Genotropin®
Ukraina
Spis treści
INSTRUKCJA dot. stosowania leku Genotropin® (GENOTROPIN®)
Skład:
substancja czynna: somatropina;
przednia komora: somatropiny 6,1 mg lub 13,8 mg;
1 ml odtworzonego roztworu zawiera somatropiny 16 JM (5,3 mg) lub 36 JM (12 mg);
substancje pomocnicze: glicyna, sorbitol (E 421), disodowy fosforan wodorotlenek bezwodny, sodowy fosforan wodorotlenek (dwunastowodór);
tylna komora (rozpuszczalnik): m-krezol, sorbitol (E 421), woda do wstrzykiwań.
Postać leku. Proszek liofilizowany i rozpuszczalnik do roztworu do wstrzykiwań.
Główne właściwości fizykochemiczne: liofilizowana jednorodna substancja białego koloru. Rozpuszczalnik powinien być praktycznie wolny od zanieczyszczeń mechanicznych.
Grupa farmakoterapeutyczna. Hormony przysadki mózgowej i ich analogi. Somatropina i agonisty somatropiny. Somatropina. Kod ATC H01A C01.
Właściwości farmakologiczne.
Farmakodynamika.
Somatropina to silny hormon metaboliczny odgrywający ważną rolę w metabolizmie lipidów, węglowodanów i białek. Somatropina jest wytwarzana w komórkach Escherichia coli metodą rekombinowanego DNA. U dzieci z niedoborem endogennego hormonu wzrostu somatropina przyspiesza liniowy wzrost szkieletu oraz tempo wzrostu. Zarówno u dzieci, jak i dorosłych, somatropina wspiera normalny skład ciała poprzez zwiększenie retencji azotu, przyspieszenie wzrostu mięśni szkieletowych oraz mobilityzację tłuszczu w organizmie. Szczególnie wrażliwą tkanką na działanie somatropiny jest tłuszcz wisceralny. Oprócz stymulacji lipolizy, somatropina zmniejsza napływ trójglicerydów do zasobów tłuszczu. Stężenia IGF-1 (insulinopodobnego czynnika wzrostu, typ 1) oraz IGFBP-3 (białka wiążącego insulinopodobny czynnik wzrostu, typ 3) w surowicy krwi wzrastają pod wpływem somatropiny. Ponadto wykazano poniższe działania.
- Metabolizm lipidów. Somatropina stymuluje receptory lipoprotein o niskiej gęstości (LDL) w wątrobie i wpływa na profil lipidów oraz lipoproteinów w surowicy krwi. Ogólnie stosowanie somatropiny u pacjentów z niedoborem hormonu wzrostu prowadzi do obniżenia stężenia LDL i apolipoproteiny B. Może również dojść do obniżenia poziomu cholesterolu ogólnego.
- Metabolizm węglowodanów. Somatropina zwiększa poziom insuliny, jednak poziom glukozy na czczo zazwyczaj nie ulega zmianie. U dzieci z hipopitularyzmem może występować hipoglikemia na czczo. Somatropina odwraca ten stan.
- Gospodarka wodno-elektrolitowa. Niedobór hormonu wzrostu wiąże się ze zmniejszeniem objętości osocza krwi i płynu tkankowego. Oba te wskaźniki szybko rosną po leczeniu somatropiną. Somatropina sprzyja zatrzymaniu w organizmie sodu, potasu i fosforu.
- Metabolizm kostny. Somatropina stymuluje odnowę tkanki kostnej szkieletu. U pacjentów z niedoborem hormonu wzrostu i osteoporozą długotrwałe leczenie somatropiną prowadzi do wzrostu zawartości mineralnej i gęstości kości w odcinkach obciążonych.
- Sprawność fizyczna. Długotrwałe leczenie somatropiną zwiększa siłę mięśni i wytrzymałość fizyczną. Somatropina zwiększa również rzut serca, jednak mechanizm tego efektu nie został jeszcze w pełni wyjaśniony. Niektórą rolę może odgrywać zmniejszenie obwodowego oporu naczyniowego.
W badaniach klinicznych z udziałem dzieci o niskim wzroście, urodzonych z małą masą ciała dla wieku ciążarności, stosowano dawki od 0,033 do 0,067 mg/kg masy ciała na dobę aż do osiągnięcia ostatecznego wzrostu. U 56 pacjentów, którzy byli leczeni ciągle i osiągnęli (lub prawie osiągnęli) ostateczny wzrost, wartość odchylenia standardowego (SDS) dla średniej zmiany wzrostu od początku leczenia wynosiła +1,90 SDS przy dawce 0,033 mg/kg na dobę i +2,19 SDS przy dawce 0,067 mg/kg na dobę. Opublikowane dane dotyczące dzieci urodzonych z małą masą ciała dla wieku ciążarności, które nie otrzymywały leczenia i nie osiągnęły spontanicznie normalnego wzrostu, sugerują późny wzrost w granicach 0,5 SDS.
Farmakokinetyka.
Absorpcja. Biodostępność somatropiny podanej podskórnie wynosi około 80% zarówno u zdrowych ochotników, jak i u pacjentów z niedoborem hormonu wzrostu. Dawka 0,035 mg/kg somatropiny podana podskórnie prowadzi do następujących zakresów wartości Cmax i tmax we krwi: 13–35 ng/ml i 3–6 godzin odpowiednio.
Eliminacja. Średni okres półtrwania po podaniu somatropiny dożylnie dorosłym pacjentom z niedoborem hormonu wzrostu wynosi około 0,4 godziny. Natomiast po podaniu podskórnym okres półtrwania może wynosić do 2–3 godzin. Obserwowana różnica może być spowodowana powolną absorpcją z miejsca iniekcji po podaniu podskórnym.
Podgrupy populacyjne. Bezwzględna biodostępność somatropiny po podaniu podskórnym jest taka sama u kobiet i mężczyzn.
Brak lub niepełne dane dotyczące farmakokinetyki somatropiny u osób starszych i dzieci, u pacjentów różnych ras oraz u pacjentów z zaburzeniami funkcji nerek i wątroby lub niewydolnością serca.
Dane przedkliniczne dotyczące bezpieczeństwa.
W badaniach toksyczności ogólnej, tolerancji miejscowej oraz toksyczności rozrodczej nie zaobserwowano żadnych klinicznie istotnych działań.
Badania in vitro i in vivo dotyczące genotoksyczności w odniesieniu do mutacji genów oraz indukcji aberracji chromosomowych dały wynik negatywny.
W jednym z badań in vitro na limfocytach pobranych od pacjentów po długotrwałym leczeniu somatropiną i kolejnym zastosowaniu dodatkowego leku radioimietycznego bleomycyny zaobserwowano zwiększoną kruchliwość chromosomów. Kliniczne znaczenie tego faktu nie jest jasne.
W innym badaniu na limfocytach pobranych od pacjentów po długotrwałym leczeniu somatropiną nie stwierdzono aberracji chromosomowych.
Charakterystyka kliniczna.
Wskazania.
Dzieci.
Niedorostanie spowodowane niedostateczną sekrecją hormonu wzrostu (niedobór hormonu wzrostu).
Niedorostanie związane z zespołem Turnera lub przewlekłą niewydolnością nerek.
Niedorostanie (obecna wysokość odchyleń standardowych mniejsza niż –2,5 i genetycznie uwarunkowana wysokość odchyleń standardowych mniejsza niż –1) u dzieci o niskim wzroście urodzonych z niewielką masą ciała dla wieku ciążowego, z odchyleniem standardowym masy ciała i/lub długości ciała mniejszym niż –2, które nie osiągnęły kompensacji wzrostu (odchylenie standardowe tempa wzrostu mniejsze niż 0 w ciągu ostatniego roku) do ukończenia 4. roku życia lub później.
