Liquestia
Ukraine
Table of Contents
INSTRUCTION FOR MEDICAL USE OF THE MEDICINAL PRODUCT LIQUESTIA® (LIQUESTIA)
Composition:
Active substance: febuxostat;
One film-coated tablet contains febuxostat 80 mg or 120 mg;
Excipients: microcrystalline cellulose, lactose monohydrate, hydroxypropylcellulose, sodium lauryl sulfate, sodium croscarmellose, anhydrous lactose, colloidal anhydrous silicon dioxide, magnesium stearate;
Film coating: polyvinyl alcohol (E 1203), titanium dioxide (E 171), macrogol 3350 (E 1521), talc (E 553b), yellow iron oxide (E 172).
Pharmaceutical form. Film-coated tablets.
Main physicochemical properties:
80 mg film-coated tablets: elongated, biconvex, film-coated tablets, from pale yellow to yellow, with "80" embossed on one side and smooth on the other;
120 mg film-coated tablets: elongated, biconvex, film-coated tablets, from pale yellow to yellow, with "120" embossed on one side and smooth on the other.
Pharmacotherapeutic group.
Medicinal products for the treatment of gout. Medicinal products that inhibit uric acid production. ATC code M04A A03.
Pharmacological Properties.
Pharmacodynamics.
Mechanism of action
Uric acid is the end product of purine metabolism in humans and is formed during the following reaction: hypoxanthine → xanthine → uric acid. Xanthine oxidase catalyzes both steps of this reaction. Febuxostat, a derivative of 2-arylthiazole, exerts its therapeutic effect by reducing serum uric acid concentrations through selective inhibition of xanthine oxidase. Febuxostat is a potent and selective non-purine inhibitor of xanthine oxidase (NP-SIXO), with an inhibition constant (Ki) in vitro of less than 1 nanomolar. Febuxostat has been shown to significantly inhibit the activity of both oxidized and reduced forms of xanthine oxidase. At therapeutic concentrations, febuxostat does not affect other enzymes involved in the metabolism of purines or pyrimidines, such as guanidine deaminase, hypoxanthine-guanine phosphoribosyltransferase, orotate phosphoribosyltransferase, orotidine monophosphate decarboxylase, or purine nucleoside phosphorylase.
Clinical efficacy and safety
Gout. The efficacy of febuxostat was confirmed in three pivotal Phase III clinical trials (two pivotal trials, APEX and FACT, and an additional trial, CONFIRMS, described below), which included 4101 patients with hyperuricemia and gout. In each of these pivotal Phase III studies, febuxostat demonstrated superior efficacy compared to allopurinol in lowering and maintaining serum uric acid concentrations at target levels. The primary efficacy endpoint in the APEX and FACT studies was the proportion of patients who achieved serum uric acid concentrations ≤ 6.0 mg/dL (357 µmol/L) during the last three monthly visits. In the additional Phase III CONFIRMS study, the results of which became available after the initial approval of febuxostat, the primary efficacy endpoint was the proportion of patients with serum uric acid concentrations ≤ 6.0 mg/dL at the last visit. Patients who had undergone organ transplantation were not included in these studies (see section "Special precautions for use").
APEX study: The Phase III Allopurinol and Placebo-Controlled Efficacy Study of Febuxostat (APEX) was a randomized, double-blind, multicenter trial of 28 weeks’ duration. A total of 1072 patients were randomized: placebo (n=134), febuxostat 80 mg once daily (n=267), febuxostat 120 mg once daily (n=269), febuxostat 240 mg once daily (n=134), and allopurinol 300 mg once daily (n=258) for patients with baseline serum creatinine concentration ≤ 1.5 mg/dL or 100 mg once daily (n=10) for patients with baseline serum creatinine concentration > 1.5 mg/dL and ≤ 2.0 mg/dL. Febuxostat 240 mg (twice the maximum recommended dose) was administered to assess safety.
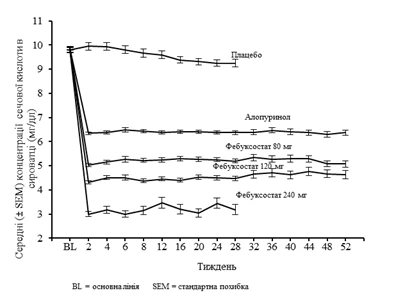
The APEX study demonstrated statistically significant superiority of both treatment regimens—febuxostat 80 mg once daily and febuxostat 120 mg once daily—compared to allopurinol at the standard dose of 300 mg (n=258)/100 mg (n=10) in reducing serum uric acid concentrations to below 6 mg/dL (357 µmol/L) (see Table 1 below). Figure 1 shows mean serum uric acid concentrations over time for each treatment group in both pivotal Phase III studies.
Figure 1. Mean serum uric acid concentrations from combined pivotal studies (Phase III)
**
**
Note: 509 patients received allopurinol 300 mg once daily; 10 patients with serum creatinine concentration > 1.5 mg/dL and < 2.0 mg/dL received allopurinol 100 mg once daily (10 out of 268 patients in the APEX study). Febuxostat 240 mg was administered to evaluate safety at a dose twice the maximum recommended.
FACT study: The Phase III Febuxostat Allopurinol Controlled Trial (FACT) was a randomized, double-blind, multicenter study of 52 weeks’ duration. A total of 760 patients were randomized: febuxostat 80 mg once daily (n=256), febuxostat 120 mg once daily (n=251), and allopurinol 300 mg once daily (n=253).
The FACT study demonstrated statistically significant superiority of both regimens—febuxostat 80 mg once daily and febuxostat 120 mg once daily—compared to allopurinol 300 mg once daily in reducing and maintaining serum uric acid concentrations below 6 mg/dL (357 µmol/L).
Table 1 presents the results for the assessment of the primary efficacy endpoint.
