Izifri®
Ukraina
Spis treści
INSTRUKCJA dotyczÄ cza stosowania leku Izifri® (IziFri)
SkÅ ad:
substancja czynna: bromek tiotropium;
1 blister zawiera: tiotropium bromku monohydrat – 0,016 mg (co odpowiada tiotropium – 0,013 mg); po 10 µg/dawka, po 1 dawce w blisterze;
substancja pomocnicza: laktoza, monohydrat.
PostaÄ c leku. Proszek do inhalacji, dawkowany.
GÅ owne fizykochemiczne wÅ aÅ ciwoÅ ci: biaÅ e plastikowe urzÄ dzenie zawierajÄ ce 30 oddzielnych aluminiowych blisterów, wypeÅ nionych biaÅ ym proszkiem. UrzÄ dzenie umieszczone w aluminiowym opakowaniu zawierajÄ cym Å rodki osuszajÄ ce.
Grupa farmakoterapeutyczna. Å rodki do leczenia chorób obturacyjnych dróg oddechowych, Å rodki do inhalacji. Å rodki antycholinergiczne.
Kod ATC R03B B04.
Właściwości farmakologiczne.
Farmakodynamika.
Mechanizm działania. Tiotropium jest specyficznym, długodziałającym lekiem przeciwblokującym receptory cholinergiczne. Tiotropium wykazuje podobne powinowactwo do wszystkich podtypów receptorów muskarynowych (od M1 do M5). W drogach oddechowych inhibicja receptorów M3 prowadzi do rozluźnienia mięśni gładkich. W badaniach przedklinicznych in vitro i in vivo efekt bronchoprotekcyjny był zależny od dawki i trwał ponad 24 godziny.
Długość działania wynika z bardzo powolnego uwolnienia z receptorów M3; okres półtrwania tiotropium jest znacznie dłuższy niż ipratropium. Jako N-czworczynowy przeciwblokownik cholinergiczny, tiotropium wykazuje działanie miejscowe (broncho-) selektywne przy inhalacyjnym stosowaniu i wykazuje odpowiedni zakres działania terapeutycznego przed pojawieniem się systemowych efektów antycholinergicznych. Dysocjacja z receptorów M2 jest szybsza niż z M3 w funkcjonalnych badaniach in vitro. Receptor M3 jest bardziej odpowiedni (kinetycznie kontrolowany) pod względem selektywności podtypu niż M2. Wysoka aktywność i powolna dysocjacja z receptorów korelują klinicznie z istotnym i długotrwałym rozszerzeniem oskrzeli u pacjentów z przewlekłymi chorobami obturacyjnymi płuc (POChP).
Efekty farmakodynamiczne. Rozszerzenie oskrzeli po inhalacji tiotropium jest przede wszystkim efektem lokalnym na drogi oddechowe, a nie systemowym.
Po stosowaniu tiotropium raz na dobę obserwowano istotne poprawienie funkcji płuc (zwiększenie objętości przepływu wydechowego w pierwszej sekundzie (FEV1) i przewidywanej pojemności życiowej płuc) już w ciągu 30 minut po pierwszej dawce, a efekt ten trwał 24 godziny. Stan farmakodynamiczny stabilizuje się w ciągu tygodnia. U większości pacjentów rozszerzenie oskrzeli pojawia się trzeciego dnia.
Na podstawie codziennych pomiarów tiotropium istotnie poprawia maksymalną prędkość wydechu rano i wieczorem.
Poprawa funkcji płuc utrzymuje się bez objawów tolerancji.
Rozszerzenie oskrzeli utrzymuje się przez 24-godzinny interwał dawkowania w porównaniu z placebo. Nie miało znaczenia, czy tiotropium było stosowane rano czy wieczorem.
Tiotropium istotnie zmniejsza duszność; poprawa stanu utrzymywała się przez cały okres leczenia.
Tiotropium istotnie zmniejsza liczbę zaostrzeń POChP i wydłuża czas do pierwszego ciężkiego zaostrzenia.
Tiotropium istotnie poprawia jakość życia związaną ze zdrowiem; poprawa utrzymywała się przez cały okres leczenia.
