących febuksostat zaobserwowano niewielkie zmiany parametrów wątrobowych (5,0%). Częstotliwość tych zmian była podobna do tej przyjmowanych z alopurynolem (4,2%) (patrz sekcja „Szczególne ostrzeżenia i środki ostrożności”). W długoterminowych otwartych badaniach rozszerzonych u pacjentów przyjmujących febuksostat (5,5%) lub alopurynol (5,8%) przez dłuższy czas zaobserwowano wzrost poziomu TSH [hormon tyreotropowy] (> 5,5 µIU/ml) (patrz sekcja „Szczególne ostrzeżenia i środki ostrożności”).
Badania długoterminowe po rejestracji. Badanie CARES było wieloośrodkowym, randomizowanym, podwójnie ślepym badaniem braku mniejszej skuteczności, w którym porównywano wyniki chorób układu sercowo-naczyniowego przy stosowaniu febuksostatu i alopurynolu u pacjentów z podagrą i istotnymi chorobami układu sercowo-naczyniowego w wywiadzie, w tym zawałem mięśnia sercowego, hospitalizacją z powodu niestabilnej dławicy piersiowej, zabiegiem rewaskularyzacji wieńcowej lub mózgowej, udarem mózgu, hospitalizacją z powodu przejściowego ataku niedokrwiennego, chorobą naczyń obwodowych lub cukrzycą z objawami mikroangiopatii lub makroangiopatii. W celu osiągnięcia poziomu kwasu moczowego we krwi (sUA) poniżej 6 mg/dl dawkę febuksostatu dopasowywano od 40 mg do 80 mg (niezależnie od funkcji nerek), a dawkę alopurynolu dopasowywano krokowo od 300 do 600 mg dla pacjentów z normalną czynnością nerek i niewydolnością nerek lekkiego stopnia oraz od 200 do 400 mg dla pacjentów z niewydolnością nerek umiarkowanego stopnia.
Pierwotnym punktem końcowym w badaniu CARES był czas do pierwszego wystąpienia poważnych zdarzeń sercowo-naczyniowych (PZSN), składających się z niemortalnego zawału mięśnia sercowego, niemortalnego udaru mózgu, śmierci z powodu chorób sercowo-naczyniowych i niestabilnej dławicy piersiowej z nagłym zabiegiem rewaskularyzacji wieńcowej.
Wskaźniki końcowe (pierwotne i wtórne) oceniano metodą analizy ITT (ITT – ang. intention-to-treat), włączając wszystkich podmiotów, którzy zostali zrandomizowani i otrzymali co najmniej jedną dawkę leku w trakcie badania podwójnie ślepego.
Ogółem 56,6% pacjentów przedwcześnie zakończyło leczenie badawcze, a 45% pacjentów nie ukończyło wszystkich wizyt badawczych.
Ogółem 6190 pacjentów było pod obserwacją przez 32 miesiące, średnia długość ekspozycji wyniosła 728 dni u pacjentów w grupie febuksostatu (n = 3098) i 719 dni u pacjentów w grupie alopurynolu (n = 3092).
Pierwotny punkt końcowy obserwowano z podobnymi wskaźnikami w grupach leczenia febuksostatem i alopurynolem (10,8% vs 10,4% pacjentów odpowiednio; stosunek ryzyka [SR] 1,03; dwustronny powtarzany 95% przedział ufności [PU] 0,89–1,21).
W analizie poszczególnych PZSN częstość zgonów z powodu chorób sercowo-naczyniowych była wyższa w grupie febuksostatu niż alopurynolu (4,3% vs 3,2% pacjentów; SR 1,34; 95% PU 1,03–1,73). Częstość innych PZSN była podobna w grupach febuksostatu i alopurynolu: niemortalnego zawału mięśnia sercowego (3,6% vs 3,8% pacjentów; SR 0,93; 95% PU 0,72–1,21), niemortalnego udaru mózgu (2,3% vs 2,3% pacjentów; SR 1,01; 95% PU 0,73–1,41) i nagłej rewaskularyzacji z powodu niestabilnej dławicy piersiowej (1,6% vs 1,8% pacjentów; SR 0,86; 95% PU 0,59–1,26).
Częstość zgonów z wszelkich przyczyn była również wyższa w grupie febuksostatu niż alopurynolu (7,8% vs 6,4% pacjentów; SR 1,22; 95% PU 1,01–1,47), co przede wszystkim wynikało z wyższego poziomu zgonów z powodu chorób sercowo-naczyniowych w tej grupie (patrz sekcja „Szczególne ostrzeżenia i środki ostrożności”).
Wskaźniki hospitalizacji z powodu niewydolności serca, hospitalizacji z powodu arytmii niezwiązanej z niedokrwieniem, zdarzeń zakrzepowo-embolicznych żylnej i hospitalizacji z powodu przejściowych napadów niedokrwiennych były porównywalne w grupach febuksostatu i alopurynolu.
Badanie FAST było prospektywnym, randomizowanym, otwartym, z zasłonięciem punktu końcowego, w którym porównywano profil bezpieczeństwa febuksostatu i alopurynolu w chorobach sercowo-naczyniowych u pacjentów z przewlekłą hiperurykemią (w warunkach, gdy osadzanie się uranów już miało miejsce) i czynnikami ryzyka sercowo-naczyniowego (tj. pacjenci w wieku powyżej 60 lat z co najmniej jednym czynnikiem ryzyka sercowo-naczyniowego). Pacjenci spełniający kryteria otrzymywali leczenie alopurynolem przed randomizacją, a w razie potrzeby wymagana była korekta dawki zgodnie z oceną kliniczną, rekomendacjami EULAR [European League Against Rheumatism – Europejska Liga Walki z Reumatyzmem] i zatwierdzoną dawką. Na końcu wstępnej fazy przyjmowania alopurynolu pacjentów ze stężeniem sUA < 0,36 mmol/l (<6 mg/dl), którzy otrzymywali maksymalną tolerowaną dawkę lub maksymalną dozwoloną dawkę alopurynolu, zrandomizowano w stosunku 1:1, aby otrzymać leczenie febuksostatem lub alopurynolem. Pierwotnym punktem końcowym badania FAST był czas do pierwszego wystąpienia jakiegokolwiek zjawiska skumulowanego punktu końcowego, który obejmował: 1) hospitalizację z powodu zawału mięśnia sercowego (ZMS), który nie zagrożował życiu / pozytywnego biomarkera ostrego zespołu wieńcowego (OZW); 2) udar mózgu, który nie zagrożał życiu; 3) śmierć z powodu zaburzeń sercowo-naczyniowych. Pierwotna analiza opierała się na analizie OT (OT – on-treatment).