Table 1
Proportion of patients with serum uric acid concentration < 6.0 mg/dL (357 µmol/L) during the last three monthly visits
**
| Study |
Febuxostat 80 mg once daily |
Febuxostat 120 mg once daily |
Allopurinol 300/100 mg once daily1 |
| APEX (28 weeks) |
48 %* (n=262) |
65 %*, # (n=269) |
22 % (n=268) |
| FACT (52 weeks) |
53 %* (n=255) |
62 %* (n=250) |
21 % (n=251) |
| Pooled results |
51 %* (n=517) |
63 %*, # (n=519) |
22 % (n=519) |
| 1 Results from patients receiving 100 mg once daily (n = 10: patients with baseline serum creatinine > 1.5 mg/dL and ≤ 2.0 mg/dL) or 300 mg once daily (n=509) were combined during analysis. * p < 0.001 vs allopurinol. # p < 0.001 vs 80 mg dose. |
|||
The ability of febuxostat to rapidly reduce serum uric acid concentration was rapid and sustained. Reduction in serum uric acid concentration to < 6.0 mg/dL (357 µmol/L) was observed as early as the second week of the study and persisted throughout treatment.
CONFIRMS study: The CONFIRMS study was a randomized, controlled phase III trial lasting 26 weeks, designed to evaluate the safety and efficacy of febuxostat at doses of 40 mg and 80 mg compared to allopurinol at doses of 300 mg and 200 mg in patients with gout and hyperuricemia. A total of 2269 patients were randomized: febuxostat 40 mg once daily (n=757), febuxostat 80 mg once daily (n=756), and allopurinol 300/200 mg once daily (n=756). At least 65% of patients had mild to moderate renal impairment (with creatinine clearance of 30–89 mL/min). Prophylaxis of gout flares was mandatory throughout the 26 weeks.
The proportion of patients achieving serum uric acid concentration < 6.0 mg/dL (357 µmol/L) at the last visit was 45% in the febuxostat 40 mg group, 67% in the febuxostat 80 mg group, and 42% in the allopurinol 300/200 mg group.
Primary endpoint in the subgroup of patients with renal impairment. The APEX study evaluated efficacy in a subgroup of 40 patients with renal impairment (i.e., baseline serum creatinine concentration > 1.5 mg/dL and ≤ 2.0 mg/dL). Patients randomized to the allopurinol group received a reduced dose of 100 mg once daily. The primary efficacy endpoint was achieved in febuxostat groups in 44% of patients (80 mg once daily), 45% (120 mg once daily), and 60% (240 mg once daily), compared to 0% in the allopurinol 100 mg once daily and placebo groups.
No clinically significant differences in the percentage reduction of serum uric acid concentration were observed in healthy volunteers regardless of renal function status (58% in the group with normal renal function and 55% in the group with severe renal impairment).
A prospective analysis conducted in patients with gout and renal impairment using the CONFIRMS study demonstrated that febuxostat was significantly more effective: serum uric acid levels decreased to < 6.0 mg/dL compared to allopurinol 300 mg/200 mg in patients with gout and mild to moderate renal impairment (65% of subjects).
Primary endpoint in the subgroup of patients with baseline serum uric acid concentration ≥ 10 mg/dL. Baseline serum uric acid concentration ≥ 10 mg/dL was observed in approximately 40% of patients (combined APEX and FACT studies). Among these patients, the primary efficacy endpoint (serum uric acid concentration < 6 mg/dL at the last three visits) was achieved in the febuxostat subgroups: 41% of patients (80 mg once daily), 48% of patients (120 mg once daily), and 66% of patients (240 mg once daily), compared to 9% in the allopurinol 300 mg/100 mg once daily group and 0% in the placebo group.
According to the CONFIRMS study, the proportion of patients achieving the primary efficacy endpoint (serum uric acid concentration < 6.0 mg/dL at the last visit) in the subgroup of patients with baseline serum uric acid concentration ≥ 10 mg/dL was 27% (66/249) in the febuxostat 40 mg once daily group, 49% (125/254) in the febuxostat 80 mg once daily group, and 31% (72/230) in the allopurinol 300 mg/200 mg once daily group.
Clinical outcomes: percentage of patients requiring treatment for gout flares
APEX study: During the 8-week prophylactic period, the proportion of patients requiring treatment for gout flares was 36% in the febuxostat 120 mg therapeutic group, compared to 28% in the febuxostat 80 mg group, 23% in the allopurinol 300 mg group, and 20% in the placebo group. The flare frequency was higher after the prophylactic period and gradually decreased over time. From weeks 8 to 28, 46% to 55% of patients required treatment for gout flares. Gout flares occurring during the last 4 weeks of the trial (weeks 24–28) were observed in 15% of patients (febuxostat 80 mg, 120 mg), 14% of patients (allopurinol 300 mg), and 20% of patients (placebo).
FACT study: During the 8-week prophylactic period, the proportion of patients requiring treatment for gout flares was 36% in the febuxostat 120 mg therapeutic group, compared to 22% in the febuxostat 80 mg group and 21% in the allopurinol 300 mg group. During the 8-week prophylactic period, flare frequency increased and then gradually decreased over time (64% and 70% of patients received treatment for gout flares from weeks 8 to 52). Gout flares during the last 4 weeks of the trial (weeks 49–52) were observed in 6–8% of patients (febuxostat 80 mg, 120 mg) and 11% of patients (allopurinol 300 mg).
The proportion of patients requiring treatment for gout flares (APEX and FACT studies) was lower in groups where the mean serum uric acid concentration after treatment decreased to < 6.0 mg/dL, < 5.0 mg/dL, or < 4.0 mg/dL compared to groups where the mean uric acid level was ≥ 6.0 mg/dL during the last 32 weeks of treatment (from weeks 20–24 to weeks 49–52).
During the CONFIRMS study, the proportion of patients requiring treatment for gout flares (1 day every 6 months) was 31% and 25% in the febuxostat 80 mg and allopurinol groups, respectively. No differences were observed in the proportion of patients requiring treatment for gout flares between the febuxostat 80 mg and 40 mg groups.
