Izifri®
Ucraina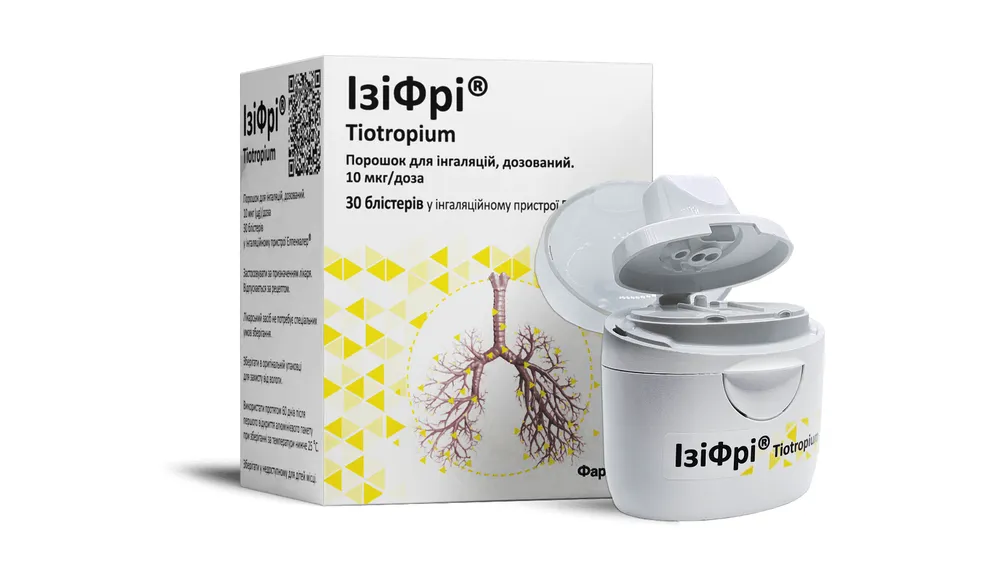
Indice
ISTRUZIONI PER L'USO MEDICO DEL MEDICINALE Izifri® (IziFri)
Composizione:
Principio attivo: bromuro di tiotropio;
1 blister contiene: monoidrato di bromuro di tiotropio - 0,016 mg (equivalente a tiotropio - 0,013 mg); 10 mcg/dose, 1 dose per blister;
Eccipiente: lattosio monoidrato.
Forma farmaceutica. Polvere per inalazione, dosata.
Principali caratteristiche fisico-chimiche: dispositivo plastico bianco contenente 30 blister singoli in alluminio riempiti con polvere bianca. Il dispositivo è inserito in un sacchetto di alluminio contenente un essiccante.
Gruppo farmacoterapeutico. Farmaci per il trattamento delle malattie ostruttive delle vie respiratorie, agenti inalatori. Agenti anticolinergici.
Codice ATC R03B B04.
Proprietà farmacologiche.
Farmacodinamica.
Meccanismo d'azione. Tiotropio è un agente anticolinergico specifico a lunga durata d'azione. Tiotropio presenta un'affinità simile per tutti i sottotipi di recettori muscarinici (da M1 a M5). Nelle vie respiratorie, l'inibizione dei recettori M3 determina il rilassamento della muscolatura liscia. Negli studi preclinici in vitro e in vivo, l'effetto broncoprotettivo era dose-dipendente e durava oltre 24 ore.
La durata dell'effetto è dovuta al lento rilascio dai recettori M3; il tempo di dimezzamento di tiotropio è significativamente più lungo rispetto a quello dell'ipratropio. Come agente anticolinergico di tipo N-quaternario, tiotropio è localmente (bronco-) selettivo quando somministrato per inalazione e mostra un ampio indice terapeutico prima della comparsa di effetti anticolinergici sistemici. La dissociazione dai recettori M2 è più rapida rispetto a quella dai recettori M3 negli studi funzionali in vitro. Il recettore M3 è pertanto il sottotipo di recettore più rilevante (controllato cineticamente) rispetto all'M2. L'elevata attività e la lenta dissociazione dai recettori si correlano clinicamente con una marcata e prolungata broncodilatazione nei pazienti con malattia polmonare ostruttiva cronica (BPCO).