Ogółem zrandomizowano 6128 pacjentów: 3063 przyjmujących febuksostat i 3065 przyjmujących alopurynol.
W pierwotnej analizie OT febuksostat nie ustępował alopurynolowi pod względem częstości pierwotnego punktu końcowego, który zaobserwowano u 172 pacjentów (1,72/100 pacjentów-roku), którzy przyjmowali febuksostat, w porównaniu z 241 pacjentami (2,05/100 pacjentów-roku), którzy przyjmowali alopurynol, z dostosowanym SR 0,85 (95% PU: 0,70, 1,03), p < 0,001. Analiza OT dla pierwotnego punktu końcowego w podgrupie pacjentów z ZMS, udarem mózgu lub OZW w wywiadzie nie wykazała istotnej różnicy między grupami leczenia: w grupie febuksostatu wystąpiło 65 (9,5%) zdarzeń niepożądanych i 83 (11,8%) pacjentów – w grupie alopurynolu; dostosowane SR 1,02 (95% PU: 0,74–1,42); p = 0,202.
Leczenie febuksostatem nie było skojarzone ze zwiększeniem śmiertelności sercowo-naczyniowej ani ze śmiertelnością z wszelkich przyczyn ogółem i w podgrupie pacjentów z ZMS, udarem mózgu lub OZW w wywiadzie. Ogółem w grupie febuksostatu było mniej zgonów (62 przypadki śmiertelności sercowo-naczyniowej i 108 przypadków śmiertelności z wszelkich przyczyn) niż w grupie alopurynolu (82 przypadki śmiertelności sercowo-naczyniowej i 174 przypadki śmiertelności z wszelkich przyczyn).
Zaobserwowano większy spadek poziomu kwasu moczowego przy leczeniu febuksostatem w porównaniu z leczeniem alopurynolem.
Zespół lizy guza (ZLG). Skuteczność i bezpieczeństwo stosowania febuksostatu w profilaktyce i leczeniu ZLG oceniano w badaniu FLORENCE (FLO-01). Febuksostat wykazał lepsze i szybsze działanie wobec obniżenia poziomu uranów w porównaniu z alopurynolem.
FLORENCE było randomizowanym (1:1), podwójnie ślepym, kontrolowanym badaniem fazy III, przeprowadzonym w celu porównania febuksostatu w dawce 120 mg 1 raz dziennie i alopurynolu w dawce 200–600 mg dziennie (średnia dzienna dawka alopurynolu ± odchylenie standardowe: 349,7 ± 112,90 mg) w warunkach kontroli stężenia kwasu moczowego we krwi. Wybrani pacjenci powinni byli być kandydatami do leczenia alopurynolem lub nie mieć dostępu do rasburikazy. Pierwotne punkty końcowe stanowiły pole pod krzywą stężenia kwasu moczowego we krwi (AUC sUA1–8) i zmianę poziomu kreatyniny we krwi (sC) od pierwszego do ósmego dnia każdego.
Do badania włączono 346 pacjentów z hematologicznymi nowotworami złośliwymi, którzy otrzymywali chemioterapię i mieli średnie/wysokie ryzyko rozwoju ZLG. Średnia wartość AUC sUA1–8 (mg×godz/dl) była istotnie niższa przy przyjmowaniu febuksostatu (514,0 ± 225,71 w porównaniu z 708,0 ± 234,42; średnie najmniejsze kwadraty dla różnicy: –196,794 [95% PU: –238,600; –154,988]; p < 0,0001). Ponadto, średni poziom stężenia kwasu moczowego we krwi był istotnie niższy przy stosowaniu febuksostatu, począwszy od pierwszych 24 godzin leczenia i w dowolnym późniejszym czasie. Nie zaobserwowano istotnych różnic statystycznie pod względem średniego stężenia kreatyniny we krwi (%) między febuksostatem i alopurynolem (–0,83 ± 26,98 w porównaniu z –4,92 ± 16,70 odpowiednio; średnie najmniejsze kwadraty dla różnicy: 4,0970 [95% PU: –0,6467; 8,8406]; p = 0,0903). Biorąc pod uwagę wtórne punkty końcowe, nie zaobserwowano istotnych różnic statystycznie pod względem częstości rozwoju laboratorycznie potwierdzonego ZLG (8,1% i 9,2% dla febuksostatu i alopurynolu odpowiednio; SR: 0,875 [95% PU: 0,4408; 1,7369]; p = 0,8488) i zespołu rozpadu guza (1,7% i 1,2% dla febuksostatu i alopurynolu odpowiednio; SR: 0,994 [95% PU: 0,9691; 1,0199]; p = 1,0000). Częstość wszystkich objawów i zdarzeń niepożądanych występujących w trakcie leczenia wyniosła 67,6% w porównaniu z 64,7% i 6,4% w porównaniu z 6,4% dla febuksostatu i alopurynolu odpowiednio. W badaniu FLORENCE febuksostat wykazał lepsze i szybsze działanie wobec obniżenia poziomu kwasu moczowego we krwi w porównaniu z alopurynolem. Dane dotyczące porównania febuksostatu i rasburikazy na ten moment są nieobecne. Skuteczność i bezpieczeństwo stosowania febuksostatu nie zostały ustalone u pacjentów z ostrym ciężkim ZLG, np. u pacjentów, u których inne formy terapii obniżające poziom uranów nie działają.
Farmakokinetyka.