Long-term extension open-label studies. EXCEL study (C02-021): EXCEL was a 3-year, open-label, multicenter, randomized, extension, allopurinol-controlled phase III safety study conducted to evaluate safety in patients who had completed the main phase III studies (APEX or FACT). A total of 1086 patients were included in the study, receiving: febuxostat 80 mg once daily (n = 649), febuxostat 120 mg once daily (n = 292), or allopurinol 300/100 mg once daily (n = 145). Approximately 69% of patients did not require dose adjustment to achieve final stable treatment. Patients whose serum uric acid concentration exceeded 6.0 mg/dL in three consecutive measurements were excluded from the study.
Serum uric acid levels remained stable over time (e.g., 91% and 93% of patients initially receiving febuxostat at 80 mg and 120 mg doses, respectively, had serum uric acid levels below 6.0 mg/dL at month 36).
Based on 3-year follow-up data, in less than 4% of patients requiring treatment for flares, a reduction in gout flare frequency was observed at 16–24 months and 30–36 months (i.e., more than 96% of patients did not require flare treatment).
Complete disappearance of the primary palpable tophus from baseline to the last visit was observed in 46% and 38% of patients receiving final stable treatment with febuxostat at 80 mg and 120 mg once daily, respectively.
The FOCUS study (TMX-01-005) was a 5-year, open-label, multicenter, phase II extension safety study conducted in patients who completed a 4-week double-blind febuxostat dosing period in the TMX-00-004 trial. The study included 116 patients who initially received febuxostat 80 mg once daily. In 62% of patients, dose adjustment was not required to maintain serum uric acid concentration below 6.0 mg/dL, while 38% of patients required dose adjustment to achieve final stable concentration.
The proportion of patients with serum uric acid concentration below 6.0 mg/dL (357 µmol/L) at the last visit was over 80% (81–100%) in each febuxostat dose group.
In phase III clinical trials, minor changes in liver parameters were observed in patients receiving febuxostat (5.0%). The frequency of these changes was similar to that with allopurinol (4.2%) (see section "Special precautions"). In long-term open-label extension studies, elevated TSH [thyrotropin] levels (> 5.5 µIU/mL) were observed in patients receiving febuxostat (5.5%) or allopurinol (5.8%) over a prolonged period (see section "Special precautions").
Post-marketing long-term studies. The CARES study was a multicenter, randomized, double-blind, non-inferiority trial comparing cardiovascular outcomes with febuxostat and allopurinol in patients with gout and a history of major cardiovascular diseases, including myocardial infarction, hospitalization for unstable angina, coronary or cerebral revascularization procedures, stroke, hospitalization for transient ischemic attack, peripheral vascular disease, or diabetes with microangiopathy or macroangiopathy. To achieve serum uric acid (sUA) levels below 6 mg/dL, febuxostat dose was titrated from 40 mg to 80 mg (regardless of renal function), and allopurinol dose was titrated in 100 mg increments from 300 to 600 mg in patients with normal renal function and mild renal impairment, and from 200 to 400 mg in patients with moderate renal impairment.
The primary endpoint in the CARES study was time to first occurrence of major adverse cardiovascular events (MACE), a composite of non-fatal myocardial infarction, non-fatal stroke, cardiovascular death, and unstable angina requiring urgent coronary revascularization.
Endpoints (primary and secondary) were evaluated using intention-to-treat (ITT) analysis, including all subjects who were randomized and received at least one dose of the study drug during the double-blind trial.
Overall, 56.6% of patients discontinued the trial treatment prematurely, and 45% of patients did not complete all study visits.
A total of 6190 patients were followed for 32 months; the mean duration of exposure was 728 days in the febuxostat group (n = 3098) and 719 days in the allopurinol group (n = 3092).
The primary endpoint occurred at similar rates in the febuxostat and allopurinol treatment groups (10.8% vs. 10.4% of patients, respectively; hazard ratio [HR] 1.03; two-sided repeated 95% confidence interval [CI] 0.89–1.21).
In the analysis of individual MACE components, the rate of cardiovascular mortality was higher in the febuxostat group than in the allopurinol group (4.3% vs. 3.2% of patients; HR 1.34; 95% CI 1.03–1.73). The rates of other MACE were similar in the febuxostat and allopurinol groups: non-fatal myocardial infarction (3.6% vs. 3.8% of patients; HR 0.93; 95% CI 0.72–1.21), non-fatal stroke (2.3% vs. 2.3% of patients; HR 1.01; 95% CI 0.73–1.41), and urgent revascularization due to unstable angina (1.6% vs. 1.8% of patients; HR 0.86; 95% CI 0.59–1.26).
The all-cause mortality rate was also higher in the febuxostat group than in the allopurinol group (7.8% vs. 6.4% of patients; HR 1.22; 95% CI 1.01–1.47), primarily due to the higher rate of cardiovascular mortality in this group (see section "Special precautions").
Hospitalization rates for heart failure, hospitalization for non-ischemic arrhythmia, venous thromboembolic events, and hospitalization for transient ischemic attacks were comparable between the febuxostat and allopurinol groups.
The FAST study was a prospective, randomized, open-label, endpoint-blinded trial comparing the safety profile of febuxostat and allopurinol in cardiovascular diseases in patients with chronic hyperuricemia (in conditions where urate deposition has already occurred) and cardiovascular risk factors (i.e., patients aged 60 years or older with at least one cardiovascular risk factor). Eligible patients received allopurinol treatment prior to randomization, and dose adjustment was required as needed based on clinical assessment, EULAR [European League Against Rheumatism] recommendations, and approved dosing. At the end of the initial allopurinol treatment phase, patients with sUA levels < 0.36 mmol/L (<6 mg/dL) receiving the maximum tolerated or maximum allowed allopurinol dose were randomized in a 1:1 ratio to receive febuxostat or allopurinol. The primary endpoint of the FAST study was time to first occurrence of any event in the composite endpoint, which included: 1) hospitalization for non-fatal myocardial infarction (MI) / positive biomarker acute coronary syndrome (ACS); 2) non-fatal stroke; 3) death due to cardiovascular disorders. The primary analysis was based on on-treatment (OT) analysis.