Tiotropium istotnie skraca liczbę hospitalizacji pacjentów z zaostrzeniami POChP i opóźnia czas do pierwszej hospitalizacji.
W dwóch badaniach tiotropium istotnie poprawił tolerancję na obciążenie fizyczne ograniczone objawami choroby o 19,7% i 28,3%.
W badaniu zastosowania tiotropium w dawkach 18 µg i 54 µg (trzy razy po 18 µg) przez 12 dni nie wywołało wydłużenia interwału QT w badaniu elektrokardiograficznym.
W czteroletnim badaniu z udziałem 5993 pacjentów tiotropium utrzymywał poprawę wartości FEV1 przez cały okres, jednak nie wpływał na roczny wskaźnik spadku FEV1.
Podczas leczenia ryzyko śmiertelności zmniejszyło się o 16%. Ogólna częstość zgonów wynosiła 4,79 na 100 pacjentów-roku w grupie placebo w porównaniu do 4,10 na 100 pacjentów-roku w grupie tiotropium (stosunek ryzyka (tiotropium/placebo) 0,84, 95% CI – 0,73; 0,97). Leczenie tiotropium zmniejszyło ryzyko niewydolności oddechowej o 19% (2,09 w porównaniu z 1,68 przypadków na 100 pacjentów-roku, względne ryzyko (tiotropium/placebo) – 0,81, 95% CI – 0,65; 1,00).
Farmakokinetyka.
Tiotropium jest związkiem amonowym czwartorzędowym, który umiarkowanie rozpuszcza się w wodzie. Tiotropium stosuje się w postaci suchego proszku do inhalacji. Zazwyczaj przy inhalacyjnym sposobie stosowania większa część dawki osiąda w przewodzie pokarmowym, a mniejsza część w płucach.
Wchłanianie. Po inhalacji suchego proszku absolutna biodostępność wynosi 19,5%, co wskazuje na wysoką biodostępność frakcji docierającej do płuc. Absolutna biodostępność roztworu tiotropium do doustnego stosowania wynosi 2-3%. Maksymalna stężenie tiotropium w osoczu krwi obserwuje się 5-7 minut po inhalacji.
W stanie stacjonarnym maksymalne stężenie tiotropium w osoczu krwi u pacjentów z POChP wynosi 12,9 pg/ml i szybko maleje zgodnie z modelem wielokomorowym. Minimalne stężenie tiotropium w osoczu krwi w stanie stacjonarnym wynosi 1,71 pg/ml. Wpływ systemowy po inhalacji tiotropium w postaci proszku był podobny do wpływu po inhalacji tiotropium w postaci roztworu.
Rozkład. 72% leku wiąże się z białkami osocza krwi. Objętość rozkładu wynosi 32 l/kg. Lokalne stężenie w płucach jest nieznane, ale na podstawie sposobu stosowania zakłada się wysokie stężenie w płucach. W badaniach na zwierzętach wykazano, że tiotropium nie przenika w znaczącej ilości przez barierę krew-mózg.
Biota transformacja. Stopień biotransformacji jest niewielki, ponieważ 74% niezmienionej substancji wydalało się z moczem po wewnątrzżylnym podaniu zdrowym ochotnikom. Jako złożony ester tiotropium ulega niel enzymatycznemu rozkładowi do alkoholu N-metyloskopinu i kwasu ditienioglikolowego, które nie wiążą się z receptorami muskarynowymi.
W dalszych badaniach in vitro na mikrosomach wątrobowych i hepatocytach tiotropium (<20% dawki po wewnątrzżylnym podaniu) metabolizuje się poprzez zależne od cytochromu P450 utlenianie i dalszą koniugację z glutationem do różnych metabolitów fazy II. Ten szlak enzymatyczny może być hamowany przez inhibitory CYP450 2D6 (i 3A4), chinidynę, ketokonazol i gestoden. Wskazane CYP450 2D6 i 3A4 biorą udział w przemianach metabolicznych odpowiadających za wydalenie mniejszej części dawki. Tiotropium nawet w stężeniach supratherapeutycznych nie hamuje cytochromu P450 1A1, 1A2, 2B6, 2C9, 2C19, 2D6, 2E1 i 3A w mikrosomach wątrobowych.