Zespół Pradera–Williego, w celu poprawy wzrostu i składu ciała. Rozpoznanie zespołu Pradera–Williego powinno być potwierdzone odpowiednimi badaniami genetycznymi.
Genotropin® (somatropina [pochodzenia rdDNA] do wstrzykiwań) wskazany jest w leczeniu idiopatycznego niedorostania, tj. niedorostania bez niedoboru hormonu wzrostu, zdefiniowanego współczynnikiem odchylenia standardowego < –2,25 (tempa wzrostu, przy których mało prawdopodobne jest osiągnięcie normalnego zakresu wzrostu w dorosłości), u dzieci z niezamkniętymi płytami wzrostowymi, u których ocena diagnostyczna wyklucza inne przyczyny niedorostania wymagające obserwacji lub leczenia innymi środkami.
Dorośli.
Terapia zastępcza u dorosłych z wyraźnym niedoborem hormonu wzrostu.
Wystąpienie niedoboru hormonu wzrostu w wieku dorosłym. Pacjenci z ciężkim niedoborem hormonu wzrostu związanym z wieloma niedoborami hormonalnymi wynikającymi ze znanego uszkodzenia podwzgorza lub przysadki mózgowej, a także pacjenci z niedoborem co najmniej jednego z hormonów przysadki, z wyjątkiem prolaktyny. U tych pacjentów należy przeprowadzić odpowiedni dynamiczny test w celu ustalenia obecności lub braku niedoboru hormonu wzrostu.
Wystąpienie niedoboru hormonu wzrostu w dzieciństwie. Pacjenci, u których niedobór hormonu wzrostu wystąpił w dzieciństwie z powodu przyczyn dziedzicznych, genetycznych, nabytych lub nieznanych. U pacjentów z niedoborem hormonu wzrostu, który wystąpił w dzieciństwie, należy przeprowadzić ponowne badanie zdolności sekrecji hormonu po zakończeniu wzrostu długości ciała. U pacjentów z wysokim prawdopodobieństwem trwałego niedoboru hormonu wzrostu (np. z powodu przyczyn dziedzicznych lub wtórnego niedoboru hormonu wzrostu spowodowanego chorobą podwzgórzowo-przysadkową lub udarem mózgu) wartość średniego odchylenia czynnika wzrostu podobnego do insuliny typu 1 (IGF-1) mniejsza niż –2 bez leczenia hormonem wzrostu przez co najmniej 4 tygodnie powinna być uznawana za wystarczającą podstawę do rozpoznania niedoboru hormonu wzrostu.
U wszystkich pozostałych pacjentów należy przeprowadzić analizę IGF-1 oraz jeden test stymulacji hormonu wzrostu.
Przeciwwskazania.
Podwyższona wrażliwość na substancję czynną lub którykolwiek ze składników pomocniczych leku.
Somatropinę należy zabronić stosować w przypadku obecności jakichkolwiek oznak aktywności guza. Guzy wewnątrzczaszkowe powinny być nieaktywne, a ponadto leczenie przeciwnowotworowe powinno zostać zakończone przed rozpoczęciem terapii hormonem wzrostu. W przypadku wystąpienia jakichkolwiek oznak wzrostu guza leczenie należy przerwać.
Genotropin® nie powinien być stosowany w celu stymulacji wzrostu u dzieci z zamkniętymi płytami wzrostowymi.
Leczenie lekiem Genotropin® jest przeciwwskazane u pacjentów przebywających w ostrym stanie krytycznym w wyniku powikłań po operacji serca, operacji jamy brzusznej, wielokrotnych urazów, ostrej niewydolności oddechowej lub innych podobnych stanów (informacje dotyczące pacjentów otrzymujących terapię zastępczą znajdują się w sekcji „Szczególne ostrzeżenia i środki ostrożności podczas stosowania”).
Interakcje z innymi lekami i inne rodzaje interakcji.
Jednoczesne stosowanie z glikokortykosteroidami hamuje stymulujący wpływ leków somatropiny na tempo wzrostu. Pacjentom z niedoborem hormonu adrenokortykotropowego należy starannie dobrać zastępcze leczenie glikokortykosteroidami, aby uniknąć jakiegokolwiek hamującego wpływu na wzrost.
Dlatego należy starannie monitorować wzrost pacjentów otrzymujących leczenie glikokortykosteroidami, aby móc ocenić potencjalny wpływ stosowania glikokortykosteroidów na wzrost.
Hormon wzrostu zmniejsza przekształcanie kortyzonu w kortyzol i może prowadzić do ujawnienia wcześniej niezdiagnozowanej centralnej niedoczynności nadnerczy lub uczynić niskie dawki glikokortykosteroidów stosowane w leczeniu zastępczym nieskutecznymi (patrz sekcja „Szczególne ostrzeżenia i środki ostrożności podczas stosowania”).
Dane uzyskane w badaniu interakcji leków przeprowadzonym u dorosłych pacjentów z niedoborem hormonu wzrostu wskazują, że stosowanie somatropiny może zwiększać klirens związków metabolizowanych przez izoenzymy cytochromu P450. Klirens związków metabolizowanych przez cytochrom P450 3A4 (takich jak steroidy płciowe, kortykosteroidy, leki przeciwpadaczkowe i cyklosporyna) może być nadmiernie zwiększony, co prowadzi do zmniejszenia stężenia tych substancji w osoczu krwi. Znaczenie kliniczne tego faktu jest nieznane.
Dodatkowe informacje dotyczące cukrzycy i dysfunkcji tarczycy znajdują się w sekcji „Szczególne ostrzeżenia i środki ostrożności podczas stosowania”.
Kobietom otrzymującym zastępcze leczenie estrogenami doustnie może być potrzebna wyższa dawka hormonu wzrostu w celu osiągnięcia celu terapeutycznego (patrz sekcja „Szczególne ostrzeżenia i środki ostrożności podczas stosowania”).
Właściwości stosowania.
Diagnozę, rozpoczynanie terapii lekiem Genotropin® oraz dalszą kontrolę powinni prowadzić wykwalifikowani lekarze posiadający doświadczenie w rozpoznawaniu i leczeniu pacjentów zgodnie z wskazaniami do stosowania.
Miozyt – bardzo rzadkie działanie niepożądane, które może być spowodowane działaniem konserwantu m-krezolu zawartego w leku. W przypadku wystąpienia miagii lub zwiększonego bólu w miejscu iniekcji należy podejrzewać rozwój miozytu. W przypadku potwierdzenia miozytu należy stosować formę leku Genotropin®, która nie zawiera m-krezolu.
Nie należy przekraczać maksymalnej zalecanej dawki dobowej (patrz dział „Sposób stosowania i dawki”).
Wrażliwość na insulinę.
Somatropina może obniżać wrażliwość na insulinę. U pacjentów z cukrzycą po rozpoczęciu terapii somatropiną może być konieczna korekta dawki insuliny. Podczas terapii somatropiną należy kontrolować stan pacjentów z cukrzycą, nietolerancją glukozy lub dodatkowymi czynnikami ryzyka rozwoju cukrzycy.
Funkcja tarczycy.
Hormon wzrostu przyspiesza obwodowe przekształcanie T4 w T3, co może prowadzić do obniżenia stężenia T4 w surowicy i wzrostu stężenia T3 w surowicy. Podczas gdy stężenia obwodowe hormonów tarczycy pozostają w normie u większości zdrowych ochotników, teoretycznie możliwy jest rozwój hipotyreozu u pacjentów z postacią subkliniczną hipotyreozu. Dlatego u wszystkich pacjentów należy kontrolować funkcję tarczycy. U pacjentów z hipopitularyzmem, którzy otrzymują standardową terapię zastępczą, należy dokładnie monitorować możliwy wpływ terapii hormonem wzrostu na funkcję tarczycy.
Niedoczynność nadnerczy.