Effetti farmacodinamici. La broncodilatazione dopo l'inalazione di tiotropio è principalmente un effetto locale sulle vie respiratorie, non sistemico.
Con l'uso di tiotropio una volta al giorno, si osserva un significativo miglioramento della funzionalità polmonare (incremento del volume espiratorio forzato nel primo secondo (FEV1) e della capacità vitale forzata polmonare) entro 30 minuti dalla prima dose, con effetto che dura 24 ore. Lo stato farmacodinamico stazionario si raggiunge entro una settimana. Nella maggior parte dei pazienti, la broncodilatazione si manifesta entro il terzo giorno.
In base alle misurazioni quotidiane, tiotropio migliora significativamente la massima velocità espiratoria mattutina e serale.
Il miglioramento della funzionalità polmonare si mantiene senza segni di tolleranza.
La broncodilatazione persiste per l'intero intervallo di 24 ore rispetto al placebo. Ciò vale indipendentemente dal fatto che tiotropio venga somministrato al mattino o alla sera.
Tiotropio riduce significativamente la dispnea; il miglioramento clinico si mantiene per tutta la durata del trattamento.
Tiotropio riduce significativamente il numero di riacutizzazioni della BPCO e aumenta il tempo fino alla prima riacutizzazione grave.
Tiotropio migliora significativamente la qualità della vita correlata alla salute; il miglioramento si mantiene per tutta la durata del trattamento.
Tiotropio riduce significativamente il numero di ospedalizzazioni per riacutizzazioni della BPCO e ritarda il tempo alla prima ospedalizzazione.
In due studi, tiotropio ha migliorato significativamente la tolleranza all'esercizio fisico limitato dai sintomi della malattia del 19,7% e del 28,3%.
In uno studio con somministrazione di tiotropio a dosi di 18 µg e 54 µg (tre dosi da 18 µg) per 12 giorni, non si è verificato allungamento dell'intervallo QT all'elettrocardiogramma.
In uno studio di quattro anni su 5993 pazienti, tiotropio ha mantenuto un miglioramento del parametro FEV1 per tutta la durata dello studio, anche se non ha modificato la riduzione annuale media del FEV1.
Durante il trattamento, il rischio di mortalità è diminuito del 16%. L'incidenza complessiva di eventi fatali è stata di 4,79 per 100 pazienti-anno nel gruppo placebo rispetto a 4,10 per 100 pazienti-anno nel gruppo tiotropio (rapporto di rischio (tiotropio/placebo) 0,84, IC 95% – 0,73; 0,97). Il trattamento con tiotropio ha ridotto il rischio di insufficienza respiratoria del 19% (2,09 rispetto a 1,68 casi per 100 pazienti-anno, rischio relativo (tiotropio/placebo) – 0,81, IC 95% - 0,65; 1,00).
Farmacocinetica.
Tiotropio è un composto ammonio quaternario moderatamente solubile in acqua. Viene somministrato come polvere secca per inalazione. Generalmente, con la via inalatoria, la maggior parte della dose rilasciata si deposita nel tratto gastrointestinale e una minore quantità nei polmoni.
Assorbimento. Dopo inalazione della polvere secca, la biodisponibilità assoluta è del 19,5%, indicativo di un'elevata biodisponibilità della frazione che raggiunge i polmoni. La biodisponibilità assoluta della soluzione orale di tiotropio è del 2-3%. La concentrazione massima di tiotropio nel plasma sanguigno si osserva 5-7 minuti dopo l'inalazione.
A regime stazionario, il livello massimo di tiotropio nel plasma sanguigno nei pazienti con BPCO è di 12,9 pg/ml e diminuisce rapidamente secondo un modello multicompartmentale. La concentrazione minima di tiotropio nel plasma a regime stazionario è di 1,71 pg/ml. L'effetto sistemico dopo inalazione di tiotropio come polvere è simile a quello dopo inalazione di tiotropio come soluzione.