U zdrowych ochotników maksymalne stężenie w osoczu (Cmax) i pole pod krzywą (AUC) zwiększały się proporcjonalnie do dawki po jednorazowym i wielokrotnym podaniu febuksostatu w dawkach od 10 mg do 120 mg. W dawkach od 120 mg do 300 mg wzrost AUC był większy niż proporcjonalny do dawki. Po podawaniu dawek 10–240 mg co 24 godziny nie obserwowano akumulacji febuksostatu. Przewidywany średni okres półtrwania terminalnego (t1/2) febuksostatu wynosił około 5–8 godzin. Przeprowadzono populacyjną analizę farmakokinetyki/farmakodynamiki na podstawie danych z 211 pacjentów z hiperurykemią i podagrą, którzy przyjmowali febuksostat w dawkach 40–240 mg 1 raz dziennie. Ogólnie uzyskane wartości parametrów farmakokinetycznych były podobne do tych u zdrowych ochotników, co stanowi dobrą model do oceny farmakokinetyki/farmakodynamiki leku u pacjentów z podagrą.
Wchłanianie. Febuksostat jest szybko (tmax (czas do osiągnięcia maksymalnego stężenia) 1,0–1,5 godziny) i dobrze (co najmniej 84%) wchłaniany. Po jednorazowym i wielokrotnym podaniu febuksostatu doustnie w dawkach 80 mg lub 120 mg 1 raz dziennie Cmax wynosi odpowiednio 2,8–3,2 µg/ml i 5,0–5,3 µg/ml. Bezpośredniej bio dostępności tabletek febuksostatu nie analizowano. Po wielokrotnym podaniu w dawce 80 mg 1 raz dziennie lub po jednorazowym podaniu w dawce 120 mg w połączeniu z tłustym posiłkiem Cmax zmniejszało się o 49% i 38%, a AUC – o 18% i 16% odpowiednio. Jednak nie towarzyszyło temu klinicznie istotne zmiany stopnia obniżenia poziomu kwasu moczowego w osoczu krwi (po wielokrotnym podaniu w dawce 80 mg). Zatem febuksostat można stosować niezależnie od spożycia pokarmu.
Rozkład. Przewidywany objętość rozkładu w stanie równowagi (Vss/F) febuksostatu zmienia się od 29 do 75 l po doustnym podaniu w dawce 10–300 mg. Stopień wiązania z białkami osocza krwi (głównie z albuminą) wynosi 99,2% i nie zmienia się przy zwiększeniu dawki od 80 mg do 120 mg. Dla aktywnych metabolitów febuksostatu stopień wiązania z białkami osocza krwi waha się od 82 do 91%.
Metabolizm. Febuksostat jest aktywnie metabolizowany poprzez koniugację z udziałem uridylo-difosfo-glukuronilotransferazy (UDP-glukuronilotransferazy) i utlenianie z udziałem enzymów układu cytochromu P 450 (CYP). Opisano łącznie 4 farmakologicznie aktywne hydroksylowe metabolity febuksostatu, z których 3 zostały wykryte u człowieka w osoczu krwi. Badania in vitro na mikrosomach wątroby człowieka wykazały, że te utlenione metabolity powstają głównie pod działaniem CYP1A1, CYP1A2, CYP2C8 i CYP2C9, podczas gdy glukuronid febuksostatu powstaje głównie pod działaniem UDP-glukuronilotransferazy 1A1, 1A8, 1A9.
Wydalanie. Febuksostat jest wydalany z organizmu przez wątrobę i nerki. Po doustnym podaniu 14C-febuksostatu w dawce 80 mg około 49% wydalało się z moczem w postaci niezmienionego febuksostatu (3%), acylglukuronidu substancji czynnej (30%), znanych metabolitów utlenionych i ich koniugatów (13%) oraz innych nieznanych metabolitów (3%). Oprócz wydalania nerkowego, około 45% wydalało się z kałem w postaci niezmienionego febuksostatu (12%), acylglukuronidu substancji czynnej (1%), znanych metabolitów utlenionych i ich koniugatów (25%) oraz innych nieznanych metabolitów (7%).
Niewydolność nerek. Po wielokrotnym podaniu febuksostatu w dawce 80 mg nie zaobserwowano zmian Cmax febuksostatu u pacjentów z lekką, umiarkowaną lub ciężką niewydolnością nerek w porównaniu z pacjentami z normalną czynnością nerek. Średnie całkowite AUC febuksostatu zwiększało się o około 1,8 raza od 7,5 µg×godz/ml u pacjentów z normalną czynnością nerek do 13,2 µg×godz/ml u pacjentów z ciężką niewydolnością nerek. Cmax i AUC aktywnych metabolitów wzrastały odpowiednio 2 i 4 razy. Jednak pacjentom z niewydolnością nerek lekkiego lub umiarkowanego stopnia nie wymaga się korekty dawki leku.
Niewydolność wątroby. Po wielokrotnym podaniu febuksostatu w dawce 80 mg nie zaobserwowano istotnych zmian Cmax i AUC febuksostatu i jego metabolitów u pacjentów z lekką (klasa A wg skali Childa-Pugh) i umiarkowaną (klasa B wg skali Childa-Pugh) niewydolnością wątroby w porównaniu z pacjentami z normalną czynnością wątroby. Badanie leku u pacjentów z ciężką niewydolnością wątroby (klasa C wg skali Childa-Pugh) nie przeprowadzono.
Wiek. Po wielokrotnym doustnym podaniu febuksostatu nie zaobserwowano istotnych zmian AUC febuksostatu i jego metabolitów u pacjentów starszych w porównaniu z młodymi zdrowymi ochotnikami.
Płeć. Po wielokrotnym doustnym podaniu febuksostatu Cmax i AUC febuksostatu u kobiet były o 24% i 12% wyższe niż u mężczyzn. Jednak Cmax i AUC skorygowane o masę ciała były podobne w obu grupach, dlatego nie wymaga się zmiany dawki febuksostatu w zależności od płci.
Dane kliniczne.
Wskazania.
Likwestia® 80 mg i Likwestia® 120 mg
Leczenie przewlekłej hiperurykemii w chorobach towarzyszących odkładaniu się kryształów moczanów, w tym w obecności tofusów i/lub artretyzmu dny moczanowej obecnego lub w wywiadzie.
Likwestia® 120 mg
Leczenie i zapobieganie hiperurykemii u dorosłych pacjentów poddawanych chemioterapii z powodu hematologicznych nowotworów złośliwych z umiarkowanym lub wysokim ryzykiem zespołu lizy guza (ZLG).