Wprowadzenie leczenia somatropiną może prowadzić do hamowania 11βHSD-1 i obniżenia stężenia kortyzolu w surowicy. U pacjentów, którzy otrzymywali somatropinę, może ujawnić się wcześniej nierozpoznana centralna (wtórna) niedoczynność nadnerczy, i mogą oni wymagać terapii zastępczej glukokortykosteroidami. Ponadto pacjenci, którzy otrzymywali terapię zastępczą glukokortykosteroidami z powodu wcześniej zdiagnozowanej niedoczynności nadnerczy, mogą wymagać zwiększenia dawek podtrzymujących lub zwiększenia dawek po rozpoczęciu leczenia somatropiną (patrz dział „Interakcje z innymi lekami i inne rodzaje interakcji”).
Stosowanie łącznie z terapią doustną estrogenami.
Jeśli kobieta stosująca somatropinę rozpoczyna doustną terapię estrogenami, może być konieczne zwiększenie dawki somatropiny w celu utrzymania stężenia IGF-1 w surowicy w granicach normy wiekowej. I odwrotnie, jeśli kobieta stosująca somatropinę przestaje stosować doustną terapię estrogenami, może być konieczne zmniejszenie dawki somatropiny, aby uniknąć nadmiaru hormonu wzrostu i/lub działań niepożądanych (patrz dział „Interakcje z innymi lekami i inne rodzaje interakcji”).
W przypadku wtórnego niedoboru hormonu wzrostu spowodowanego leczeniem chorób nowotworowych zaleca się zwracać uwagę na objawy nawrotu choroby nowotworowej. U osób, które przebyły chorobę nowotworową w dzieciństwie, donoszono o zwiększonego ryzyka rozwoju nowotworu wtórnego u pacjentów leczonych somatropiną po pierwotnym nowotworze.
Najczęstsze nowotwory wtórne u pacjentów, którzy otrzymywali radioterapię głowy z powodu pierwotnego nowotworu, to guzy wewnątrzczaszkowe, w szczególności meningiomy.
U pacjentów z zaburzeniami endokrynnymi, w szczególności z niedoborem hormonu wzrostu, może występować częstsze wypadanie główki kości udowej niż w ogólnej populacji. Dzieci, które kulawią podczas terapii somatropiną, powinny zostać poddane badaniu klinicznemu.
Łagodne wewnątrzczaszkowe nadciśnienie tętnicze.
W przypadku ciężkiego lub częstego bólu głowy, zaburzeń widzenia, nudności i/lub wymiotów zaleca się przeprowadzenie oftalmoskopii w celu wykrycia obrzęku tarczy nerwu wzrokowego. Jeśli potwierdza się obecność obrzęku tarczy nerwu wzrokowego, należy rozważyć rozpoznanie łagodnego wewnątrzczaszkowego nadciśnienia tętniczego i w razie potrzeby przerwać leczenie hormonem wzrostu. Obecnie brakuje wystarczających danych, aby sformułować zalecenia dotyczące kontynuacji terapii hormonem wzrostu u pacjentów po ustąpieniu wewnątrzczaszkowego nadciśnienia tętniczego. Po wznowieniu terapii hormonem wzrostu należy dokładnie monitorować objawy wewnątrzczaszkowego nadciśnienia tętniczego.
Białaczka.
Donoszono o przypadkach białaczki u niewielkiej liczby pacjentów z niedoborem hormonu wzrostu, niektórzy z nich otrzymywali terapię somatropiną. Jednak nie ma dowodów na zwiększoną częstość występowania białaczki u pacjentów otrzymujących hormon wzrostu, którzy nie mają predyspozycji do tej choroby.
Przeciwciała.
Jak w przypadku wszystkich leków zawierających somatropinę, u niewielkiej części pacjentów mogą powstawać przeciwciała przeciwko lekowi Genotropin®. Po podaniu leku Genotropin® przeciwciała powstają u około 1% pacjentów. Przeciwciała te charakteryzują się słabą zdolnością wiązania i nie wpływają na tempo wzrostu. W przypadku niewystarczającej odpowiedzi na leczenie (której nie można wyjaśnić innymi przyczynami) pacjentowi należy przeprowadzić test na obecność przeciwciał przeciwko somatropinie.
Pacjenci w podeszłym wieku.
Doświadczenie stosowania u pacjentów w wieku 80 lat jest ograniczone. Pacjenci w podeszłym wieku mogą być bardziej wrażliwi na działanie leku Genotropin®, a tym samym bardziej skłonni do występowania działań niepożądanych.
Stan krytyczny.
Skuteczność leku Genotropin® podczas rekonwalescencji była badana w dwóch badaniach z grupą placebo obejmujących 522 pacjentów przebywających w stanie krytycznym z powodu powikłań po operacji serca, operacji jamy brzusznej, wielokrotnych urazów lub ostrej niewydolności oddechowej. Śmiertelność wśród pacjentów, którzy otrzymywali 5,3 lub 8 mg leku Genotropin® dziennie, była wyższa niż w grupie placebo: 42% w porównaniu do 19%. Na podstawie tych informacji pacjentów tych nie należy leczyć lekiem Genotropin®. Ze względu na brak informacji o bezpieczeństwie terapii zastępczej hormonem wzrostu u pacjentów w ostrym stanie krytycznym, w tej sytuacji należy dokładnie rozważyć korzyści z kontynuacji leczenia i związane potencjalne ryzyko.
U wszystkich pacjentów, u których wystąpił inny lub podobny ostry stan krytyczny, należy rozważyć możliwą korzyść z leczenia lekiem Genotropin® i związane potencjalne ryzyko.
Zapalenie trzustki.
Chociaż zapalenie trzustki może występować rzadko, należy rozważyć możliwość jego rozwoju u każdego pacjenta leczonego somatropiną, szczególnie u dzieci, u których występuje ból brzucha.
Zespół Pradera-Williego.
Leczenie pacjentów ze zespołem Pradera-Williego należy zawsze łączyć z dietą o niskiej zawartości kalorii.
Donoszono o przypadkach śmiertelnych związanych ze stosowaniem hormonu wzrostu u dzieci ze zespołem Pradera-Williego, które miały jeden lub więcej czynników ryzyka: ciężkie otyłość (pacjenci ze stosunkiem masy ciała do wzrostu przekraczającym 200%), wcześniejsze zaburzenia oddechowe lub bezdech senny lub niezidentyfikowaną infekcję dróg oddechowych. Pacjenci z jednym lub większą liczbą wymienionych czynników mogą należeć do grupy zwiększonego ryzyka.
Przed rozpoczęciem leczenia somatropiną u pacjentów ze zespołem Pradera-Williego należy ocenić obecność objawów obturacji dróg oddechowych, bezdechu sennego lub infekcji dróg oddechowych.
Jeśli podczas oceny przepływu powietrza przez drogi oddechowe górne stwierdza się patologię, dziecko należy skierować do laryngologa w celu leczenia i usunięcia zaburzeń oddechowych przed rozpoczęciem leczenia hormonem wzrostu.
Przed rozpoczęciem terapii hormonem wzrostu należy ocenić obecność bezdechu sennego za pomocą standardowych metod polisomnografii lub nocnej oksymetrii i monitorować jego ewentualny rozwój.
Jeśli podczas leczenia somatropiną u pacjentów wystąpią objawy obturacji dróg oddechowych (w szczególności pojawienie się lub nasilenie chrapania), leczenie należy przerwać i przeprowadzić ponowne badanie laryngologiczne.
Wszystkich pacjentów ze zespołem Pradera-Williego należy monitorować w przypadku podejrzenia rozwoju bezdechu sennego.
Pacjentów należy badać pod kątem objawów infekcji dróg oddechowych, które należy rozpoznawać jak najwcześniej i aktywnie leczyć.
U wszystkich pacjentów ze zespołem Pradera-Williego należy również dokładnie monitorować masę ciała przed rozpoczęciem i w trakcie leczenia hormonem wzrostu.