A total of 6128 patients were randomized: 3063 receiving febuxostat and 3065 receiving allopurinol.
In the primary OT analysis, febuxostat was non-inferior to allopurinol regarding the frequency of the primary endpoint, which occurred in 172 patients (1.72/100 patient-years) receiving febuxostat compared to 241 patients (2.05/100 patient-years) receiving allopurinol, with an adjusted HR of 0.85 (95% CI: 0.70, 1.03), p < 0.001. The OT analysis for the primary endpoint in the subgroup of patients with a history of MI, stroke, or ACS showed no significant difference between treatment groups: 65 (9.5%) patients experienced adverse events in the febuxostat group and 83 (11.8%) patients in the allopurinol group; adjusted HR 1.02 (95% CI: 0.74–1.42); p = 0.202.
Treatment with febuxostat was not associated with increased cardiovascular mortality or all-cause mortality overall and in the subgroup of patients with a history of MI, stroke, or ACS. Overall, fewer deaths occurred in the febuxostat group (62 cases of cardiovascular death and 108 cases of all-cause mortality) compared to the allopurinol group (82 cases of cardiovascular death and 174 cases of all-cause mortality).
Greater reduction in uric acid levels was observed with febuxostat treatment compared to allopurinol treatment.
Tumor lysis syndrome (TLS). The efficacy and safety of febuxostat for the prevention and treatment of TLS were evaluated in the FLORENCE (FLO-01) study. Febuxostat demonstrated superior and faster action in reducing urate levels compared to allopurinol.
FLORENCE was a randomized (1:1), double-blind, active-controlled phase III trial comparing febuxostat 120 mg once daily with allopurinol 200–600 mg daily (mean daily allopurinol dose ± standard deviation: 349.7 ± 112.90 mg) under controlled serum uric acid concentration conditions. Eligible patients were candidates for allopurinol treatment or had no access to rasburicase. Primary endpoints were the area under the serum uric acid concentration-time curve (AUC sUA1–8) and change in serum creatinine (sCr) from day 1 to day 8.
A total of 346 patients with hematologic malignancies receiving chemotherapy and at intermediate/high risk of developing TLS were included in the study. The mean AUC sUA1–8 (mg×h/dL) was significantly lower with febuxostat (514.0 ± 225.71 vs. 708.0 ± 234.42; least squares mean difference: –196.794 [95% CI: –238.600; –154.988]; p < 0.0001). Additionally, the mean serum uric acid level was significantly lower with febuxostat starting from the first 24 hours of treatment and at any subsequent time point. No statistically significant differences were observed in mean serum creatinine (%) between febuxostat and allopurinol (–0.83 ± 26.98 vs. –4.92 ± 16.70, respectively; least squares mean difference: 4.0970 [95% CI: –0.6467; 8.8406]; p = 0.0903). Regarding secondary endpoints, no statistically significant differences were observed in the incidence of laboratory-confirmed TLS (8.1% and 9.2% for febuxostat and allopurinol, respectively; HR: 0.875 [95% CI: 0.4408; 1.7369]; p = 0.8488) and clinical tumor lysis syndrome (1.7% and 1.2% for febuxostat and allopurinol, respectively; HR: 0.994 [95% CI: 0.9691; 1.0199]; p = 1.0000). The frequency of all treatment-emergent adverse events and adverse reactions was 67.6% vs. 64.7% and 6.4% vs. 6.4% for febuxostat and allopurinol, respectively. In the FLORENCE study, febuxostat demonstrated superior and faster action in reducing serum uric acid levels compared to allopurinol. Data comparing febuxostat with rasburicase are currently lacking. The efficacy and safety of febuxostat have not been established in patients with acute severe TLS, such as patients in whom other urate-lowering therapies are ineffective.
Pharmacokinetics.
In healthy volunteers, maximum plasma concentration (Cmax) and area under the curve (AUC) increased proportionally with dose after single and multiple doses of febuxostat ranging from 10 mg to 120 mg. At doses from 120 mg to 300 mg, the increase in AUC was greater than proportional to dose. With repeated administration of 10–240 mg every 24 hours, accumulation of febuxostat was not observed. The predicted mean terminal elimination half-life (t1/2) of febuxostat was approximately 5–8 hours. A population pharmacokinetic/pharmacodynamic analysis was conducted based on data from 211 patients with hyperuricemia and gout receiving febuxostat at doses of 40–240 mg once daily. Overall, the obtained pharmacokinetic parameter values were similar to those in healthy volunteers, providing a good model for evaluating the pharmacokinetics/pharmacodynamics of the drug in patients with gout.
Absorption. Febuxostat is rapidly (tmax [time to maximum concentration] 1.0–1.5 hours) and well absorbed (at least 84%). After single and multiple oral doses of 80 mg or 120 mg once daily, Cmax was 2.8–3.2 µg/mL and 5.0–5.3 µg/mL, respectively. The absolute bioavailability of febuxostat tablets was not analyzed. With repeated administration of 80 mg once daily or single administration of 120 mg with a high-fat meal, Cmax decreased by 49% and 38%, and AUC decreased by 18% and 16%, respectively. However, this was not associated with clinically significant changes in the degree of plasma uric acid reduction (with repeated administration of 80 mg). Therefore, febuxostat can be administered regardless of food intake.
Distribution. The predicted steady-state volume of distribution (Vss/F) of febuxostat ranges from 29 to 75 L after oral administration of 10–300 mg. The extent of plasma protein binding (primarily to albumin) is 99.2% and does not change with increasing dose from 80 mg to 120 mg. The extent of plasma protein binding for active metabolites of febuxostat ranges from 82% to 91%.