Lek Likwestia® wskazany jest dla dorosłych pacjentów.
Przeciwwskazania.
Nadwrażliwość na substancję czynną lub którykolwiek ze składników pomocniczych leku wymienionych w sekcji „Skład”.
Współdziałanie z innymi lekami i inne rodzaje interakcji.
Merkaptopuryna/azatiopryna.
Ze względu na mechanizm działania febuksostat hamuje ksantynooksydazę, dlatego nie zaleca się jednoczesnego stosowania. Hamowanie ksantynooksydazy może prowadzić do zwiększenia stężenia obu leków we krwi, co może spowodować reakcję mielotoksyczną.
W przypadku jednoczesnego stosowania z febuksostatem dawkę merkaptopuryny/azatiopryny należy zmniejszyć do 20 % lub mniej w porównaniu z wcześniejszą dawką (patrz sekcja „Szczególne wskazania dotyczące stosowania”).
Skuteczność proponowanej korekty dawki, opartej na modelowaniu i analizie symulacyjnej danych przedklinicznych uzyskanych na szczurach, potwierdzono wynikami badania klinicznego oceniającego interakcję leków przeprowadzonego u zdrowych ochotników otrzymujących azatioprynę 100 mg oddzielnie oraz zmniejszoną dawkę azatiopryny (25 mg) w połączeniu z febuksostatem (40 lub 120 mg).
Brak danych dotyczących bezpieczeństwa stosowania febuksostatu podczas cytotoksycznej chemioterapii.
Badania interakcji febuksostatu z innymi cytotoksycznymi lekami przeciwnowotworowymi nie były prowadzone.
Rosiglitazon / substraty CYP2C8.
Febuksostat jest słabym inhibitorem CYP2C8 in vitro. W badaniu jednoczesne podawanie 120 mg febuksostatu raz dziennie oraz jednorazowej dawki doustnej 4 mg rosiglitazonu u zdrowych ochotników nie wpływało na farmakokinetykę rosiglitazonu ani jego metabolitu N-detylorosiglitazonu, co pokazuje, że febuksostat nie hamuje enzymu CYP2C8 in vivo. W związku z tym jednoczesne stosowanie febuksostatu i rosiglitazonu lub innych substratów CYP2C8 nie wymaga korekty dawki tych leków.
Teofilina. Przeprowadzono badanie interakcji febuksostatu z udziałem zdrowych ochotników w celu oceny wpływu hamowania ksantynooksydazy na wzrost stężenia teofiliny we krwi krążącej, obserwowany przy innych inhibitorach ksantynooksydazy. Jednoczesne stosowanie febuksostatu w dawce 80 mg i teofiliny w dawce 400 mg nie powodowało żadnych interakcji farmakokinetycznych ani wpływu na bezpieczeństwo teofiliny. W związku z tym febuksostat w dawce 80 mg można stosować jednocześnie z teofiliną bez szczególnych ostrzeżeń. Brak danych dotyczących dawki febuksostatu 120 mg.
Naproxen i inne inhibitory glukuronidacji.
Metabolizm febuksostatu zależy od aktywności enzymu UDP-glukuronozylotransferazy. Leki hamujące proces glukuronidacji, np. niesteroidowe leki przeciwzapalne (NLPZ) i probenecyd, teoretycznie mogą wpływać na wydalanie febuksostatu. U zdrowych ochotników jednoczesne stosowanie febuksostatu i naproxenu w dawce 250 mg dwa razy dziennie powodowało zwiększenie działania febuksostatu (Cmax o 28 %, AUC o 41 %, t1/2 (okres półtrwania) o 26 %). W trakcie badań klinicznych stosowanie naproxenu i innych NLPZ / inhibitorów COX-2 nie wiązało się z klinicznie istotnym wzrostem reakcji niepożądanych.
Febuksostat można stosować jednocześnie z naproxenem bez zmiany dawek obu leków.
Induktory glukuronidacji.
Silne induktory UDP-glukuronozylotransferazy mogą nasilać metabolizm i zmniejszać skuteczność febuksostatu. U pacjentów stosujących silne induktory glukuronidacji zaleca się kontrolę stężenia kwasu moczowego we krwi po 1–2 tygodniach jednoczesnej terapii. Po odstawieniu induktora glukuronidacji możliwe jest zwiększenie stężenia febuksostatu we krwi.
Kolcharyna/indometacyna/hydrochlorotiazyd/warfaryna.
Febuksostat można stosować jednocześnie z kolcharyną lub indometacyną bez zmiany dawek leków.
Nie trzeba również zmieniać dawki febuksostatu przy jednoczesnym stosowaniu z hydrochlorotiazydem.
Jednoczesne stosowanie febuksostatu z warfaryną nie wymaga zmiany dawki tego ostatniego. Stosowanie febuksostatu (80 mg lub 120 mg raz dziennie) z warfaryną u zdrowych ochotników nie wpływało na farmakokinetykę warfaryny. Jednoczesne stosowanie z febuksostatem nie wpływało również na INR (międzynarodowe znormalizowane stosunki) i aktywność czynnika VII.
Desipramina / substraty CYP2D6.
Dane uzyskane in vitro wskazują, że febuksostat jest słabym inhibitorem CYP2D6. W badaniach przeprowadzonych u zdrowych ochotników otrzymujących 120 mg febuksostatu raz dziennie zaobserwowano wzrost AUC desipraminy (substrat CYP2D6) o 22 %, co świadczy o słabym działaniu hamującym febuksostatu na enzym CYP2D6 in vivo.
W związku z tym przy jednoczesnym stosowaniu febuksostatu i substratów CYP2D6 nie ma potrzeby zmiany ich dawek.
Leki przeciwwskazowe (antacida).
Przy jednoczesnym stosowaniu z lekami przeciwwskazowymi zawierającymi wodorotlenek magnezu i wodorotlenek glinu obserwuje się opóźnienie wchłaniania febuksostatu (około 1 godzinę) oraz zmniejszenie Cmax o 32 %, jednak AUC febuksostatu istotnie się nie zmienia, dlatego febuksostat można łączyć z lekami przeciwwskazowymi.