U pacjentów ze zespołem Pradera-Williego często występuje skrzywienie kręgosłupa. U niektórych dzieci w związku z szybkim wzrostem skrzywienie kręgosłupa może się nasilać. W trakcie leczenia należy monitorować objawy skrzywienia kręgosłupa.
Doświadczenie długotrwałego stosowania hormonu wzrostu w leczeniu dorosłych i pacjentów ze zespołem Pradera-Williego jest ograniczone.
Dzieci urodzone małe w stosunku do czasu ciąży.
Przed rozpoczęciem leczenia dzieci urodzonych małe w stosunku do czasu ciąży należy wykluczyć możliwość wpływu innych przyczyn medycznych lub metod leczenia na zaburzenia wzrostu.
Przed rozpoczęciem leczenia dzieci urodzonych małe w stosunku do czasu ciąży zaleca się oznaczenie stężenia insuliny i glukozy we krwi na czczo i powtarzać to badanie co roku. Pacjentom z wysokim ryzykiem rozwoju cukrzycy (np. z rodziną chorego na cukrzycę, otyłością, wyraźną opornością na insulinę, czerniakiem acanthosis nigricans) należy przeprowadzić doustny test tolerancji glukozy. Jeśli rozpoznano cukrzycę, hormon wzrostu nie powinien być stosowany.
Przed rozpoczęciem leczenia dzieci urodzonych małe w stosunku do czasu ciąży zaleca się oznaczenie stężenia IGF-1 i powtarzać to badanie dwa razy w roku. Jeśli po ponownym oznaczeniu odchylenie standardowe stężenia IGF-1 przekracza +2 SD w porównaniu z normą wiekową i dojrzewaniem płciowym, należy rozważyć stosunek IGF-1/IGFBP-3 przy podejmowaniu decyzji o konieczności korekty dawki.
Doświadczenie leczenia bezpośrednio przed rozpoczęciem dojrzewania płciowego u dzieci urodzonych małe w stosunku do czasu ciąży jest ograniczone. Dlatego nie zaleca się rozpoczynania leczenia bezpośrednio przed rozpoczęciem dojrzewania płciowego. Doświadczenie leczenia pacjentów z zespołem Silvera-Russella jest ograniczone.
Osiągnięty przyrost wzrostu u dzieci urodzonych małe w stosunku do czasu ciąży w wyniku leczenia hormonem wzrostu może ulec utracie, jeśli leczenie zostanie przerwane przed osiągnięciem ostatecznego wzrostu.
Przewlekła niewydolność nerek.
W przypadku przewlekłej niewydolności nerek funkcja nerek przed rozpoczęciem leczenia powinna być niższa niż 50% normy. Aby potwierdzić objawy zaburzeń wzrostu, należy monitorować wzrost przez rok przed rozpoczęciem terapii. W tym okresie należy rozpocząć zachowawcze leczenie zaburzeń funkcji nerek (w tym kontrolę zakwaszenia, hiperparatyreoidyzmu i odżywiania) i kontynuować je podczas terapii hormonem wzrostu. Leczenie należy przerwać w przypadku przeszczepienia nerki.
Obecnie brakuje danych dotyczących osiągnięcia ostatecznego wzrostu u pacjentów z przewlekłymi zaburzeniami funkcji nerek, u których stosowano lek Genotropin®.
Zawartość sodu.
Ten lek zawiera mniej niż 1 mmol sodu (23 mg) w jednej dawce. Pacjentom przestrzegającym diety o ograniczonej zawartości soli można przekazać, że lek ten może być uważany za niezawierający sodu.
Specjalne ostrzeżenia dotyczące postępowania z lekiem i utylizacji resztek.
Roztwór przygotowuje się przez skręcanie sekcji wstępnie napełnionej strzykawki do mieszania rozpuszczalnika z proszkiem w dwukomorowym kartuszu. Proszek rozpuszcza się ostrożnie przekładając go naprzód i wstecz. Nie wolno intensywnie wstrząsać. Może to spowodować denaturację substancji czynnej. Odtworzony roztwór do wstrzykiwań powinien być bezbarwny lub lekko opalescencyjny. Odtworzony roztwór do wstrzykiwań należy przejrzeć przed zastosowaniem i należy stosować wyłącznie klarowny roztwór bez cząsteczek.
Każdy nieużywany lek lub odpady należy utylizować zgodnie z obowiązującymi przepisami. Nigdy nie wolno ponownie napełniać pustych wstępnie napełnionych strzykawek; należy je utylizować.
Stosowanie w okresie ciąży lub karmienia piersią.
Ciąża.
Nie istnieje wystarczająca ilość danych uzyskanych w badaniach na zwierzętach dotyczących wpływu leku Genotropin® na ciążę, rozwój embrionalny, poród lub rozwój poporodowy (patrz podrozdział „Farmakokinetyka”). Brakuje badań klinicznych dotyczących stosowania leku w okresie ciąży. Dlatego leków zawierających somatropinę nie zaleca się przepisywać kobietom w ciąży ani kobietom w wieku rozrodczym, które nie stosują środków antykoncepcyjnych.
Karmienie piersią.
Nie przeprowadzono badań klinicznych dotyczących stosowania leków zawierających somatropinę u kobiet karmiących piersią. Nie wiadomo, czy somatropina przenika do mleka matki, ale wchłanianie całkowitego białka z przewodu pokarmowego niemowlęcia jest bardzo mało prawdopodobne. Dlatego leki zawierające somatropinę należy stosować z ostrożnością u kobiet karmiących piersią.
Wpływ na zdolność prowadzenia pojazdów lub obsługiwania maszyn.
Genotropin® nie wpływa na szybkość reakcji podczas prowadzenia pojazdów lub pracy z innymi maszynami.
Sposób stosowania i dawki.
Dawkowanie i schemat stosowania należy dobrać indywidualnie.
Wstrzyknięcie należy wykonywać podskórnie, zmieniając miejsce wstrzykiwania w celu zapobiegania lipodystrofii.
Opóźnienie wzrostu spowodowane niedoborem hormonu wzrostu u dzieci. Zwykle zalecana dawka wynosi 0,025–0,035 mg/kg masy ciała na dobę lub 0,7–1,0 mg/m² powierzchni ciała na dobę. Istnieje doświadczenie z zastosowania nawet wyższych dawek.
Jeśli niedobór hormonu wzrostu wystąpił w dzieciństwie i utrzymuje się w okresie dojrzewania, leczenie należy kontynuować aż do osiągnięcia pełnego rozwoju somatycznego (tzn. budowy ciała, masy kości). W okresie przejściowym jednym z celów terapeutycznych była kontrola osiągnięcia normalnego szczytu masy kości, określonego jako wartość wskaźnika T > –1 (tzn. uśredniony szczyt masy kości dorosłego, mierzony metodą dwuenergetycznej rentgenowskiej absorpcjometrii, z uwzględnieniem płci i pochodzenia etnicznego pacjenta). Instrukcje dotyczące dawkowania u dorosłych opisano poniżej.
Zespół Pradera-Williego, w celu poprawy wzrostu i budowy ciała u dzieci. Zwykle należy podawać 0,035 mg/kg masy ciała na dobę (1,0 mg/m² powierzchni ciała na dobę). Nie należy przekraczać dziennej dawki 2,7 mg. Leczenia nie należy stosować u dzieci z prędkością wzrostu mniejszą niż 1 cm na rok oraz w wieku, w którym rozpoczyna się zamknięcie stref wzrostowych w trzonach kości.
Zespół Turnera. Zalecana dawka wynosi 0,045–0,050 mg/kg masy ciała na dobę lub 1,4 mg/m² powierzchni ciała na dobę.