Distribuzione. Il 72% del farmaco si lega alle proteine plasmatiche. Il volume di distribuzione è di 32 l/kg. La concentrazione locale nei polmoni non è nota, ma, considerando la via di somministrazione, si presume un'elevata concentrazione nei polmoni. Negli studi sugli animali è stato dimostrato che tiotropio non attraversa in misura significativa la barriera ematoencefalica.
Biotrasformazione. Il grado di biotrasformazione è basso, poiché il 74% della sostanza invariata viene escreto con le urine dopo somministrazione endovenosa a volontari sani. Tiotropio, essendo un estere complesso, si decompone non enzimaticamente in alcool N-metilscopina e acido ditienilglicolico, che non si legano ai recettori muscarinici.
Ulteriori studi in vitro su microsomi epatici ed epatociti hanno mostrato che tiotropio (< 20% della dose dopo somministrazione endovenosa) viene metabolizzato attraverso ossidazione dipendente dal citocromo P450 e successiva coniugazione con glutatione a diversi metaboliti di fase II. Questa catena enzimatica può essere inibita dagli inibitori del CYP450 2D6 (e 3A4), chinidina, chetoconazolo e gestodene. I citocromi CYP450 2D6 e 3A4 sono coinvolti nelle trasformazioni metaboliche responsabili dell'eliminazione di una piccola parte della dose. Tiotropio, anche a concentrazioni sovra-terapeutiche, non inibisce i citocromi P450 1A1, 1A2, 2B6, 2C9, 2C19, 2D6, 2E1 e 3A nei microsomi epatici.
Eliminazione. Il tempo di dimezzamento efficace di tiotropio varia tra 27 e 45 ore dopo somministrazione a pazienti con BPCO. La clearance totale è stata di 880 ml/min dopo somministrazione endovenosa a giovani volontari sani. Dopo somministrazione endovenosa, tiotropio viene principalmente escreto invariato con le urine. Dopo inalazione della polvere secca, l'escrezione urinaria è del 7% (1,3 µg) della quantità invariata entro 24 ore; il resto non viene assorbito dall'intestino ed è eliminato con le feci. La clearance renale di tiotropio supera la clearance della creatinina, indicando un'eliminazione urinaria. Dopo somministrazione inalatoria quotidiana prolungata a pazienti con BPCO, lo stato farmacocinetico stazionario viene raggiunto entro 7 giorni senza ulteriore cumulo.
Linearità/non linearità. Tiotropio ha dimostrato proprietà farmacocinetiche lineari nell'intervallo terapeutico, indipendentemente dalla forma farmaceutica.
Farmacocinetica nei pazienti anziani. Come per tutti gli altri farmaci eliminati principalmente attraverso le urine, con l'uso di tiotropio nei pazienti anziani si osserva una riduzione della clearance renale (365 ml/min nei pazienti con BPCO di età < 65 anni rispetto a 271 ml/min nei pazienti con BPCO di età ≥ 65 anni). Ciò non comporta un aumento corrispondente dei valori di AUC0-6,ss o Cmax,ss.
Farmacocinetica nei pazienti con compromissione renale. L'inalazione di tiotropio una volta al giorno a regime stazionario in pazienti con BPCO e lieve insufficienza renale (clearance della creatinina 50-80 ml/min) ha portato a un lieve aumento dei valori di AUC0-6,ss (da 1,8 a 30%) e a livelli simili di Cmax rispetto ai pazienti con funzione renale normale (clearance della creatinina > 80 ml/min).
Nei pazienti con BPCO e compromissione renale moderata o grave (clearance della creatinina <50 ml/min), la somministrazione endovenosa di tiotropio porta a un raddoppio dell'esposizione totale (l'indice AUC0-4h è più alto dell'82%, l'indice Cmax è più alto del 52%) rispetto ai pazienti con funzione renale normale, confermato dai dati di concentrazione plasmatica dopo inalazione della polvere secca.
Farmacocinetica nei pazienti con compromissione epatica. L'insufficienza epatica non ha un impatto significativo sulla farmacocinetica di tiotropio. Tiotropio viene principalmente eliminato attraverso l'eliminazione renale (fino al 74% in giovani volontari sani) e attraverso la semplice scissione non enzimatica dell'estere in prodotti che non si legano ai recettori muscarinici.