Eliminacja. Efektywny okres półtrwania tiotropium wynosi od 27 do 45 godzin po podaniu pacjentom z POChP. Całkowity klirens wynosił 880 ml/min po wewnątrzżylnym podaniu młodym zdrowym ochotnikom. Po wewnątrzżylnym podaniu tiotropium wydalane jest głównie z moczem w niezmienionej formie. Po inhalacji suchym proszkiem wydalenie z moczem wynosi 7% (1,3 µg) niezmienionej ilości w ciągu 24 godzin, reszta nie jest wchłaniana przez jelita i wydala się z kałem. Klirens nerkowy tiotropium przekracza klirens kreatyniny, co wskazuje na wydalenie z moczem. Po stałym codziennym stosowaniu inhalacyjnym u pacjentów z POChP stan farmakokinetyczny stacjonarny osiągany był po 7 dniach bez dalszej kumulacji.
Linowość/nielinowość. Tiotropium wykazał liniowe właściwości farmakokinetyczne w zakresie terapeutycznym niezależnie od postaci leku.
Farmakokinetyka u pacjentów w podeszłym wieku. Jak w przypadku wszystkich innych leków, które głównie wydzielane są z moczem, przy stosowaniu tiotropium u pacjentów w podeszłym wieku obserwuje się obniżenie klirensu nerkowego (365 ml/min u pacjentów z POChP w wieku <65 lat w porównaniu do 271 ml/min u pacjentów z POChP w wieku ≥65 lat). Nie prowadzi to do odpowiedniego wzrostu wartości AUC0-6,ss ani Cmax,ss.
Farmakokinetyka u pacjentów z zaburzeniami funkcji nerek. Inhalacyjne stosowanie tiotropium raz na dobę u pacjentów z POChP w stanie równowagi z łagodną niewydolnością nerek (klirens kreatyniny 50-80 ml/min) prowadziło do nieznacznego wzrostu wartości AUC0-6,ss (o 1,8-30%) i uzyskania podobnych wartości Cmax w porównaniu z pacjentami z normalną funkcją nerek (klirens kreatyniny >80 ml/min).
U pacjentów z POChP ze średnim lub ciężkim zaburzeniem funkcji nerek (klirens kreatyniny <50 ml/min) wewnątrzżylne podanie tiotropium prowadzi do podwojenia całkowitego narażenia (wartość AUC0-4h wyższa o 82%, wartość Cmax wyższa o 52%) w porównaniu z pacjentami z normalną funkcją nerek, co potwierdzają dane dotyczące stężenia w osoczu krwi po inhalacji suchego proszku.
Farmakokinetyka u pacjentów z zaburzeniami funkcji wątroby. Niewydolność wątroby nie ma istotnego wpływu na farmakokinetykę tiotropium. Tiotropium wydala się głównie drogą nerkową (do 74% u młodych zdrowych ochotników) oraz poprzez proste niel enzymatyczne rozszczepienie estru do produktów, które nie wiążą się z receptorami muskarynowymi.
Pacjenci z POChP japońskiej narodowości. W trakcie badania krzyżowego średnie maksymalne stężenie tiotropium w osoczu krwi 10 minut po podaniu w stanie równowagi było o 20-70% wyższe u Japończyków w porównaniu z Europejczykami po inhalacji tiotropium, jednak nie stwierdzono zwiększonej śmiertelności ani ryzyka powikłań sercowych u pacjentów japońskich w porównaniu z Europejczykami. W odniesieniu do innych ras lub grup etnicznych brakuje wystarczających danych farmakokinetycznych.
Związek farmakokinetyka/farmakodynamika. Nie ma bezpośredniego związku między farmakokinetyką a farmakodynamiką.
Właściwości kliniczne.
Wskazania.
Wsparcie broncholityczne w celu złagodzenia objawów przewlekłej obturacyjnej choroby płuc (POChP).
Przeciwwskazania.
Lek jest przeciwwskazany u pacjentów ze znaną nadwrażliwością na bromek tiotropium, atropinę lub jej pochodne (na ipratropium lub oksytropium) albo na inne składniki leku.
Interakcje z innymi lekami i inne rodzaje interakcji.