Idiopatyczna niskorosłość. Zalecana dawka wynosi do 0,47 mg/kg masy ciała w ciągu tygodnia, tj. do 0,067 mg/kg masy ciała na dobę.
Opóźnienie wzrostu u pacjentów z przewlekłą niewydolnością nerek. Zalecana dawka wynosi 0,045–0,050 mg/kg masy ciała na dobę (1,4 mg/m² powierzchni ciała na dobę). Niewystarczająca prędkość wzrostu może wymagać podania wyższej dawki. Korekta dawki może być konieczna po 6 miesiącach leczenia.
Opóźnienie wzrostu u małych dzieci urodzonych z małą masą ciała dla wieku ciążenia. Zwykle zalecana dawka wynosi 0,035 mg/kg masy ciała na dobę (1 mg/m² powierzchni ciała na dobę) aż do osiągnięcia ostatecznego wzrostu (patrz sekcja „Właściwości farmakologiczne”).
Leczenie należy przerwać po pierwszym roku, jeśli wartość odchylenia standardowego dla prędkości wzrostu jest mniejsza niż +1. Leczenie należy przerwać, jeśli prędkość wzrostu jest mniejsza niż 2 cm na rok i (w razie potrzeby potwierdzenia) wiek kostny przekracza 14 lat u dziewcząt lub 16 lat u chłopców, co odpowiada wiekowi zamknięcia stref wzrostowych w trzonach kości.
Tabela 1
Zalecenia dotyczące dawkowania u dzieci
| Wskazania |
mg/kg masy ciała |
mg/m2 powierzchni ciała |
| dawka dobową |
||
| Deficyt hormonu wzrostu |
0,025–0,035 |
0,7–1,0 |
| Zespół Pradera-Williego |
0,035 |
1,0 |
| Zespół Turnera |
0,045–0,050 |
1,4 |
| Przewlekła niewydolność nerek |
0,045–0,050 |
1,4 |
| Opóźnienie wzrostu u dzieci urodzonych z małą masą ciała dla wieku ciążowego |
0,035 |
1,0 |
| Idiopatyczna karłowatość |
do 0,067 |
do 2 |
Pacjenci dorośli z niedoborem hormonu wzrostu. U pacjentów kontynuujących leczenie hormonem wzrostu po wystąpieniu niedoboru hormonu wzrostu w dzieciństwie, zalecana dawka wynosi 0,2–0,5 mg na dobę. Dawkę należy stopniowo zwiększać lub zmniejszać w zależności od indywidualnych potrzeb pacjenta, które określa stężenie IGF-1.
U pacjentów, u których niedobór hormonu wzrostu wystąpił w wieku dorosłym, leczenie należy rozpocząć od niskiej dawki: 0,15–0,3 mg na dobę. Dawkę należy stopniowo zwiększać w zależności od indywidualnych potrzeb pacjenta, które określa stężenie IGF-1.
W obu przypadkach celem leczenia jest osiągnięcie stężenia IGF-1 w granicach wartości odchylenia standardowego od średniej normy wiekowej, które wynosi 2. Pacjentom z początkowym stężeniem IGF-1 w normie należy podawać hormon wzrostu w dawce, która zwiększy stężenie IGF-1 do górnej granicy normy, ale nie więcej niż o wartość odchylenia standardowego, która wynosi 2. Dawkę należy dobrać również z uwzględnieniem efektu klinicznego i działań niepożądanych. Wiadomo, że u niektórych pacjentów z niedoborem hormonu wzrostu, pomimo odpowiedniej odpowiedzi klinicznej, poziomy IGF-1 nie ulegają normalizacji; u takich pacjentów nie należy zwiększać dawki. Dawkę utrzymania rzadko przekracza 1,0 mg na dobę. U kobiet może być konieczne stosowanie wyższych dawek niż u mężczyzn, ponieważ mężczyźni z czasem wykazują większą wrażliwość na IGF-1. Oznacza to, że u kobiet, szczególnie tych przyjmujących doustną terapię zastępczą estrogenami, istnieje ryzyko niewystarczającego efektu klinicznego, a u mężczyzn – nadmiernego. Dlatego dokładność dawki hormonu wzrostu należy kontrolować co 6 miesięcy. Ponieważ z wiekiem występuje fizjologiczne zmniejszanie się produkcji hormonu wzrostu, można zmniejszyć dawkę leku. U pacjentów w wieku powyżej 60 lat leczenie należy rozpocząć od dawki 0,1–0,2 mg na dobę; dawkę należy powoli zwiększać w zależności od indywidualnych potrzeb pacjenta. Należy stosować minimalne dawki skuteczne. Dawkę utrzymania u tych pacjentów rzadko przekracza 0,5 mg na dobę.
Informacja dla pacjenta
Instrukcja stosowania wstępnie napełnionej ruchki do wstrzykiwań o dawkowaniu 5,3 mg lub 12 mg
Lek Genotropin® stosuje się za pomocą jednorazowej wielodawkowej wstępnie napełnionej ruchki (szprycety-ruchki „GoQuick”) do wstrzykiwań, zawierającej 5,3 mg lub 12 mg somatropiny. Za pomocą ruchki zawierającej 5,3 mg można podawać dawki od 0,1 mg do 1,5 mg leku. Każde obrócenie czarnego pierścienia zmienia dawkę o 0,05 mg. Za pomocą ruchki zawierającej 12 mg można podawać dawki od 0,3 mg do 4,5 mg leku. Każde obrócenie czarnego pierścienia zmienia dawkę o 0,15 mg. Lek w ruchce mieszany jest tylko raz, przed rozpoczęciem stosowania nowej ruchki. Nie trzeba wymieniać kartuszy. Po wyczerpaniu leku w ruchce rozpoczyna się stosowanie leku z nowej ruchki. Ruchka posiada funkcję zapamiętania dawki. Dawkę ustawia się raz na początku stosowania. Po tym czasie ruchka umożliwia dozowanie tej samej ustalonej dawki przy każdej iniekcji. Zapobiega to przekroczeniu ustalonej dawki.
Lek Genotropin® w dawkowaniu 5,3 mg jest dostarczany w wstępnie napełnionej ruchce z przyciskiem do wstrzykiwania, logo i oznaczeniami w kolorze niebieskim, w dawkowaniu 12 mg – w kolorze purpurowym.
Ważna informacja
- Nie mieszać proszku i cieczy w wstępnie napełnionej ruchce, jeśli ruchka nie ma założonej igły.
- Nie przechowywać ruchki z założoną igłą. Lek może wyciekać z ruchki, a w kartuszu mogą powstawać pęcherzyki powietrza. Zawsze należy zdjąć igłę i założyć pokrywkę lub osłonę ochronną igły przed przechowywaniem.
- Należy zachować ostrożność, aby nie upuścić ruchki. Jeśli upuściłeś ruchkę i jakakolwiek jej część jest złamana lub uszkodzona, nie należy jej używać. Skontaktuj się z lekarzem lub pielęgniarką w celu uzyskania innej ruchki. Jeśli upuściłeś ruchkę, ale nie jest ona uszkodzona ani złamana, należy wykonać przygotowanie do pracy, zgodnie z opisem w Krok 6 (patrz podrozdział „Nastawianie i stosowanie nowej ruchki GoQuick” poniżej).
- Oczyść ruchkę wilgotną ściereczką. Nie wkładaj ruchki do wody.
- Zawsze stosuj nową igłę przy każdej iniekcji. Nie pozwalaj nikomu korzystać z Twoich igieł do ruchki.
- Skala pozostałej objętości wzdłuż bocznej strony uchwytu kartusza służy jako wskazówka do wyświetlania objętości pozostałego Genotropinu® w Twojej ruchce.
Przechowywanie i utylizacja
- Nie stosuj ruchki po upływie terminu ważności.
- Po 28 dniach od momentu zmieszania należy wyrzucić (utylizować) ruchkę, nawet jeśli w leku pozostał pewien nadmiar leku.