Pazienti giapponesi con BPCO. Nell'analisi incrociata, la concentrazione media massima di tiotropio nel plasma sanguigno 10 minuti dopo la somministrazione a regime stazionario era del 20-70% più alta nei giapponesi rispetto agli europei dopo inalazione di tiotropio, ma non sono state osservate evidenze di aumento della mortalità o del rischio di complicanze cardiache nei pazienti giapponesi rispetto agli europei. Per altre razze o gruppi etnici, i dati farmacocinetici sono insufficienti.
Relazione farmacocinetica/farmacodinamica. Non esiste una relazione diretta tra farmacocinetica e farmacodinamica.
Caratteristiche cliniche.
Indicazioni.
Terapia broncodilatatrice di mantenimento per alleviare i sintomi nella malattia polmonare ostruttiva cronica (BPCO).
Controindicazioni.
Controindicato nei pazienti con ipersensibilità nota al bromuro di tiotropio, all'atropina o ai suoi derivati (come ipratropio o ossitropio) o ad altri componenti del medicinale.
Interazioni con altri medicinali e altre forme di interazione.
Nonostante non siano stati effettuati studi formali sulle interazioni con altri medicinali, il bromuro di tiotropio è stato utilizzato in associazione ad altri farmaci (broncodilatatori simpaticomimetici, metilxantine, corticosteroidi orali e inalatori utilizzati nel trattamento della BPCO) senza evidenze cliniche di interazioni con altri medicinali.
Non è stato dimostrato che l'uso di agonisti dei recettori beta-adrenergici a lunga durata o di corticosteroidi inalatori modifichi l'esposizione al tiotropio.
Tuttavia, l'associazione di Izifri® con altri medicinali anticolinergici non è stata studiata, pertanto tale utilizzo non è raccomandato.
Caratteristiche di impiego.
Izifri® è un broncodilatatore da somministrare una volta al giorno per la terapia di mantenimento e non deve essere utilizzato per il trattamento iniziale di attacchi acuti di broncospasmo.
Dopo l'uso di Izifri® sono possibili reazioni di ipersensibilità di tipo immediato.
Come per altri farmaci anticolinergici, Izifri® deve essere usato con cautela nei pazienti con glaucoma ad angolo chiuso, iperplasia prostatica o ostruzione al collo della vescica (vedere il paragrafo «Effetti indesiderati»).
I farmaci inalatori possono causare broncospasmo indotto per inalazione.
Il farmaco deve essere usato con cautela nei seguenti pazienti: coloro che hanno recentemente avuto un infarto miocardico (<6 mesi); pazienti con aritmia instabile o potenzialmente letale, o con aritmia che ha richiesto intervento o modifica della terapia nell'anno precedente; pazienti ospedalizzati per insufficienza cardiaca (classe NYHA III o IV) nell'ultimo anno. Tali pazienti sono stati esclusi dagli studi clinici. In queste condizioni, l'effetto anticolinergico potrebbe nuocere al paziente.
Poiché la concentrazione plasmatica di bromuro di tiotropio aumenta nei pazienti con insufficienza renale da moderata a grave (clearance della creatinina ≤ 50 ml/min), Izifri® deve essere utilizzato solo se il beneficio atteso supera il rischio potenziale. Non sono disponibili dati sull'uso prolungato di Izifri® nei pazienti con insufficienza renale (vedere il paragrafo «Farmacocinetica»).
I pazienti devono evitare il contatto della polvere con gli occhi. Tale contatto può causare precipitazione o peggioramento del glaucoma ad angolo chiuso, dolore o fastidio oculare, offuscamento temporaneo della vista, sensazione di aloni o macchie colorate davanti agli occhi, in combinazione con arrossamento dell'occhio sotto forma di iperemia della congiuntiva o della cornea.
In caso di comparsa di tali sintomi in qualsiasi combinazione, si deve interrompere immediatamente l'uso di bromuro di tiotropio e consultare senza indugio un medico specializzato.
La secchezza orale osservata durante la terapia anticolinergica può essere associata, nel tempo, a carie dentaria.