Mimo że nie przeprowadzono formalnych badań interakcji z innymi lekami, bromek tiotropium stosowano łącznie z innymi lekami (sympatomyketykami rozszerzającymi oskrzela, metyloksantynami, sterydami doustnymi i inhalacyjnymi stosowanymi w leczeniu POChP) bez klinicznych dowodów interakcji z innymi lekami.
Nie stwierdzono, że stosowanie agonistów β-adrenoreceptorów o długim działaniu lub sterydów inhalacyjnych wpływa na ekspozycję na tiotropium.
Jednak nie badano stosowania Izifri® w połączeniu z innymi lekami antycholinergicznymi, dlatego nie zaleca się takiego połączenia.
Szczególne ostrzeżenia i środki ostrożności dotyczące stosowania.
Izifri® jest bronchodilatatorem, który należy stosować raz dziennie jako leczenie wspomagające, i nie należy go stosować do wstępnego leczenia ostrych napadów bronchospazmu.
Po zastosowaniu Izifri® możliwe są natychmiastowe reakcje nadwrażliwości.
Tak jak inne leki antycholinergiczne, Izifri® należy stosować z ostrożnością u pacjentów z jaskrą zamkniętostanową, przerostem prostaty lub zwężeniem szyjki pęcherza moczowego (patrz dział «Działania niepożądane»).
Leki do inhalacji mogą powodować indukowany inhalacją bronchospazm.
Lek należy stosować z ostrożnością u następujących pacjentów: u osób, które niedawno (w ciągu ostatnich 6 miesięcy) przeżyły zawał mięśnia sercowego; u osób z niestabilną lub zagrażającą życiu arytmią lub arytmią wymagającą interwencji lub zmiany terapii w ciągu ostatniego roku; u hospitalizowanych z niewydolnością serca (klasa NYHA III lub IV) w ciągu ostatniego roku. Pacjenci ci byli wykluczeni z badań klinicznych. W takich stanach działanie antycholinergiczne może im zaszkodzić.
Ponieważ stężenie tiotropiumu bromku we krwi wzrasta u pacjentów z niewydolnością nerek od umiarkowanej do ciężkiej (klirens kreatyniny ≤ 50 ml/min), Izifri® należy stosować tylko wtedy, gdy oczekiwana korzyść przewyższa potencjalne ryzyko. Brak danych dotyczących długotrwałego stosowania Izifri® u pacjentów z niewydolnością nerek (patrz dział «Farmakokinetyka»).
Pacjenci powinni unikać dostania się proszku do oczu. Może to prowadzić do wywołania lub nasilenia jaskry zamkniętostanowej, bólu lub dyskomfortu w oczach, tymczasowej nieostrości widzenia, uczucia pojawienia się aureoli lub kolorowych plam przed oczami w połączeniu z zaczerwienieniem oka w postaci hiperemii spojówek lub rogówki.
W przypadku wystąpienia wskazanych objawów w dowolnej kombinacji należy natychmiast przerwać stosowanie tiotropiumu bromku i bezzwłocznie skontaktować się z lekarzem.
Suchość w ustach, która występuje przy terapii antycholinergicznej, może w dalszej perspektywie wiązać się z próchnicą zębów.
Izifri® nie należy stosować więcej niż raz dziennie.
Izifri® zawiera laktozę. Jeśli u pacjenta stwierdzono nietolerancję niektórych cukrów, należy skonsultować się z lekarzem przed przyjęciem tego leku.
Stosowanie w czasie ciąży lub karmienia piersią.
Ciąża. Dane dotyczące stosowania tiotropiumu u ciężarnych kobiet są bardzo ograniczone. Badania przedkliniczne nie wykazały bezpośredniego lub pośredniego szkodliwego wpływu związanego z toksycznością rozrodczą w dawkach klinicznie istotnych. Jako środek ostrożności należy unikać stosowania Izifri® w czasie ciąży.
Karmienie piersią. Nie wiadomo, czy tiotropiumu bromek przenika do mleka matki. Pomimo wyników badań przeprowadzonych na gryzoniach, które wykazały przenikanie tiotropiumu bromku do mleka matki jedynie w niewielkiej ilości u gryzoni, stosowanie leku w czasie karmienia piersią nie jest zalecane.