Szczególne środki ostrożności dotyczące stosowania.
Choroby układu sercowo-naczyniowego.
U pacjentów z poważnymi chorobami układu sercowo-naczyniowego (np. zawałem mięśnia sercowego, udarem mózgu lub niestabilną dławicą piersiową) w trakcie badań przedrejestracyjnych oraz w jednym z badań pozwoleniowych (CARES) obserwowano większą liczbę zagrażających życiu zaburzeń sercowo-naczyniowych przy stosowaniu febuksostatu w porównaniu z allopurynolem.
Jednak kolejne badanie pozwoleniowe (FAST) wykazało, że stosowanie febuksostatu nie ustępowało allopurynolowi pod względem częstości występowania zaburzeń sercowo-naczyniowych, zarówno zagrażających, jak i niezagrażających życiu.
Leczenie tej grupy pacjentów należy prowadzić z ostrożnością oraz regularnie monitorować ich stan. Szczegółowe informacje dotyczące bezpieczeństwa stosowania febuksostatu w odniesieniu do układu sercowo-naczyniowego zawarte są w punktach „Farmakodynamika” oraz „Działania niepożądane”.
Profilaktyka i leczenie hiperurykemii u pacjentów z ryzykiem wystąpienia zespołu lizy nowotworowej (ZLN).
Pacjenci poddawani chemioterapii z powodu hematologicznych nowotworów złośliwych z umiarkowanym lub wysokim ryzykiem ZLN, stosujący lek Likwestia®, powinni być pod opieką kardiologa, jeśli istnieją wskazania kliniczne.
Alergia/reakcje nadwrażliwości na leki.
W ramach nadzoru po wprowadzeniu na rynek zgłaszano rzadkie przypadki poważnych reakcji alergicznych/reakcji nadwrażliwości, w tym zagrażające życiu przypadki zespołu Stevensa-Johnsona, toksycznego nekrolizy epidermalnej (TEN) oraz ostrych reakcji anafilaktycznych/szoku. Zazwyczaj reakcje te pojawiały się w ciągu pierwszego miesiąca stosowania febuksostatu. U części, ale nie wszystkich pacjentów, stwierdzano zaburzenia funkcji nerek i/lub nadwrażliwość na allopurynol w wywiadzie. Ciężkie reakcje nadwrażliwości, w tym reakcje lekowe z eozynofilią i objawami systemowymi (zespołu DRESS), w niektórych przypadkach były związane z gorączką, zaburzeniami hematologicznymi, niewydolnością nerek lub wątroby.
Pacjentów należy poinformować o objawach i oznakach nadwrażliwości/alergii oraz należy monitorować ich pod kątem rozwoju takich reakcji. W przypadku wystąpienia poważnych reakcji alergicznych/reakcji nadwrażliwości, w szczególności zespołu Stevensa-Johnsona, stosowanie febuksostatu należy natychmiast przerwać, ponieważ wcześniejsze przerwanie leczenia poprawia rokowanie. Jeśli u pacjenta wystąpiła reakcja alergiczna/reakcja nadwrażliwości, w tym zespół Stevensa-Johnsona, oraz ostre reakcje anafilaktyczne/szok, ponowne stosowanie febuksostatu jest przeciwwskazane.
Zaostrzenie (napad) podagry.
Leczenie febuksostatem należy rozpoczynać dopiero w okresie między napadami choroby. Febuksostat może wywołać napad podagry na początku leczenia z powodu zmiany stężenia kwasu moczowego we krwi spowodowanej uwalnianiem się moczanów z zasobników. Na początku leczenia febuksostatem zaleca się podawanie leków przeciwbólowych niesteroidowych (NLPZ) lub kolchicyny przez co najmniej 6 miesięcy w celu zapobiegania napadom podagry.
W przypadku wystąpienia napadu podagry podczas stosowania febuksostatu leczenie należy kontynuować. Jednocześnie należy prowadzić odpowiednią terapię indywidualną w celu leczenia zaostrzenia podagry. Przy długotrwałym stosowaniu febuksostatu częstość i ciężkość napadów podagry zmniejszają się.
Odkładanie ksenotynów.
U pacjentów z przyspieszonym tworzeniem się moczanów (np. na tle nowotworów złośliwych i ich leczenia lub przy zespole Lescha-Nyhana) może dojść do istotnego wzrostu całkowitego stężenia ksenotynów w moczu, co w rzadkich przypadkach może prowadzić do ich odkładania się w drogach moczowych. Nie obserwowano tego w podstawowym badaniu klinicznym febuksostatu w ZLN. Z uwagi na ograniczone doświadczenie w stosowaniu febuksostatu nie wskazuje się go pacjentom z zespołem Lescha-Nyhana.
6-merkaptopuryna/azytiopryna.
Nie zaleca się stosowania febuksostatu pacjentom, którzy jednocześnie przyjmują 6-merkaptopurynę/azytioprynę, ponieważ hamowanie ksantynooksydazy przez febuksostat może prowadzić do wzrostu stężenia 6-merkaptopuryny/azytiopriny w osoczu krwi i spowodować ciężką toksyczność.
Jeśli nie można uniknąć tej kombinacji, stan pacjentów należy dokładnie monitorować. Zaleca się zmniejszenie dawki 6-merkaptopuryny lub azytiopriny do 20% lub mniej w stosunku do poprzednio przepisanej dawki w celu uniknięcia możliwych działań hematologicznych (patrz punkt „Współdziałanie z innymi lekami i inne formy interakcji”). Pacjentów należy dokładnie monitorować, a dawkę 6-merkaptopuryny/azytiopriny należy odpowiednio dostosować w zależności od odpowiedzi terapeutycznej i możliwych działań toksycznych.
Pacjenci po przeszczepie narządów.
Nie ma doświadczenia w stosowaniu febuksostatu u tej grupy pacjentów, dlatego stosowanie leku nie jest wskazane.
Teofilina.
Jednorazowe jednoczesne podanie febuksostatu w dawce 80 mg i teofiliny w dawce 400 mg nie wykazało żadnych interakcji farmakokinetycznych. Febuksostat w dawce 80 mg można stosować jednocześnie z teofliną bez ryzyka wzrostu stężenia teofiliny w osoczu krwi. Brak danych dotyczących dawki febuksostatu 120 mg.