Metabolism. Febuxostat is extensively metabolized via conjugation by uridine diphosphate glucuronosyltransferases (UDP-glucuronosyltransferases) and oxidation by cytochrome P450 (CYP) enzymes. Four pharmacologically active hydroxyl metabolites of febuxostat have been described, three of which were detected in human plasma. In vitro studies using human liver microsomes showed that these oxidized metabolites are formed primarily by CYP1A1, CYP1A2, CYP2C8, and CYP2C9, while febuxostat glucuronide is formed mainly by UDP-glucuronosyltransferase 1A1, 1A8, and 1A9.
Excretion. Febuxostat is eliminated via the liver and kidneys. After oral administration of 14C-febuxostat at a dose of 80 mg, approximately 49% was excreted in urine as unchanged febuxostat (3%), active moiety acylglucuronide (30%), known oxidized metabolites and their conjugates (13%), and other unknown metabolites (3%). In addition to renal excretion, approximately 45% was excreted in feces as unchanged febuxostat (12%), active moiety acylglucuronide (1%), known oxidized metabolites and their conjugates (25%), and other unknown metabolites (7%).
Renal impairment. With repeated administration of febuxostat at a dose of 80 mg, no changes in Cmax of febuxostat were observed in patients with mild, moderate, or severe renal impairment compared to patients with normal renal function. The mean total AUC of febuxostat increased approximately 1.8-fold from 7.5 µg×h/mL in patients with normal renal function to 13.2 µg×h/mL in patients with severe renal impairment. Cmax and AUC of active metabolites increased 2-fold and 4-fold, respectively. However, dose adjustment of the drug is not required in patients with mild or moderate renal impairment.
Hepatic impairment. With repeated administration of febuxostat at a dose of 80 mg, no significant changes in Cmax and AUC of febuxostat and its metabolites were observed in patients with mild (Child-Pugh class A) and moderate (Child-Pugh class B) hepatic impairment compared to patients with normal liver function. The drug has not been studied in patients with severe hepatic impairment (Child-Pugh class C).
Age. With repeated oral administration of febuxostat, no significant changes in AUC of febuxostat and its metabolites were observed in elderly patients compared to young healthy volunteers.
Gender. With repeated oral administration of febuxostat, Cmax and AUC of febuxostat in women were 24% and 12% higher, respectively, than in men. However, Cmax and AUC adjusted for body weight were similar in both groups; therefore, dose adjustment of febuxostat based on gender is not required.
Clinical characteristics.
Indications.
Lyquestia® 80 mg and Lyquestia® 120 mg
Treatment of chronic hyperuricemia associated with diseases characterized by urate crystal deposition, including presence of tophi and/or current or past history of gouty arthritis.
Lyquestia® 120 mg
Treatment and prevention of hyperuricemia in adult patients undergoing chemotherapy for hematologic malignancies with moderate or high risk of tumor lysis syndrome (TLS).
Lyquestia® is indicated for adult patients.
Contraindications.
Hypersensitivity to the active substance or to any of the excipients listed in the section "Composition".
Interaction with other medicinal products and other forms of interaction.
Mercaptopurine/azathioprine.
Due to its mechanism of action, febuxostat inhibits xanthine oxidase; therefore, concomitant use is not recommended. Inhibition of xanthine oxidase may increase plasma concentrations of both drugs, potentially causing myelotoxic reactions.
If co-administration with febuxostat is necessary, the dose of mercaptopurine/azathioprine should be reduced to 20% or less of the previously prescribed dose (see section "Special precautions for use").
The adequacy of this proposed dose adjustment, based on modeling and simulation analysis of preclinical data in rats, was confirmed by results of a clinical drug interaction study in healthy volunteers who received 100 mg azathioprine alone and a reduced dose of azathioprine (25 mg) in combination with febuxostat (40 or 120 mg).
There are no data on the safety of febuxostat use during cytotoxic chemotherapy.
Studies on the interaction of febuxostat with other cytotoxic chemotherapeutic agents have not been conducted.
Rosiglitazone / CYP2C8 substrates.
Febuxostat is a weak inhibitor of CYP2C8 in vitro. In a clinical study, concomitant administration of 120 mg febuxostat once daily and a single 4 mg oral dose of rosiglitazone in healthy volunteers did not affect the pharmacokinetics of rosiglitazone or its metabolite N-desmethylrosiglitazone, demonstrating that febuxostat does not inhibit the CYP2C8 enzyme in vivo. Therefore, concomitant administration of febuxostat with rosiglitazone or other CYP2C8 substrates does not require dose adjustment of these drugs.
Theophylline. A drug interaction study in healthy volunteers was conducted to evaluate the potential of febuxostat to increase circulating theophylline levels due to xanthine oxidase inhibition, as observed with other xanthine oxidase inhibitors. Concomitant administration of febuxostat 80 mg and theophylline 400 mg did not result in any pharmacokinetic interactions or safety concerns with theophylline. Thus, febuxostat 80 mg can be administered concomitantly with theophylline without special precautions. Data for febuxostat 120 mg are not available.
Naproxen and other inhibitors of glucuronidation.
Febuxostat metabolism depends on the activity of the enzyme UDP-glucuronosyltransferase. Medicinal products that inhibit glucuronidation, such as nonsteroidal anti-inflammatory drugs (NSAIDs) and probenecid, may theoretically alter febuxostat elimination. In healthy volunteers, concomitant administration of febuxostat and naproxen 250 mg twice daily resulted in enhanced febuxostat exposure (Cmax increased by 28%, AUC by 41%, and t1/2 [half-life] by 26%). In clinical trials, the use of naproxen and other NSAIDs/COX-2 inhibitors was not associated with clinically significant increases in adverse reactions.
Febuxostat can be co-administered with naproxen without dose adjustment of either drug.
Inducers of glucuronidation.
Potent inducers of UDP-glucuronosyltransferase may enhance febuxostat metabolism and reduce its efficacy. In patients receiving potent glucuronidation inducers, plasma uric acid levels should be monitored 1–2 weeks after initiation of concomitant therapy. Upon discontinuation of the glucuronidation inducer, plasma levels of febuxostat may increase.
Colchicine/indomethacin/hydrochlorothiazide/warfarin.