Izifri® non deve essere utilizzato più di una volta al giorno.
Izifri® contiene lattosio. Se il paziente ha una nota intolleranza ad alcuni zuccheri, deve consultare il medico prima di assumere questo medicinale.
Uso durante la gravidanza o l’allattamento.
Gravidanza. I dati sull'uso di tiotropio in donne in gravidanza sono molto limitati. Studi preclinici non hanno evidenziato effetti dannosi diretti o indiretti legati alla tossicità riproduttiva a dosi clinicamente rilevanti. Come misura precauzionale, si raccomanda di evitare l'uso di Izifri® durante la gravidanza.
Allattamento. Non è noto se il bromuro di tiotropio passi nel latte materno. Nonostante gli studi condotti sui roditori abbiano mostrato solo una minima escrezione del bromuro di tiotropio nel latte, l'uso del farmaco durante l’allattamento non è raccomandato.
Il bromuro di tiotropio è un composto a lunga durata d'azione. La decisione se continuare o interrompere l’allattamento o continuare o interrompere il trattamento con Izifri® deve essere presa valutando attentamente i benefici dell’allattamento per il neonato e quelli della terapia per la madre.
Fertilità. Non sono disponibili dati clinici sull'effetto del tiotropio sulla fertilità. Gli studi preclinici non hanno evidenziato alcun effetto indesiderato sulla fertilità.
Capacità di guidare veicoli o di usare macchinari.
Non sono stati effettuati studi sull'effetto del medicinale sulla capacità di guidare veicoli o di usare macchinari. La comparsa di capogiri, cefalea o offuscamento della vista può influire sulla capacità di guidare veicoli o di usare macchinari.
Modalità e dosaggio di somministrazione.
Il medicinale è destinato esclusivamente all'inalazione.
La dose raccomandata di Izifri® è l'inalazione del contenuto di una striscia blister una volta al giorno mediante l'utilizzo del dispositivo inalatorio Elpenhaler®. L'inalazione deve essere effettuata sempre allo stesso orario della giornata.
Non si deve superare la dose raccomandata.
Le strisce blister sono destinate esclusivamente all'inalazione e non devono essere assunte per via orale. Le strisce blister di Izifri® non devono essere ingerite.
Il medicinale deve essere utilizzato esclusivamente con il dispositivo inalatorio Elpenhaler®. Il dispositivo Elpenhaler® deve essere conservato nella confezione per proteggerlo dall'umidità ed estratto immediatamente prima del primo utilizzo.
Gruppi particolari
Nei pazienti di età avanzata, Izifri® deve essere utilizzato esclusivamente alla dose raccomandata dal medico.
Nei pazienti con insufficienza renale, Izifri® deve essere utilizzato secondo la dose raccomandata dal medico. Informazioni sull'uso di Izifri® nei pazienti con insufficienza renale da moderata a grave (clearance della creatinina ≤ 50 ml/min) sono riportate nelle sezioni «Avvertenze speciali e precauzioni di impiego» e «Farmacocinetica».
Nei pazienti con insufficienza epatica, Izifri® può essere utilizzato secondo la dose raccomandata dal medico (vedere sezione «Farmacocinetica»).
Modalità di somministrazione
Per garantire un uso corretto del medicinale, è necessario informare il paziente su come utilizzare l'inalatore.
ISTRUZIONI PER L'USO DI Elpenhaler®
Elpenhaler® è un dispositivo per l'assunzione di polvere per inalazione in dosi.
Ogni dose è contenuta in un blister di una striscia blister appositamente progettata per la suddivisione in dosi singole.
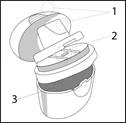
Il dispositivo Elpenhaler® è composto da 3 parti:
- L'ugello bocchino e il suo tappo protettivo (1).
- La superficie (2), sulla quale viene posizionata la striscia blister (superficie di appoggio).
- Il vano di conservazione (3), in cui viene riposta la striscia blister.
Le tre parti sono collegate tra loro e possono essere aperte separatamente.