Tiotropiumu bromek jest związkiem o długim czasie działania. Decyzję o kontynuowaniu/przerwaniu karmienia piersią lub kontynuowaniu/przerwaniu stosowania Izifri® należy podjąć po ocenie korzyści z karmienia piersią dla dziecka i korzyści z terapii dla kobiety.
Plodność. Brak dostępnych danych klinicznych dotyczących wpływu tiotropiumu na płodność. Wyniki badań przedklinicznych tiotropiumu nie wykazały niepożądanych skutków na płodność.
Wpływ na zdolność kierowania pojazdami i obsługiwania maszyn.
Nie przeprowadzono badań wpływu na zdolność kierowania pojazdami i obsługiwania maszyn. Pojawienie się zawrotów głowy, bólu głowy lub nieostrości widzenia może wpływać na zdolność kierowania pojazdami i obsługiwania maszyn.
Sposób stosowania i dawki
Lek jest przeznaczony wyłącznie do stosowania przez inhalację.
Zalecana dawka Izifri® to inhalacja zawartości jednej paski blisterowej 1 raz na dobę za pomocą inhalatora Elpenhaler®. Inhalację należy wykonywać o tej samej porze dnia.
Nie należy przekraczać zalecanej dawki.
Paski blisterowe są przeznaczone wyłącznie do inhalacji, a nie do doustnego stosowania. Pasków blisterowych Izifri® nie wolno połykać.
Lek należy stosować wyłącznie za pomocą inhalatora Elpenhaler®. Inhalator Elpenhaler® należy przechowywać w opakowaniu w celu ochrony przed wilgocią i wyjmować bezpośrednio przed pierwszym użyciem.
Grupy szczególne
Pacjentom w wieku podeszłym lek Izifri® należy stosować wyłącznie w dawce zaleconej przez lekarza.
Pacjentom z niewydolnością nerek należy stosować Izifri® zgodnie z dawką zaleconą przez lekarza. Informacje dotyczące stosowania Izifri® u pacjentów z niewydolnością nerek od umiarkowanej do ciężkiej (klirens kreatyniny ≤ 50 ml/min) znajdują się w sekcjach „Szczególne środki ostrożności” i „Farmakokinetyka”.
Pacjentom z niewydolnością wątroby można stosować Izifri® zgodnie z dawką zaleconą przez lekarza (patrz sekcja „Farmakokinetyka”).
Sposób stosowania
W celu zapewnienia właściwego stosowania leku należy poinformować pacjenta, jak korzystać z inhalatora.
INSTRUKCJA DOTYCZĄCA STOSOWANIA Elpenhaler®
Elpenhaler® to urządzenie do przyjmowania proszku do inhalacji w dawkach.
Każda dawka jest przechowywana w blisterze specjalnie zaprojektowanej paski blisterowej do dzielenia na pojedyncze dawki.
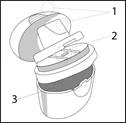
Inhalator Elpenhaler® składa się z 3 części:
- Mundystka i jego osłonki ochronnej (1).
- Powierzchni (2), na której umieszcza się paskę blisterową (powierzchnia podpierająca).
- Odbioru do przechowywania (3), w którym znajduje się paska blisterowa.
Trzy części są połączone ze sobą i mogą być otwierane osobno.
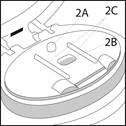
Powierzchnia podpierająca składa się z:
- Punktu mocowania (2A), do którego mocuje się paskę blisterową.
- Włóżka (2B), do którego wkłada się paskę blisterową.
- Dwóch prowadnic dla pasków (2C), które bezpiecznie mocują paskę blisterową we właściwej pozycji na powierzchni podpierającej.
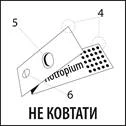
Paska blisterowa składa się z:
- Dwóch folii aluminiowych (4).
- Blistera (5), zawierającego lek.
- Otworu (6).