Febuxostat can be administered concomitantly with colchicine or indomethacin without dose adjustment of either drug.
No dose adjustment of febuxostat is required when administered concomitantly with hydrochlorothiazide.
Concomitant administration of febuxostat with warfarin does not require warfarin dose adjustment. Administration of febuxostat (80 mg or 120 mg once daily) with warfarin in healthy volunteers did not affect warfarin pharmacokinetics. Concomitant use with febuxostat also had no effect on INR (International Normalized Ratio) or factor VII activity.
Desipramine / CYP2D6 substrates.
In vitro data indicate that febuxostat is a weak inhibitor of CYP2D6. In studies involving healthy volunteers receiving 120 mg febuxostat once daily, an increase in AUC of desipramine (a CYP2D6 substrate) by 22% was observed, indicating weak inhibitory effect of febuxostat on CYP2D6 in vivo.
Therefore, when febuxostat is used concomitantly with CYP2D6 substrates, no dose adjustment is required.
Antacids.
Concomitant administration with antacids containing magnesium hydroxide and aluminum hydroxide results in delayed absorption of febuxostat (by approximately 1 hour) and a 32% reduction in Cmax; however, the AUC of febuxostat is not significantly altered. Therefore, febuxostat may be co-administered with antacids.
Special precautions for use.
Cardiovascular diseases.
In patients with serious cardiovascular diseases (e.g., myocardial infarction, stroke, or unstable angina) a higher number of life-threatening cardiovascular events were observed during drug development and in one post-marketing study (CARES) when febuxostat was used compared to allopurinol.
However, a subsequent post-marketing study (FAST) showed that febuxostat was non-inferior to allopurinol regarding the incidence of cardiovascular events, both life-threatening and non-life-threatening.
Treatment of this patient group should be carried out with caution and their condition should be monitored regularly. For more detailed information on the safety of febuxostat use with respect to the cardiovascular system, see sections "Pharmacodynamics" and "Adverse reactions".
Prevention and treatment of hyperuricaemia in patients at risk of tumour lysis syndrome (TLS).
Patients undergoing chemotherapy for haematological malignancies with moderate or high risk of TLS who are receiving the medicinal product Lekvestia® should, if clinically indicated, be under cardiologist supervision.
Allergy/hypersensitivity to medicinal products.
Rare cases of serious allergic reactions/hypersensitivity reactions, including life-threatening cases of Stevens-Johnson syndrome, toxic epidermal necrolysis, and acute anaphylactic reactions/shock, have been reported during post-marketing surveillance. These reactions mostly occurred within the first month of febuxostat treatment. Renal function impairment and/or history of allopurinol hypersensitivity were present in several, but not all, patients. Severe hypersensitivity reactions, including drug reaction with eosinophilia and systemic symptoms (DRESS syndrome), in some cases were associated with fever, haematological disorders, and renal or hepatic insufficiency.
Patients should be informed about signs and symptoms of hypersensitivity/allergy and should be monitored for the development of such reactions. If serious allergic reactions/hypersensitivity reactions occur, including Stevens-Johnson syndrome, febuxostat must be discontinued immediately, as early discontinuation improves prognosis. Re-administration of febuxostat is contraindicated if the patient has previously experienced an allergic reaction/hypersensitivity reaction, including Stevens-Johnson syndrome, or acute anaphylactic reactions/shock.
Acute gout flare (attack).
Treatment with febuxostat should only be initiated after an acute flare has subsided. Febuxostat may provoke gout flares at the beginning of treatment due to changes in serum uric acid levels caused by mobilization of urates from tissue deposits. At the start of febuxostat treatment, it is recommended to co-administer NSAIDs or colchicine for at least 6 months to prevent gout flares.
If a gout flare occurs during febuxostat treatment, the treatment should be continued. Appropriate individual therapy for the acute gout flare should be administered concomitantly. With prolonged use of febuxostat, the frequency and severity of gout flares decrease.
Xanthine deposition.
In patients with accelerated urate production (e.g., due to malignancies and their treatment or in Lesch-Nyhan syndrome), a significant increase in absolute xanthine concentration in urine may occur, which in rare cases may lead to xanthine deposition in the urinary tract. This was not observed in the pivotal clinical study of febuxostat in TLS. Due to limited experience with febuxostat use, it is not recommended for patients with Lesch-Nyhan syndrome.
Azathioprine/mercaptopurine.
Febuxostat is not recommended for patients receiving azathioprine/mercaptopurine concurrently, as inhibition of xanthine oxidase by febuxostat may lead to increased plasma concentrations of mercaptopurine/azathioprine and result in severe toxicity.
If co-administration cannot be avoided, patients should be closely monitored. A dose reduction of mercaptopurine or azathioprine to 20% or less of the previously prescribed dose is recommended to avoid potential haematological effects (see section "Interaction with other medicinal products and other forms of interaction"). Patients should be closely monitored, and the dose of mercaptopurine/azathioprine should be adjusted subsequently based on therapeutic response and possible toxic effects.
Organ transplant recipients.
There is no experience with febuxostat use in this patient group; therefore, the use of the medicinal product is not recommended.
Theophylline.
Single-dose co-administration of febuxostat 80 mg and theophylline 400 mg showed no pharmacokinetic interactions. Febuxostat 80 mg may be administered concomitantly with theophylline without risk of increasing theophylline plasma concentrations. Data for febuxostat 120 mg are not available.
Hepatic disorders.
During the combined phase III clinical trials, minor changes in liver function parameters were observed in 5.0% of patients receiving febuxostat. Therefore, it is recommended to assess liver function tests before initiating febuxostat and during treatment as clinically indicated.
Thyroid disorders.
Elevated TSH levels (> 5.5 mU/mL) were observed in 5.5% of patients receiving long-term febuxostat treatment during long-term open-label extension studies. Therefore, the medicinal product should be prescribed with caution in patients with thyroid dysfunction.
Lactose.