Choroby wątroby.
W trakcie połączonej fazy III badań klinicznych u 5,0% pacjentów przyjmujących febuksostat obserwowano nieznaczne zmiany parametrów wątrobowych, dlatego zaleca się sprawdzenie czynności wątroby przed rozpoczęciem stosowania febuksostatu oraz w trakcie leczenia, jeśli istnieją wskazania.
Choroby tarczycy.
U 5,5% pacjentów przyjmujących febuksostat przez dłuższy czas obserwowano wzrost stężenia TSH (> 5,5 mIU/ml) w trakcie długoterminowych, otwartych badań rozszerzonych. Dlatego lek należy przepisywać z ostrożnością pacjentom z zaburzeniami funkcji tarczycy.
Laktoza.
Lek zawiera laktozę. Jeśli u pacjenta stwierdzono nietolerancję niektórych cukrów, należy skonsultować się z lekarzem przed przyjęciem tego leku.
Stosowanie w czasie ciąży lub karmienia piersią.
Ciąża
Ograniczone doświadczenie w stosowaniu febuksostatu w czasie ciąży wskazuje na brak niekorzystnego wpływu leku na przebieg ciąży i zdrowie płodu/noworodka. Badania na zwierzętach nie wykazały bezpośredniego ani pośredniego szkodliwego wpływu na przebieg ciąży, rozwój embrionalny/fetalny oraz przebieg porodu. Potencjalne ryzyko dla człowieka jest nieznane. Febuksostat nie powinien być stosowany w czasie ciąży.
Karmienie piersią
Nie wiadomo, czy febuksostat przenika do mleka matki. Badania na zwierzętach wykazały, że febuksostat przenika do mleka matki i negatywnie wpływa na rozwój noworodków karmionych tym mlekiem. Ryzyko przenikania leku do mleka matki nie może być wykluczone. Februksostat nie powinien być stosowany w czasie karmienia piersią.
Plodność
Badania dotyczące płodności przeprowadzone na zwierzętach w dawce 48 mg/kg/dobę nie wykazały zależności działań niepożądanych od dawki. Działanie leku Likwestia® na funkcję rozrodczą człowieka jest nieznane.
Wpływ na zdolność prowadzenia pojazdów i obsługi mechanizmów.
Zgłaszano przypadki wystąpienia senności, zawrotów głowy, parestezji oraz zaburzeń ostrości widzenia podczas stosowania febuksostatu. Dlatego pacjentom przyjmującym lek Likwestia® zaleca się zachowanie ostrożności podczas prowadzenia pojazdów oraz pracy z innymi mechanizmami, aż do momentu, gdy będą pewni braku powyższych działań niepożądanych leku.
Sposób stosowania i dawki.
Dozowanie
Podagra. Zalecana dawka leku Likwestia® wynosi 80 mg 1 raz dziennie doustnie, niezależnie od przyjmowania pokarmu. Jeżeli po 2–4 tygodniach leczenia stężenie kwasu moczowego w surowicy przekracza 6 mg/dl (357 µmol/l), należy rozważyć zwiększenie dawki leku Likwestia® do 120 mg 1 raz dziennie. Działanie leku pojawia się dość szybko, co pozwala na ponowne oznaczenie stężenia kwasu moczowego już po 2 tygodniach. Celem leczenia jest obniżenie stężenia kwasu moczowego w surowicy i utrzymanie go na poziomie poniżej 6 mg/dl (357 µmol/l).
Czas trwania profilaktyki napadów podagry wynosi co najmniej 6 miesięcy.
Zespół lizy nowotworu (ZLN). Zalecana dawka leku Likwestia® wynosi 120 mg 1 raz dziennie doustnie, niezależnie od przyjmowania pokarmu.
Stosowanie leku Likwestia® należy rozpocząć dwa dni przed rozpoczęciem terapii cytotoksycznej i kontynuować co najmniej przez 7 dni, choć czas leczenia może być wydłużony do 9 dni, w zależności od długości chemioterapii i oceny klinicznej.
Pacjenci w podeszłym wieku. U tej grupy pacjentów nie jest wymagana korekta dawki.
Niewydolność nerek. U pacjentów z ciężkim zaburzeniem funkcji nerek (klirens kreatyniny < 30 ml/min) skuteczność i bezpieczeństwo stosowania leku nie zostały wystarczająco przebadane. U pacjentów z łagodnym lub umiarkowanym zaburzeniem funkcji nerek korekta dawki nie jest wymagana.
Niewydolność wątroby. Badania skuteczności i bezpieczeństwa febukostatu u pacjentów z ciężką niewydolnością wątroby (klasa C wg skali Childa–Pugh) nie były prowadzone.
Podagra. W przypadku łagodnego zaburzenia funkcji wątroby zalecana dawka wynosi 80 mg. Doświadczenie zastosowania leku Likwestia® u pacjentów z umiarkowanym zaburzeniem funkcji wątroby jest ograniczone.
Zespół lizy nowotworu (ZLN). Z badania III fazy (FLORENCE) wykluczono jedynie osoby z ciężką niewydolnością wątroby. U pacjentów włączonych do badania nie była wymagana korekta dawki ze względu na funkcję wątroby.
Sposób stosowania
Do stosowania doustnego.
Likwestia® stosuje się doustnie, niezależnie od przyjmowania pokarmu.
Dzieci.
Bezpieczeństwo i skuteczność stosowania leku Likwestia® u dzieci (do 18. roku życia) nie zostały ustalone. Brak danych dotyczących stosowania.
Przedawkowanie.
W przypadku przedawkowania wskazana jest terapia objawowa i wspomagająca.