STOSOWANIE Elpenhaler®
A. Przygotowanie urządzenia
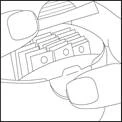
- Otworzyć odbiór do przechowywania, naciskając jak pokazano na rysunku, wyjąć paskę i ponownie zamknąć odbiór do przechowywania.
- Całkowicie otworzyć mundystek, delikatnie naciskając na pasekowany obszar.
- Odblokować i odsunąć mundystek do tyłu, aby odsłonić powierzchnię podpierającą.
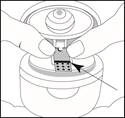
Trzymać paskę blisterową połyskującą stroną do góry, aby zobaczyć niebieską linię, jak wskazano strzałką na rysunku. Oznakowana strona paska powinna być skierowana w dół.
- Umieścić otwór paska na punkcie mocowania powierzchni podpierającej. Delikatnie naciskając, upewnić się, że pasek jest bezpiecznie zamocowany na punkcie mocowania.
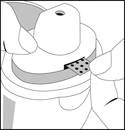
Blister paska umieszcza się we włóżku powierzchni podpierającej, a prowadnice mocują pasek we właściwej pozycji.
- Zamknąć mundystek, poziomo odciągnąć wypukły występ paska, aby go odłączyć, i wyrzucić.
- Dawka jest teraz gotowa do inhalacji.
B. Inhalacja dawki
Trzymać urządzenie z dala od ust. Całkowicie wyeksprymować. Należy uważać, aby nie wyeksprymować przez mundystek urządzenia. Przyłożyć Elpenhaler® do ust i szczelnie obejmować mundystek wargami.
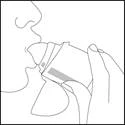
Powoli i głęboko wdychać przez usta (nie przez nos), aż płuca się wypełnią.
- Zatrzymać oddech na około 5 sekund lub tak długo, jak to wygodne, jednocześnie wyjmując urządzenie z ust.
- Wyeksprymować i kontynuować normalne oddychanie.
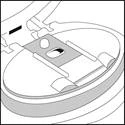
Otworzyć mundystek.
- Można zauważyć, że cały proszek został wdychany i że blister paska jest pusty.
- Usunąć pustą paskę, wyrzucić ją i przejść do kroku C.
C. Czyszczenie urządzenia
Po każdym użyciu przetrzeć mundystek i powierzchnię podpierającą suchą tkaniną lub suchym ręcznikiem papierowym. Nie należy używać wody do czyszczenia urządzenia.
Zamknąć mundystek i jego osłonkę.
Dzieci
Brak odpowiedniego doświadczenia w stosowaniu u pacjentów w wieku dziecięcym (do 18 roku życia).
Przedawkowanie
Wysokie dawki Izifri® mogą powodować objawy antycholinergiczne.
Jednak nie stwierdzono istotnych skutków ubocznych o charakterze antycholinergicznym u zdrowych ochotników po pojedynczej dawce aż do 340 µg bromku tiotropium.
Nie zaobserwowano żadnych istotnych reakcji ubocznych poza suchością w ustach po 7 dniach stosowania bromku tiotropium w dawkach do 170 µg u zdrowych ochotników.
W badaniach wielokrotnej dawki u pacjentów z POChP stosowanie maksymalnej dawki dobowej 43 µg bromku tiotropium przez 4 tygodnie nie powodowało istotnych reakcji ubocznych.
Ostra zatrucie przy doustnym stosowaniu kapsułek tiotropium jest mało prawdopodobne ze względu na niską doustną biodostępność.
Niepożądane działania.
Wiele z wymienionych efektów niepożądanych można przypisać antycholinergicznym właściwościom Izifri®.
Niepożądane działania dotyczące leku zostały ustalone na podstawie danych uzyskanych z badań klinicznych oraz spontanicznych zgłoszeń w okresie po rejestracji. Baza danych badań klinicznych obejmuje 9647 pacjentów stosujących tiotropium w 28 randomizowanych badaniach klinicznych z grupą placebo, z okresem leczenia od 4 tygodni do 4 lat.
Częstość występowania działań niepożądanych zgodnie z MedDRA: bardzo często (≥ 1/10); często (≥ 1/100, < 1/10); rzadko (≥ 1/1000, < 1/100); pojedyncze przypadki (≥ 1/10000, < 1/1000); rzadkie (<1/10000); nieznane (nie można określić na podstawie dostępnych danych).