The medicinal product contains lactose. If a patient has been diagnosed with carbohydrate intolerance, medical advice should be sought before taking this medicinal product.
Use during pregnancy or breastfeeding.
Pregnancy
Limited experience with febuxostat use during pregnancy suggests no adverse effects of the drug on pregnancy outcome or fetal/neonatal health. Animal studies have not shown any direct or indirect harmful effects on pregnancy, embryonic/foetal development, or parturition. The potential risk for humans is unknown. Febuxostat should not be used during pregnancy.
Breastfeeding period
It is unknown whether febuxostat passes into human breast milk. Animal studies have shown that febuxostat is excreted in milk and has a negative effect on the development of suckling newborns. The risk of drug transfer into breast milk cannot be excluded. Febuxostat should not be used during breastfeeding.
Fertility
Fertility studies in animals at a dose of 48 mg/kg/day did not reveal dose-dependent adverse effects. The effect of the medicinal product Lekvestia® on human reproductive function is unknown.
Ability to influence the speed of reactions when driving or operating machinery.
Cases of somnolence, dizziness, paraesthesia, and visual blurring have been reported during febuxostat treatment. Therefore, patients taking the medicinal product Lekvestia® are advised to exercise caution when driving vehicles or operating machinery until they are certain that the drug does not cause the aforementioned adverse effects.
Method of Administration and Dosage
Dosage
Gout. The recommended dose of the medicinal product Lyquexity® is 80 mg once daily orally, regardless of food intake. If serum uric acid concentration exceeds 6 mg/dL (357 µmol/L) after 2–4 weeks of treatment, increasing the dose of Lyquexity® to 120 mg once daily should be considered. The effect of the drug manifests rapidly, allowing serum uric acid levels to be re-assessed after 2 weeks. The goal of treatment is to reduce and maintain serum uric acid concentration below 6 mg/dL (357 µmol/L).
The duration of gout attack prophylaxis should be at least 6 months.
Tumor Lysis Syndrome (TLS). The recommended dose of the medicinal product Lyquexity® is 120 mg once daily orally, regardless of food intake.
Administration of Lyquexity® should be initiated two days prior to the start of cytotoxic therapy and continued for at least 7 days; however, the duration of treatment may be extended up to 9 days depending on the duration of chemotherapy and clinical assessment.
Elderly patients. Dose adjustment is not required for this patient population.
Renal impairment. The efficacy and safety of the medicinal product have not been sufficiently studied in patients with severe renal impairment (creatinine clearance < 30 mL/min). Dose adjustment is not required in patients with mild or moderate renal impairment.
Hepatic impairment. The efficacy and safety of febuxostat have not been studied in patients with severe hepatic impairment (Child-Pugh class C).
Gout. In patients with mild hepatic impairment, the recommended dose is 80 mg. Experience with the medicinal product in patients with moderate hepatic impairment is limited.
Tumor Lysis Syndrome (TLS). In the pivotal phase III study (FLORENCE), only subjects with severe hepatic impairment were excluded. For patients included in the study, dose adjustment based on hepatic function was not required.
Method of Administration
For oral use.
Lyquexity® is administered orally, independent of food intake.
Children.
The safety and efficacy of Lyquexity® in children (under 18 years of age) have not been established. Data on use are lacking.
Overdose.
In case of overdose, symptomatic and supportive therapy is indicated.
Adverse Reactions
Summary of safety profile
The most common adverse reactions observed in clinical trials (4072 patients receiving doses from 10 to 300 mg), post-marketing safety studies (FAST study: 3001 patients receiving doses from 80 to 120 mg), and during post-marketing surveillance in patients with gout were gout flares (attacks), hepatic function abnormalities, diarrhea, nausea, headache, dizziness, dyspnea, rash, pruritus, arthralgia, myalgia, limb pain, edema, and fatigue. These reactions were mostly mild to moderate in severity. During post-marketing surveillance, rare cases of serious hypersensitivity reactions to febuxostat have been reported, some of which were accompanied by systemic reactions, as well as rare cases of sudden cardiac death.
Table 2 lists the adverse reactions observed in patients treated with febuxostat. The reactions are classified by frequency as follows: common (≥ 1/100 to < 1/10), uncommon (≥ 1/1000 to < 1/100), and rare (≥ 1/10000 to < 1/1000). Frequency is based on data from clinical trials and post-marketing experience in patients with gout.
Within each frequency category, adverse reactions are listed in order of decreasing severity.
Table 2.
Adverse reactions observed during Phase III combined long-term extension studies, post-marketing safety studies, and post-marketing surveillance in patients with gout.