Reakcje niepożądane
Podsumowanie profilu bezpieczeństwa
Najczęstszymi reakcjami niepożądanymi w badaniach klinicznych (4072 pacjentów przyjmujących dawkę od 10 do 300 mg), badaniach bezpieczeństwa po rejestracji (badanie FAST: 3001 pacjentów przyjmujących dawkę od 80 do 120 mg) oraz w trakcie nadzoru posprzedażowego u pacjentów z podagrą były nasilenia (napady) podagry, zaburzenia funkcji wątroby, biegunka, nudności, ból głowy, zawroty głowy, duszność, wysypka, świąd, artregie, miaglie, ból kończyn, obrzęki oraz zmęczenie. Reakcje te miały w większości przypadków charakter łagodny lub umiarkowany. W okresie nadzoru posprzedażowego zgłaszano rzadkie przypadki ciężkich reakcji nadwrażliwości na febuksostat, niektóre z towarzyszącymi reakcjami systemowym, oraz rzadkie przypadki nagłej śmierci sercowej.
W tabeli 2 wymieniono reakcje niepożądane występujące u pacjentów podczas stosowania febuksostatu. Według częstości występowania reakcje niepożądane sklasyfikowano następująco: często (od ≥ 1/100 do < 1/10), rzadko (od ≥ 1/1000 do < 1/100) oraz bardzo rzadko (od ≥ 1/10000 do < 1/1000). Częstość występowania oparta jest na danych z badań klinicznych oraz doświadczeniu posprzedażowym u pacjentów z podagrą.
W każdej grupie według częstości występowania reakcje niepożądane uporządkowane są według malejącego nasilenia.
Tabela 2.
Reakcje niepożądane obserwowane w fazie III, połączonych, rozszerzonych, długoterminowych badań klinicznych, badaniach bezpieczeństwa po rejestracji oraz w okresie nadzoru posprzedażowego u pacjentów z podagrą.
| Z boku krwi i układu limfatycznego |
Rzadko Pancytopenia, trombocytopenia, agranulocytoza*, anemia# |
| Z boku układu odpornościowego |
Rzadko Reakcje anafilaktyczne*, nadwrażliwość na lekarstwo* |
| Z boku układu endokrynnego |
Nieczęsto Podwyższenie stężenia hormonu tyreotropowego we krwi, niedoczynność tarczycy# |
| Z boku narządów wzroku |
Nieczęsto Zamazane widzenie Rzadko Zator tętnicy siatkówki# |
| Z boku odżywiania i przemiany materii |
Często*** Wzmożenie (napady) podagry Nieczęsto Cukrzyca, hiperlipidemia, zmniejszenie apetytu, przyrost masy ciała Rzadko Ubytek masy ciała, zwiększenie apetytu, anoreksja |
| Z boku psychiki |
Nieczęsto Obniżenie libidum, bezsenność Rzadko Niespokojność, przygnębienie nastroju#, zaburzenia snu# |
| Z boku układu nerwowego i narządów zmysłów |
Często Ból głowy, zawroty głowy Nieczęsto Parastezje, hemipareza, senność, letargia#, zmiana wrażliwości smakowej, hipestezja, osłabienie węchu Rzadko Ageuzja#, uczucie pieczenia# |
| Z boku narządów słuchu i labiryntu |
Nieczęsto Szum w uszach Rzadko Vertigo# |
| Z boku serca |
Nieczęsto Fibrulacja przedsionków, uczucie kołatania serca, odchylenie od normy na EKG, blokada lewej nogi pęczka Hisa (patrz sekcja „Efekty niepożądane. Zespół lizy guza”), tachykardia zatokowa (patrz sekcja „Efekty niepożądane. Zespół lizy guza”), arytmia# Rzadko Nagła śmierć sercowa* |
| Z boku naczyń |
Nieczęsto Nadciśnienie tętnicze, napływy, napływy z uczuciem gorąca, krwawienia (patrz sekcja „Efekty niepożądane. Zespół lizy guza”) Rzadko Kolaps krążeniowy# |
| Z boku układu oddechowego |
Często Uczucie duszności Nieczęsto Przewlekłe zapalenie oskrzeli, infekcje górnych dróg oddechowych, infekcje dolnych dróg oddechowych#, kaszel, rynoroea# Rzadko Łagodne zapalenie płuc# |
| Z boku układu trawiennego |
Często Diareia**, nudności Nieczęsto Ból brzucha, ból w górnej części brzucha#, wzdęcia brzucha, choroba refluksowa przełyku, wymioty, suchość w ustach, wzdęcia, zaparcia, częste wypróżnienia, meteorizm, dyskomfort w żołądku lub jelitach, owrzodzenie jamy ustnej, obrzęk warg#, zapalenie trzustki Rzadko Przebicie przewodu pokarmowego#, zapalenie jamy ustnej# |
| Z boku wątroby i dróg żółciowych |
Często Zaburzenia funkcji wątroby** Nieczęsto Choroba kamica żółciowa Rzadko Zapalenie wątroby, żółtaczka*, uszkodzenie wątroby*, zapalenie pęcherza żółciowego# |
| Z boku skóry i tkanki podskórnej |
Często Wysypka (w tym wysypka z niższą częstością występowania, patrz niżej), swędzenie Nieczęsto Dermita, pokrzywka, zmiana zabarwienia skóry, uszkodzenie skóry, plamki krwotoczne, wysypka plamista, wysypka makularna i papularna, wysypka papularna, nadpotliwość, łysienie, egzema#, rumień, nocne pocenia się#, trądzik#, wysypka z swędzeniem# Rzadko Toxyczny epidermalny nekroliz*, zespół Stevensa-Johnsona*, obrzęk naczynioruchowy*, reakcja lekowa z eozynofilią i objawami ogólnoustrojowymi*, uogólniona wysypka (poważna)*, wysypka egzfoliatywna, wysypka folikularna, wysypka pęcherzykowa, wysypka pustularna, wysypka rumieniowa, wysypka różyczkowa |
| Z boku układu ruchu i tkanki łącznej |
Często Ból stawów, ból mięśni, ból kończyn# Nieczęsto Zapalenie stawów, ból mięśniowo-szkieletowy, osłabienie mięśni, skurcze mięśni, sztywność mięśni, sztywność stawów#, zapalenie bursy, obrzęk stawów#, ból pleców#, sztywność mięśniowo-szkieletowa# Rzadko Rabdomyoliza*, zespół rotatora#, reumatyczna polimialgia# |
| Z boku nerek i dróg moczowych |
Nieczęsto Niewydolność nerek, choroba kamica nerek, krwiomocz, częstomocz, białkomocz, naglące pозiwy do mikcji, infekcje dróg moczowych# Rzadko Nefryt naczyniowo-śródmiąższowy* |
| Z boku układu rozrodczego i gruczołów mlekowych |
Nieczęsto Dysfunkcja erektilna |
| Z boku organizmu ogólnie |
Często Obroty, zmęczenie Nieczęsto Ból w klatce piersiowej, uczucie dyskomfortu w klatce piersiowej, ból#, osłabienie# Rzadko Pragnienie, uczucie gorąca# |
| Wyniki badań laboratoryjnych |
Nieczęsto Podwyższenie stężenia amylazy we krwi, zmniejszenie liczby płytek krwi, zmniejszenie liczby leukocytów we krwi, zmniejszenie liczby limfocytów we krwi, podwyższenie stężenia kreatyny we krwi, podwyższenie stężenia kreatyniny we krwi, obniżenie stężenia hemoglobiny we krwi, podwyższenie stężenia mocznika we krwi, podwyższenie stężenia trójglicerydów we krwi, podwyższenie stężenia cholesterolu we krwi, obniżenie hematokrytu, podwyższenie stężenia dehydrogenazy mleczanowej (LDH) we krwi, podwyższenie stężenia potasu we krwi, podwyższenie MCV# Rzadko Podwyższenie stężenia glukozy we krwi, wydłużenie częściowego czasu tromboplastynowego, zmniejszenie liczby erytrocytów we krwi, podwyższenie stężenia fosfatazy zasadowej we krwi, podwyższenie stężenia kinazy kreatynowej we krwi* |
| Urazy, zatrucia i powikłania proceduralne |
Nieczęsto Siniak |
* Reakcje niepożądane wykryte w ramach analizy po wprowadzeniu na rynek.