Zaburzenia układu metabolicznego: nieznane – odwodnienie.
Zaburzenia układu nerwowego: rzadko – zawroty głowy, ból głowy, zaburzenia wrażliwości smakowej; pojedyncze przypadki – bezsenność.
Zaburzenia oka: rzadko – nieostre widzenie; pojedyncze przypadki – jaskra, podwyższone ciśnienie wewnątrzgałkowe.
Zaburzenia układu sercowo-naczyniowego: rzadko – migotanie przedsionków; pojedyncze przypadki – nadkomorowa tachykardia, tachykardia, uczucie kołatania serca.
Zaburzenia układu oddechowego, klatki piersiowej i jamy śródpiersiowej: rzadko – kaszel, dysfonia, zapalenie gardła; pojedyncze przypadki – skurcz oskrzeli, krwawienie z nosa, zapalenie krtani, zapalenie zatok.
Zaburzenia przewodu pokarmowego: często – suchość w ustach; rzadko – zaparcia, choroba refluksowa przełyku, kandydoza jamy ustnej i gardła; pojedyncze przypadki – niedrożność jelit, w tym niedrożność jelit paralityczną; dysfagia, zapalenie dziąseł, zapalenie języka, zapalenie błony śluzowej jamy ustnej, nudności; nieznane – próchnica zębów.
Zaburzenia układu immunologicznego, skóry i tkanki podskórnej: rzadko – wysypka; pojedyncze przypadki – obrzęk naczynioruchowy, nadwrażliwość (w tym reakcje alergiczne natychmiastowego typu), świąd, pokrzywka; nieznane – suchość skóry, infekcje skóry i powstawanie owrzodzeń, reakcje anafilaktyczne.
Zaburzenia układu mięśniowo-szkieletowego i tkanki łącznej: nieznane – obrzęk stawów.
Zaburzenia układu moczowego: rzadko – zatrzymanie moczu, dysuria; pojedyncze przypadki – infekcja dróg moczowych.
Opis wybranych działań niepożądanych
W kontrolowanych badaniach klinicznych obserwowano zazwyczaj antycholinergiczną reakcję niepożądaną w postaci suchości w ustach, występującą u około 4% pacjentów.
W 28 badaniach klinicznych suchość w ustach doprowadziła do przerwania przyjmowania leku u 18 z 9647 pacjentów (0,2%).
Do poważnych działań niepożądanych zaliczane są jaskra, zaparcia, niedrożność jelit, w tym niedrożność jelit paralityczną oraz zatrzymanie moczu.
Inne szczególne grupy pacjentów
Liczba działań antycholinergicznego typu może wzrosnąć z wiekiem.
Okres ważności. 2 lata.
Zużyć w ciągu 60 dni od pierwszego otwarcia folii aluminiowej przy przechowywaniu w temperaturze poniżej 25 °C.
Nie stosować leku po upływie okresu ważności wskazanego na opakowaniu.
Warunki przechowywania.
Lek nie wymaga specjalnych warunków przechowywania. Przechowywać w oryginalnym opakowaniu w celu ochrony przed wilgocią.
Przechowywać w miejscu niedostępnym dla dzieci.
Opakowanie.
Po 10 µg/dawkę, po 1 dawce w blistrze; po 30 blisterów w urządzeniu do inhalacji Elpenhaler®; po 1 urządzeniu w torebce; po 1 torebce w pudełku kartonowym.
Kategoria wydania. Na receptę.
Producent.
Elpen Pharmaceutical Co. Inc., Greece
Elpen Pharmaceutical Co., Inc., Grecja
(wyprodukowanie, opakowanie pierwotne i wtórne, kontrola jakości, wprowadzenie serii leku do obrotu).
Miejsce produkcji oraz adres siedziby działalności.
Marathonos Avenue 95, Pikermi, 190 09, Greece
Awenida Maratonos 95, Pikermi, 190 09, Grecja.
Wnioskodawca. AT «Farmak».
Siedziba wnioskodawcy. Ukraina, 04080, miasto Kijów, ul. Kirylowska, 63.