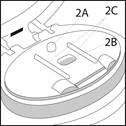
La superficie di appoggio è costituita da:
- Il punto di fissaggio (2A), al quale viene ancorata la striscia blister.
- La cavità (2B), in cui viene inserita la striscia blister.
- Due guide per la striscia (2C), che fissano saldamente la striscia blister nella corretta posizione sulla superficie di appoggio.
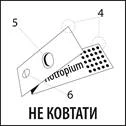
La striscia blister è composta da:
- Due fogli di alluminio (4).
- Un blister (5) contenente il medicinale.
- Un foro (6).
UTILIZZO DI Elpenhaler®
Α. Preparazione del dispositivo
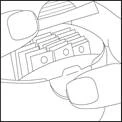
- Aprire il vano di conservazione premendo come mostrato in figura, estrarre la striscia e richiudere nuovamente il vano di conservazione.
- Aprire completamente il bocchino premendo leggermente sulla zona rigata.
- Sbloccare e spingere indietro il bocchino per esporre la superficie di appoggio.
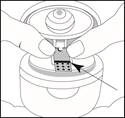
Tenere la striscia blister con la superficie lucida rivolta verso l'alto, in modo da vedere la linea blu, come indicato dalla freccia in figura. La superficie marcata della striscia deve essere rivolta verso il basso.
- Inserire il foro della striscia nel punto di fissaggio della superficie di appoggio. Applicare una leggera pressione per assicurarsi che la striscia sia fissata saldamente al punto di fissaggio.
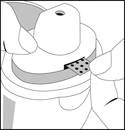
Il blister della striscia viene inserito nella cavità della superficie di appoggio e le guide fissano la striscia nella corretta posizione.
- Chiudere il bocchino, tirare orizzontalmente la linguetta rilevata per staccarla, quindi gettarla via.
- La dose è ora pronta per l'inalazione.
B. Inalazione della dose
Tenere il dispositivo lontano dalla bocca. Espirare completamente. Fare attenzione a non espirare attraverso il bocchino del dispositivo. Avvicinare Elpenhaler® alla bocca e stringere saldamente il bocchino con le labbra.
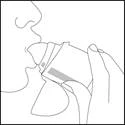
Inspirare lentamente e profondamente attraverso la bocca (non attraverso il naso) fino a quando i polmoni non saranno pieni.
- Trattenere il respiro per circa 5 secondi o per il tempo che risulti comodo, contemporaneamente estrarre il dispositivo dalla bocca.
- Espirare e riprendere a respirare normalmente.
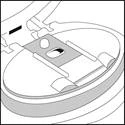
Aprire il bocchino.
- Sarà visibile che tutta la polvere è stata inalata e che il blister della striscia è vuoto.
- Rimuovere la striscia vuota, gettarla via e passare al passo C.
C. Pulizia del dispositivo
Dopo ogni utilizzo, pulire il bocchino e la superficie di appoggio con un panno asciutto o un tovagliolo di carta asciutto. Non utilizzare acqua per la pulizia del dispositivo.
Chiudere il bocchino e il suo tappo protettivo.
Pediatria.
Non vi è esperienza di utilizzo appropriato nei pazienti di età pediatrica (al di sotto dei 18 anni).
Sovradosaggio.
Alte dosi di Izifri® possono causare sintomi anticolinergici.
Tuttavia, effetti indesiderati sistemici di tipo anticolinergico sono risultati assenti in volontari sani dopo una dose singola fino a 340 mcg di bromuro di tiotropio.
Nessuna reazione avversa significativa, eccetto la secchezza orale, è stata osservata dopo 7 giorni di somministrazione di bromuro di tiotropio fino a 170 mcg in volontari sani.
Negli studi con dosaggio ripetuto in pazienti con BPCO, l'uso della dose giornaliera massima di 43 mcg di bromuro di tiotropio per 4 settimane non ha causato reazioni avverse significative.
L'intossicazione acuta dopo somministrazione orale delle capsule di tiotropio è improbabile a causa della bassa biodisponibilità orale.
Effetti indesiderati.
Molti degli effetti indesiderati elencati possono essere attribuiti alle proprietà anticolinergiche di Izifri®.