- Przy wyrzucaniu (utylizacji) ruchki należy przestrzegać obowiązujących przepisów dotyczących ochrony zdrowia i bezpieczeństwa. Jeśli nie jesteś pewien, co należy zrobić, skontaktuj się z lekarzem lub pielęgniarką.
Poniżej znajduje się szczegółowy opis kolejnych kroków stosowania ruchki do dawkowania 5,3 mg. Dla dawkowania 12 mg należy wykonać analogiczne czynności. Wygląd zewnętrzny i kompletacja igły mogą się różnić.
Części szprycety-ruchki „GoQuick”
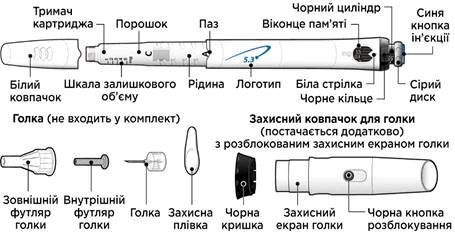
Igły do ruchki nie są dołączone do opakowania. Będziesz musiał zakupić w aptece igły o długości do 8 mm.
- Igły do stosowania z ruchką „GoQuick”:
- 31G lub 32G — „Becton Dickinson and Company”
- 31G lub 32G — „Novo Nordisk®”
- 32,5G lub 34G — „Terumo”
Nastawianie i stosowanie nowej ruchki „GoQuick”
Krok 1. Przygotowanie
- Wymyj i osusz ręce.
- Przygotuj poniższe przedmioty na czystej, równej powierzchni:
- Nowa ruchka.
- Nowa igła (nie dołączona).
- Odpowiedni pojemnik na przedmioty ostry (nie dołączony).
- Sprawdź termin ważności na etykiecie ruchki. Nie stosuj ruchki, jeśli termin ważności upłynął.
Krok 2. Wybierz miejsce iniekcji
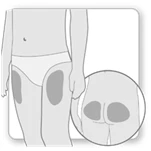
- Wybierz i oczyść miejsce iniekcji zgodnie z zaleceniami lekarza lub pielęgniarki. Za każdym razem, gdy wykonujesz sobie iniekcję, wybieraj inne miejsce. Każdą nową iniekcję należy wykonywać w odległości co najmniej 2 cm od miejsca, które było używane wcześniej.
- Unikaj obszarów z kośćmi, siniakami lub zaczerwienieniem, bolesnych lub twardych obszarów, a także obszarów skóry z bliznami lub chorobami skóry.
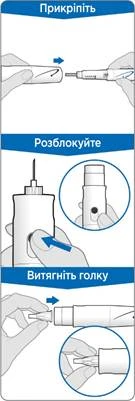
Krok 3. Załóż nową igłę
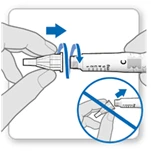
- Zdejmij białą pokrywkę z ruchki, trzymając ją prosto.
- Weź nową igłę i usuń ochronną folię.
- Delikatnie naciśnij, a następnie dokręć igłę do ruchki. Nie dokręcaj zbyt mocno.
Uwaga. Bądź ostrożny: nie zakładaj igły pod kątem — może to prowadzić do wycieku ruchki.
- Zostaw oba osłonki na igle.
Krok 4. Wymieszaj lek
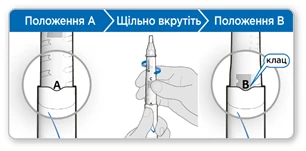
- Trzymaj ruchkę końcem igły do góry, tak aby oznaczenie A znajdowało się przed Twoją twarzą.
- Mocno dokręć uchwyt kartusza do ruchki, aż zablokuje się przy oznaczeniu B w rowku.
- Delikatnie przechylaj ruchkę z boku na bok, aby proszek całkowicie się rozpuścił. Nie wstrząsaj. Wstrząsanie może prowadzić do zmiany hormonu wzrostu.
- Upewnij się, że ciecz w kartuszu jest przejrzysta i cały proszek się rozpuścił.
- Jeśli ciecz jest mętna lub widzisz proszek, delikatnie przechylaj ruchkę z boku na bok jeszcze kilka razy.
- Jeśli ciecz nadal jest mętna lub widzisz proszek, nie stosuj tej ruchki, a weź nową ruchkę.
Krok 5. Usuń powietrze z ruchki
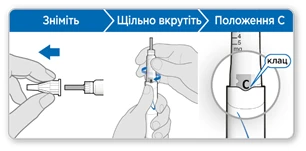
- Zdejmij zewnętrzną osłonkę igły. Zostaw ją, aby zdjąć igłę po iniekcji.
- Zostaw wewnętrzną osłonkę igły na miejscu.
Uwaga: Po zdjęciu zewnętrznej osłonki powinieneś zobaczyć wewnętrzną osłonkę igły. Jeśli jej nie widzisz, spróbuj ponownie założyć igłę.
- Trzymaj ruchkę końcem igły do góry.
- Delikatnie postukaj po uchwycie kartusza, aby przesunąć pęcherzyki powietrza do góry.
- Mocno dokręć uchwyt kartusza do ruchki, aż zablokuje się przy oznaczeniu C w rowku.
- Wokół wewnętrznej osłonki igły może pojawić się niewielka ilość cieczy. Jest to zjawisko normalne.
Krok 6. Napełnij ruchkę
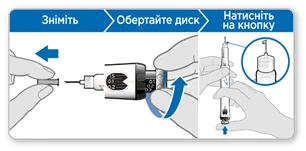
Napełnienie usuwa pozostałe powietrze, wypychając niewielką ilość cieczy z ruchki. Dawkę napełnienia stanowi 0,1 mg dla ruchki zawierającej 5,3 mg somatropiny oraz 0,3 mg dla ruchki zawierającej 12 mg somatropiny i różni się od dawki przepisanej przez lekarza lub pielęgniarkę. Wykonaj napełnienie ruchki tylko podczas pierwszego użycia.
Zdejmij wewnętrzną osłonkę igły i wyrzuć ją.
Uwaga! Nie dotykaj igły, aby uniknąć ukłucia.
- Upewnij się, że w okienku pamięci ustawiono 0,1 mg dla ruchki zawierającej 5,3 mg somatropiny lub 0,3 mg dla ruchki zawierającej 12 mg somatropiny.
- Obracaj szary dysk w kierunku strzałek, aż przestanie klikać.
- Trzymaj ruchkę tak, aby igła była skierowana prosto do góry.
- Naciśnij niebieski przycisk iniekcji do oporu.
- Sprawdź, czy pojawiła się ciecz na końcu igły. Jeśli ciecz się pojawiła, Twoja ruchka jest napełniona.
- Jeśli ciecz się nie pojawiła, powtórz kroki napełniania jeszcze dwa razy.
- Jeśli ciecz nadal się nie pojawia, nie stosuj ruchki. Skontaktuj się z lekarzem lub pielęgniarką w celu uzyskania porady.
Krok 7. Ustaw i dostrój swoją dawkę
Podczas pierwszego użycia ruchki ustawisz dawkę przepisaną przez lekarza lub pielęgniarkę.
Nie musisz ponownie ustawiać dawki, dopóki nie zaczniesz używać nowej ruchki lub dopóki lekarz lub pielęgniarka nie powie Ci inaczej.
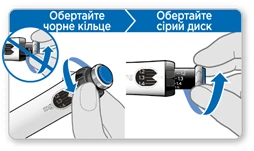
- Obracaj czarny pierścień przeciwnie do ruchu wskazówek zegara, aż Twoja dawka wyrówna się z białą strzałką w okienku pamięci. Bądź ostrożny i nie obracaj szarego dysku.
- Jeśli obróciłeś pierścień tak, że Twoja dawka przekroczyła białą strzałkę, obróć czarny pierścień z powrotem, aby ustawić właściwą dawkę.