| Blood and lymphatic system disorders |
Rare Pancytopenia, thrombocytopenia, agranulocytosis*, anemia# |
| Immune system disorders |
Rare Anaphylactic reactions*, hypersensitivity to the drug* |
| Endocrine disorders |
Uncommon Increased blood thyroid-stimulating hormone levels, hypothyroidism# |
| Eye disorders |
Uncommon Blurred vision Rare Retinal artery occlusion# |
| Nutritional and metabolism disorders |
Common*** Exacerbation (attacks) of gout Uncommon Diabetes mellitus, hyperlipidemia, decreased appetite, weight gain Rare Weight loss, increased appetite, anorexia |
| Psychiatric disorders |
Uncommon Decreased libido, insomnia Rare Nervousness, depressed mood#, sleep disorders# |
| Nervous system and sensory disorders |
Common Headache, dizziness Uncommon Paresthesia, hemiparesis, somnolence, lethargy#, altered taste sensation, hypoaesthesia, reduced sense of smell Rare Ageusia#, burning sensation# |
| Ear and labyrinth disorders |
Uncommon Tinnitus Rare Vertigo# |
| Cardiac disorders |
Uncommon Atrial fibrillation, palpitations, ECG abnormalities, left bundle branch block (see section "Adverse Reactions. Tumor Lysis Syndrome"), sinus tachycardia (see section "Adverse Reactions. Tumor Lysis Syndrome"), arrhythmia# Rare Sudden cardiac death* |
| Vascular disorders |
Uncommon Arterial hypertension, flushing, hot flushes, hemorrhage (see section "Adverse Reactions. Tumor Lysis Syndrome") Rare Circulatory collapse# |
| Respiratory system disorders |
Common Dyspnea Uncommon Bronchitis, upper respiratory tract infections, lower respiratory tract infections#, cough, rhinorrhea# Rare Pneumonia# |
| Gastrointestinal disorders |
Common Diarrhea**, nausea Uncommon Abdominal pain, upper abdominal pain#, bloating, gastroesophageal reflux disease, vomiting, dry mouth, dyspepsia, constipation, frequent defecation, flatulence, discomfort in stomach or intestine, oral ulceration, lip swelling#, pancreatitis Rare Gastrointestinal perforation#, stomatitis# |
| Hepatobiliary disorders |
Common Liver function abnormalities** Uncommon Cholelithiasis Rare Hepatitis, jaundice*, liver damage*, cholecystitis# |
| Skin and subcutaneous tissue disorders |
Common Rash (including rashes with lower frequency of occurrence, see below), pruritus Uncommon Dermatitis, urticaria, skin discoloration, skin damage, petechiae, maculopapular rash, papular rash, hyperhidrosis, alopecia, eczema#, erythema, night sweats#, psoriasis#, itchy rash# Rare Toxic epidermal necrolysis*, Stevens-Johnson syndrome*, angioedema*, drug reaction with eosinophilia and systemic symptoms (DRESS)*, generalized rash (serious)*, exfoliative rash, follicular rash, vesicular rash, pustular rash, erythematous rash, measles-like rash |
| Musculoskeletal and connective tissue disorders |
Common Arthralgia, myalgia, limb pain# Uncommon Arthritis, musculoskeletal pain, muscle weakness, muscle cramps, muscle stiffness, joint stiffness#, bursitis, joint swelling#, back pain#, musculoskeletal stiffness# Rare Rhabdomyolysis*, rotator cuff syndrome#, polymyalgia rheumatica# |
| Renal and urinary disorders |
Uncommon Renal failure, urolithiasis, hematuria, polyuria, proteinuria, urgency, urinary tract infections# Rare Tubulointerstitial nephritis* |
| Reproductive system and breast disorders |
Uncommon Erectile dysfunction |
| General disorders and administration site conditions |
Common Edema, fatigue Uncommon Chest pain, feeling of chest discomfort, pain#, weakness# Rare Thirst, feeling of heat# |
| Investigations |
Uncommon Elevated blood amylase levels, decreased platelet count, decreased white blood cell count, decreased lymphocyte count in blood, elevated creatine levels in blood, elevated creatinine levels in blood, decreased hemoglobin levels, elevated blood urea levels, elevated triglyceride levels in blood, elevated cholesterol levels in blood, decreased hematocrit, elevated lactate dehydrogenase (LDH) levels in blood, elevated potassium levels in blood, elevated MCH# Rare Elevated blood glucose levels, prolonged activated partial thromboplastin time, decreased red blood cell count, elevated alkaline phosphatase levels in blood, elevated creatine phosphokinase levels in blood* |
| Injury, poisoning and procedural complications |
Uncommon Contusion |
* Adverse reactions identified during post-marketing surveillance.
** Diarrhea requiring treatment and abnormal liver function tests observed in Phase III studies occurred more frequently in patients receiving concomitant colchicine therapy.
*** See section "Pharmacodynamics" for the frequency of gout flares observed in Phase III of individual randomized controlled trials.
Adverse reactions observed during post-authorization safety studies.
Description of selected adverse reactions.
During post-marketing surveillance, rare cases of serious hypersensitivity reactions to febuxostat have been reported, including Stevens–Johnson syndrome, toxic epidermal necrolysis, and anaphylactic reactions/shock. Stevens–Johnson syndrome and toxic epidermal necrolysis are characterized by progressive skin rash with bullous lesions of the skin or mucous membranes and mucosal irritation of the eyes. Hypersensitivity reactions to febuxostat may present with symptoms such as skin reactions characterized by infiltrated maculopapular rashes, generalized or exfoliative rashes, skin lesions, facial swelling, fever, hematological disorders such as thrombocytopenia and eosinophilia, and involvement of single or multiple organs (liver and kidneys, including tubulointerstitial nephritis).
Gout flares were commonly observed shortly after initiation of treatment and during the first months of therapy. The frequency of gout flares decreased over time. Prophylaxis for acute gout flares is recommended when initiating febuxostat therapy.
Tumor lysis syndrome (TLS).
Summary of safety profile.
In a randomized, double-blind, active-controlled Phase III study FLORENCE (FLO-01), comparing febuxostat and allopurinol in 346 chemotherapy-treated patients with hematologic malignancies at moderate or high risk of TLS, only 22 (6.4%) patients experienced adverse reactions, with 11 (6.4%) in each treatment group. The majority of adverse reactions were of mild or moderate severity.
Overall, during the FLORENCE study, no additional safety concerns were identified for febuxostat use in patients with gout, except for the three adverse reactions listed below (see Table 2).
Cardiac disorders: uncommon — left bundle branch block, sinus tachycardia.
Vascular disorders: uncommon — hemorrhage.
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after medicine authorization is important. It allows continuous monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals and patients, as well as their legal representatives, are encouraged to report any suspected adverse reactions and lack of efficacy via the Automated Pharmacovigilance Information System at: https://aisf.dec.gov.ua.
Shelf life. 3 years.
Do not use the medicinal product after the expiry date stated on the packaging.
Storage conditions. This medicinal product does not require special storage conditions.
Keep out of reach of children.
Packaging. 14 tablets in a blister. 2 or 4 blisters per carton.
Prescription status. Prescription only.
Manufacturer. JSC "Farmak" (packaging of bulk product manufactured by the company Genepharm S.A., Greece).
Manufacturer's address.
74, Kyrylivska Street, Kyiv, 04080, Ukraine.