** Diareę wymagającą leczenia oraz odchylenia wyników badań funkcji wątroby od normy, obserwowane w badaniach fazy III, rozwijały się częściej u pacjentów otrzymujących terapię współbieżną kolchicyną.
*** Zob. sekcję „Farmakodynamika” w odniesieniu do częstości napadów podagry obserwowanych w fazie III poszczególnych randomizowanych badań kontrolowanych.
Reakcje niepożądane obserwowane podczas badań bezpieczeństwa po rejestracji.
Opis poszczególnych reakcji niepożądanych.
W ramach nadzoru po wprowadzeniu na rynek zgłaszano rzadkie przypadki ciężkich reakcji nadwrażliwości na fubukostat, w tym zespół Stevensa-Johnsona, toksyczny epidermalny nekroliz oraz reakcje anafilaktyczne/szok. Zespół Stevensa-Johnsona i toksyczny epidermalny nekroliz charakteryzują się postępującym wysypem skórnym z pęcherzowatym uszkodzeniem skóry lub błon śluzowych oraz podrażnieniem spojówek. Reakcje nadwrażliwości na fubukostat mogą objawiać się takimi objawami jak reakcje skórne charakteryzujące się infiltracyjnymi wysypami makularno-papularnymi, ogólnoustrojowymi lub egfoliatywnymi wysypami, a także uszkodzeniami skóry, obrzękiem twarzy, gorączką, zaburzeniami hematologicznymi, takimi jak trombocytopenia i eozynofilia, oraz uszkodzeniem pojedynczych narządów lub wielu narządów (wątroba i nerki, w tym nerkowy zapalenie kanalików i przestrzeni międzykomórkowej).
Napady podagry zazwyczaj pojawiały się krótko po rozpoczęciu leczenia i w pierwszych miesiącach terapii. Częstość napadów podagry zmniejszała się z czasem. W trakcie stosowania fubukostatu zaleca się profilaktykę ostrych napadów podagry.
Zespół lizy guza (ZLG).
Podsumowanie profilu bezpieczeństwa.
W trakcie randomizowanego, podwójnie ślepego badania kontrolnego fazy III FLORENCE (FLO-01), w którym porównywano fubukostat i alopiurinol (346 pacjentów poddawanych chemioterapii z powodu hematologicznych nowotworów złośliwych z umiarkowanym lub wysokim ryzykiem ZLG), reakcje niepożądane wystąpiły tylko u 22 (6,4 %) pacjentów, a mianowicie u 11 (6,4 %) pacjentów w każdej grupie leczenia. Większość reakcji niepożądanych była łagodnego lub umiarkowanego nasilenia.
Ogólnie w trakcie badania FLORENCE nie stwierdzono żadnych dodatkowych podejrzeń dotyczących bezpieczeństwa stosowania fubukostatu u pacjentów z podagrą, z wyjątkiem trzech niżej wymienionych reakcji niepożądanych (zob. tabela 2).
Ze strony serca: rzadko — blokada lewej odnogi wiązki Hisa, tachykardia zatokowa.
Ze strony naczyń: rzadko — krwawienia.
Zgłaszanie podejrzewanych reakcji niepożądanych
Zgłaszanie reakcji niepożądanych po rejestracji leku ma duże znaczenie. Pozwala to na monitorowanie stosunku korzyści do ryzyka w przypadku stosowania tego leku. Pracownicy medyczni i farmaceutyczni, a także pacjenci lub ich ustawieni reprezentanci, powinni zgłaszać wszystkie przypadki podejrzewanych reakcji niepożądanych oraz braku skuteczności leku poprzez Automatyczny System Informacyjny Nadzoru Farmakologicznego pod adresem: https://aisf.dec.gov.ua.
Okres ważności. 3 lata.
Nie stosować leku po upływie okresu ważności wskazanego na opakowaniu.
Warunki przechowywania. Ten lek nie wymaga specjalnych warunków przechowywania.
Przechowywać w miejscu niedostępnym dla dzieci.
Opakowanie. Po 14 tabletek w blisterze. Po 2 lub 4 blistery w puszce.
Kategoria wydania. Na receptę.
Producent. A.T. „Farmak” (produkcja z produktu in bulk firmy producenta GenePharm S.A., Grecja).
Adres siedziby producenta i miejsce prowadzenia działalności.
Ukraina, 04080, miasto Kijów, ul. Cyrylicka, 74.