Le reazioni avverse al medicinale sono state definite sulla base di dati ottenuti da studi clinici e segnalazioni spontanee durante il periodo post-commercializzazione. Il database degli studi clinici comprende 9647 pazienti che hanno utilizzato tiotropio in 28 studi clinici controllati con placebo, con un periodo di trattamento da 4 settimane a 4 anni.
Frequenza degli effetti indesiderati secondo MedDRA: molto frequente (≥ 1/10); frequente (≥ 1/100, < 1/10); non frequente (≥ 1/1000, < 1/100); raro (≥ 1/10000, < 1/1000); molto raro (<1/10000); non noto (non può essere determinato sulla base dei dati disponibili).
Disturbi del metabolismo e della nutrizione: non noto – disidratazione.
Sistema nervoso centrale: non frequente – vertigini, cefalea, alterazioni del gusto; raro – insonnia.
Organi della vista: non frequente – offuscamento della vista; raro – glaucoma, aumento della pressione intraoculare.
Sistema cardiaco: non frequente – fibrillazione atriale; raro – tachicardia sopraventricolare, tachicardia, palpitazioni.
Apparato respiratorio, torace e mediastino: non frequente – tosse, disfonia, faringite; raro – broncospasmo, emorragia nasale, laringite, sinusite.
Apparato gastrointestinale: frequente – secchezza delle fauci; non frequente – stitichezza, malattia da reflusso gastroesofageo, candidosi orale e faringea; raro – ostruzione intestinale, inclusa l'ostruzione intestinale paralitica; disfagia, gengivite, glossite, stomatite, nausea; non noto – carie dentale.
Sistema immunitario, cute e tessuto sottocutaneo: non frequente – eruzioni cutanee; raro – angioedema, ipersensibilità (inclusi reazioni allergiche di tipo immediato), prurito, orticaria; non noto – secchezza della pelle, infezioni cutanee e formazione di ulcere, reazioni anafilattiche.
Apparato muscoloscheletrico e tessuto connettivo: non noto – edema articolare.
Apparato urinario: non frequente – ritenzione urinaria, disuria; raro – infezione delle vie urinarie.
Descrizione di singole reazioni avverse
Negli studi clinici controllati è stata generalmente osservata la reazione avversa anticolinergica rappresentata dalla secchezza delle fauci, che si è verificata in circa il 4% dei pazienti.
Nei 28 studi clinici, la secchezza delle fauci ha portato all'interruzione del trattamento in 18 su 9647 pazienti (0,2%).
Tra gli effetti indesiderati gravi: glaucoma, stitichezza, ostruzione intestinale, inclusa l'ostruzione intestinale paralitica, e ritenzione urinaria.
Altri gruppi di pazienti particolari
Il numero di effetti anticolinergici può aumentare con l'età.
Durata della validità. 2 anni.
Utilizzare entro 60 giorni dal primo apporto della busta in alluminio, se conservato a temperatura inferiore a 25 °C.
Non utilizzare il medicinale dopo la data di scadenza indicata sull’imballaggio.
Condizioni di conservazione.
Il medicinale non richiede condizioni particolari di conservazione. Conservare nell’imballaggio originale per proteggere dall’umidità.
Conservare fuori dalla portata dei bambini.
Confezione.
10 mcg/dose, 1 dose in blister; 30 blister in un dispositivo inalatorio Elpenhaler®; 1 dispositivo in un sacchetto; 1 sacchetto in una scatola di cartone.
Categoria di prescrizione. Con ricetta medica.
Produttore.
Elpen Pharmaceutical Co. Inc., Greece
Elpen Pharmaceutical Co., Inc., Grecia
(produttivo, confezionamento primario e secondario, controllo qualità, rilascio del lotto del medicinale).
Sede del produttore e indirizzo del luogo di esercizio dell’attività.
Marathonos Avenue 95, Pikermi, 190 09, Greece
Marathonos Avenue 95, Pikermi, 190 09, Grecia.
Richiedente. AТ «Farmak».
Sede del richiedente. Ucraina, 04080, Kiev, via Kyrylivska, 63.