Uwaga: jeśli nie możesz obrócić czarnego pierścienia, naciskaj niebieski przycisk iniekcji, aż przestanie klikać. Następnie spróbuj ponownie ustawić dawkę. Zwróć uwagę, że ciecz będzie wyciekać z igły.
- Obracaj szary dysk w kierunku strzałek, aż przestanie klikać.
Krok 8. Sprawdź swoją dawkę

Twoja dawka na czarnym cylindrze powinna współgrać z białą strzałką.
- Sprawdź, czy dawka ustawiona na czarnym cylindrze pokrywa się z dawką ustawioną w okienku pamięci.
- Jeśli dawki się pokrywają, Twoja ruchka jest gotowa do wykonania iniekcji.
- Jeśli dawki się nie pokrywają, upewnij się, że obracasz szary dysk w kierunku strzałek, aż przestanie klikać.
Krok 9. Wykonaj iniekcję
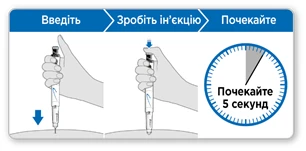
- Trzymaj ruchkę nad miejscem iniekcji.
- Wprowadź igłę prosto w skórę.
- Naciskaj na przycisk iniekcji w dół, aż przestanie klikać.
- Poczekaj 5 sekund, aby upewnić się, że wprowadzono całą dawkę. Kontynuuj lekko naciskając na niebieski przycisk iniekcji, gdy liczysz.
- Po 5 sekundach wyciągnij igłę ze skóry.
Uwaga: jeśli zobaczysz kroplę cieczy w miejscu iniekcji lub na końcu igły, podczas następnej iniekcji spróbuj dłużej naciskać na niebieski przycisk, zanim wyciągniesz igłę ze skóry.
Krok 10. Wyjmij igłę
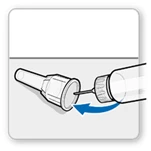
- Delikatnie przykryj igłę zewnętrzną osłonką igły.
Uwaga! Nie dotykaj igły, aby uniknąć ukłucia.
- Użyj osłonki igły, aby odkręcić igłę.
- Wyrzuć (utylizuj) igłę do odpowiedniego pojemnika na przedmioty ostre.
- Załóż białą pokrywkę na ruchkę.
- Przechowuj ruchkę w lodówce do następnej iniekcji.
Regularne (codzienne) stosowanie ruchki „GoQuick”
Krok 1. Przygotowanie
- Wymyj i osusz ręce.
- Przygotuj poniższe przedmioty na czystej, równej powierzchni:
- Ruchka „GoQuick” z wymieszanym roztworem
- Nowa igła (nie dołączona)
- Odpowiedni pojemnik na przedmioty ostre (nie dołączony).
- Sprawdź termin ważności na etykiecie ruchki. Nie stosuj ruchki, jeśli termin ważności upłynął.
- Nie stosuj ruchki, jeśli minęło 28 dni od pierwszego użycia.
Krok 2. Wybierz miejsce iniekcji
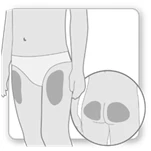
- Wybierz i oczyść miejsce iniekcji zgodnie z zaleceniami lekarza lub pielęgniarki. Za każdym razem, gdy wykonujesz sobie iniekcję, wybieraj inne miejsce. Każdą nową iniekcję należy wykonywać w odległości co najmniej 2 cm od miejsca, które było używane wcześniej.
- Unikaj obszarów z kośćmi, siniakami lub zaczerwienieniem, bolesnych lub twardych obszarów, a także obszarów skóry z bliznami lub chorobami skóry.
Krok 3. Załóż nową igłę
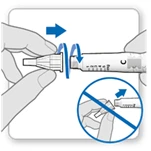
- Zdejmij białą pokrywkę z ruchki, trzymając ją prosto.
- Weź nową igłę i usuń ochronną folię.
- Delikatnie naciśnij, a następnie dokręć igłę do ruchki. Nie dokręcaj zbyt mocno.
Uwaga. Bądź ostrożny: nie zakładaj igły pod kątem — może to prowadzić do wycieku ruchki.
- Zdejmij obie osłonki igły.
- Zostaw zewnętrzną osłonkę igły, aby zdjąć ją po iniekcji.
Krok 4. Dostrój swoją dawkę
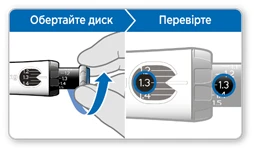
- Obracaj szary dysk w kierunku strzałek, aż przestanie klikać.
- Twoja dawka na czarnym cylindrze powinna współgrać z białą strzałką.
- Sprawdź, czy dawka ustawiona na czarnym cylindrze pokrywa się z dawką ustawioną w okienku pamięci.
- Jeśli dawki się pokrywają, Twoja ruchka jest gotowa do wykonania iniekcji.
Uwaga: jeśli dostrajana dawka jest mniejsza, oznacza to, że w Twojej ruchce nie ma wystarczającej dawki Genotropinu®.
Postępuj zgodnie z wskazówkami lekarza lub pielęgniarki, jeśli w Twojej ruchce nie ma pełnej dawki. Albo skontaktuj się z lekarzem lub pielęgniarką w celu uzyskania porady.
Krok 5. Wykonaj iniekcję
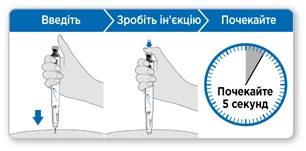
- Trzymaj ruchkę nad miejscem iniekcji.
- Wprowadź igłę prosto w skórę.
- Naciskaj na niebieski przycisk iniekcji w dół, aż przestanie klikać.
- Poczekaj 5 sekund, aby upewnić się, że wprowadzono całą dawkę. Kontynuuj lekko naciskając na niebieski przycisk iniekcji, gdy liczysz.
- Po 5 sekundach wyciągnij igłę ze skóry.
Uwaga: jeśli zobaczysz kroplę cieczy w miejscu iniekcji lub na końcu igły, podczas następnej iniekcji spróbuj dłużej naciskać na niebieski przycisk, zanim wyciągniesz igłę ze skóry.
Krok 6. Wyjmij igłę
- Delikatnie przykryj igłę zewnętrzną osłonką igły.
Uwaga! Nie dotykaj igły, aby uniknąć ukłucia.
- Użyj osłonki igły, aby odkręcić igłę.
- Wyrzuć (utylizuj) igłę do odpowiedniego pojemnika na przedmioty ostre.
- Załóż białą pokrywkę na ruchkę.
- Przechowuj ruchkę w lodówce do następnej iniekcji.
Stosowanie osłony ochronnej igły (opcjonalnie)
Osłona ochronna igły to dodatkowy element, który nie jest dołączony do opakowania, służący do zasłonięcia igły podczas iniekcji.
Ustaw osłonę ochronną igły:
Przyłącz osłonę ochronną igły po Krok 5 (patrz podrozdział „Nastawianie i stosowanie nowej ruchki GoQuick” powyżej), aby uniknąć zaklinowania igły.
- Zdejmij czarną pokrywkę z osłony ochronnej igły.
- Jeśli osłona ochronna igły wysuwa się, włóż ją z powrotem do osłony ochronnej igły, aż zablokuje się.
- Wyrównaj logo na osłonie ochronnej igły z logiem na ruchce. Delikatnie załóż osłonę ochronną igły na ruchkę, aż zablokuje się.
- Po Krok 6 (patrz podrozdział „Nastawianie i stosowanie nowej ruchki GoQuick” powyżej) naciśnij czarny przycisk, aby odblokować osłonę ochronną osłony ochronnej igły.
- Postępuj zgodnie z wskazówkami podanymi w Krok 7 (patrz podrozdział „Nastawianie i stosowanie nowej ruchki GoQuick” powyżej).
Aby wyjąć igłę z założoną osłoną ochronną igły:
- Włóż zewnętrzną osłonkę igły na koniec osłony ochronnej igły.
- Za pomocą zewnętrznej osłonki igły przesuń osłonę ochronną igły, aż zablokuje się.
- Za pomocą osłonki igły odkręć igłę i wyrzuć (utylizuj) ją do odpowiedniego pojemnika na przedmioty ostre.
- Zostaw osłonę ochronną igły na ruchce.
- Załóż czarną pokrywkę na osłonę ochronną igły. Przechowuj ruchkę w lodówce.
Aby zdjąć osłonę ochronną igły:
- Najpierw wyjmij igłę, a następnie delikatnie zdjąć osłonę ochronną igły z ruchki.
- Nie wyrzucaj osłony ochronnej igły. Może ona być ponownie użyta z następną ruchką.
Dzieci.
Lek można stosować w praktyce pediatrycznej.
Przedawkowanie.
Objawy. Ostra przedawkowanie może początkowo prowadzić do hipoglikemii, a następnie do hiperglikemii.
Przedawkowanie długotrwałe może prowadzić do objawów i objawów odpowiadających znanym efektom nadmiaru hormonu wzrostu człowieka.
Działania niepożądane.
U pacjentów z niedoborem hormonu wzrostu charakterystyczny jest niedobór płynu pozakomórkowego. Po rozpoczęciu leczenia somatropiną następuje szybka kompensacja tego niedoboru płynu. U dorosłych pacjentów często występują następujące działania niepożądane związane z zatrzymaniem płynu: obrzęki obwodowe, obrzęk twarzy, sztywność kończyn, artralgia, mialgia i parestezje. Ogólnie te działania niepożądane są łagodne lub umiarkowane, pojawiają się w pierwszych miesiącach leczenia i ustępują spontanicznie lub po zmniejszeniu dawki.
Częstość występowania tych działań niepożądanych zależy od dawki leku, wieku pacjenta oraz prawdopodobnie jest odwrotnie proporcjonalna do wieku, w którym wystąpił niedobór hormonu wzrostu. U dzieci takie działania niepożądane pojawiają się rzadko.
Genotropin® powoduje powstawanie przeciwciał u około 1% pacjentów. Przeciwciała te charakteryzują się słabym powinowactwem wiązania, a ich powstawanie nie prowadziło do żadnych zmian klinicznych (patrz sekcja „Szczególne ostrzeżenia i środki ostrożności podczas stosowania”).
Lista podejrzewanych działań niepożądanych.
Poniżej przedstawiono listę działań niepożądanych według klasyfikacji układowo-organowej pod względem częstości występowania u dzieci i dorosłych: bardzo często (≥1/10); często (od ≥1/100 do <1/10); rzadko (od ≥1/1000 do <1/100); bardzo rzadko (od ≥1/10000 do <1/1000); bardzo rzadko (<1/10000); częstość nieznana (nie można ustalić na podstawie dostępnych danych).
Dobroczynne, złośliwe i nieokreślone nowotwory (w tym torbiele i polipy). Rzadko: białaczka† (dzieci).
Zaburzenia metabolizmu i przemiany materii. Częstość nieznana: cukrzyca typu 2 (dorośli i dzieci).
Zaburzenia ze strony układu nerwowego. Często: parestezje* (dorośli), zespół cieśni nadgarstka (dorośli); rzadko: doboczną nadciśnienie wewnątrzczaszkowe (dzieci), parestezje* (dzieci); częstość nieznana: doboczną nadciśnienie wewnątrzczaszkowe (dorośli), ból głowy (dorośli i dzieci).
Zaburzenia ze strony skóry i tkanek podskórnych. Rzadko: wysypka**, świąd**, pokrzywka** (dzieci); częstość nieznana: wysypka**, świąd**, pokrzywka** (dorośli).
Zaburzenia ze strony układu mięśniowo-szkieletowego i tkanki łącznej. Bardzo często: artralgia* (dorośli); często: mialgia* (dorośli), sztywność kończyn* (dorośli), artralgia* (dzieci); rzadko: mialgia* (dzieci); częstość nieznana: sztywność kończyn* (dzieci).
Zaburzenia ze strony układu rozrodczego i gruczołów mlekowych. Rzadko: ginekomastia (dorośli i dzieci).
Zaburzenia ogólne i reakcje w miejscu wstrzyknięcia. Bardzo często: obrzęki obwodowe* (dorośli), reakcje w miejscu wstrzyknięcia$ (dzieci); rzadko: obrzęki obwodowe* (dzieci); częstość nieznana: obrzęk twarzy* (dorośli i dzieci), reakcje w miejscu wstrzyknięcia$ (dorośli).
Badania. Częstość nieznana: obniżenie stężenia kortyzolu we krwi‡ (dorośli i dzieci).
* Ogólnie te działania niepożądane są łagodne lub umiarkowane, pojawiają się w pierwszych miesiącach leczenia i ustępują spontanicznie lub po zmniejszeniu dawki. Częstość tych działań niepożądanych zależy od dawki leku i wieku pacjenta. Prawdopodobnie częstość jest odwrotnie proporcjonalna do wieku, w którym wystąpił niedobór hormonu wzrostu.
** Działania niepożądane zidentyfikowane w badaniach pozarejestrowych.
$ U dzieci zgłaszano krótkotrwałe reakcje w miejscu wstrzyknięcia.
‡ Znaczenie kliniczne nieznane.
† Zgłoszono wystąpienie tej reakcji u dzieci z niedoborem hormonu wzrostu leczonych somatropiną. Prawdopodobnie częstość jej występowania nie różni się od częstości u dzieci bez niedoboru hormonu wzrostu.
Obniżenie stężenia kortyzolu w surowicy krwi.
Zgłaszano, że somatropina obniża stężenie kortyzolu w surowicy krwi, prawdopodobnie poprzez wpływ na białka transportowe lub zwiększenie klirensu wątrobowego. Znaczenie kliniczne tych danych może być ograniczone. Mimo to, należy zoptymalizować leczenie zastępcze kortykosteroidami przed rozpoczęciem terapii lekiem Genotropin®.
Zespół Pradera–Williego.
W okresie pozarejestrowym zgłaszano pojedyncze przypadki nagłej śmierci u pacjentów z zespołem Pradera–Williego leczonych somatropiną, jednak nie udowodniono związku przyczynowego z leczeniem.
Białaczka.
U dzieci z niedoborem hormonu wzrostu, z których niektóre otrzymywały leczenie somatropiną, zgłaszano przypadki białaczki, obserwowane również w okresie pozarejestrowym. Nie ma jednak dowodów na zwiększone ryzyko rozwoju białaczki w przypadku braku czynników sprzyjających, takich jak napromienienie mózgu lub głowy.
Wypuknięcie głowy kości udowej lub choroba Legga–Calvégo–Perthesa.
Zgłaszano wypuknięcie głowy kości udowej oraz chorobę Legga–Calvégo–Perthesa u dzieci leczonych hormonem wzrostu. Wypuknięcie głowy kości udowej występuje najczęściej przy zaburzeniach endokrynologicznych, a choroba Legga–Calvégo–Perthesa najczęściej obserwowana jest przy niskim wzroście. Nie wiadomo jednak, czy te dwie patologie występują częściej podczas leczenia somatropiną. Diagnozę tę należy rozważyć u dziecka z dolegliwościami lub bólem w okolicy uda lub kolana.
Inne działania niepożądane związane z lekiem.
Inne działania niepożądane, które mogą być uważane za efekty klasy leków somatropiny, obejmują m.in. możliwą hiperglikemię spowodowaną zmniejszoną wrażliwością na insulinę, obniżenie stężenia wolnego tyroksyny oraz łagodne nadciśnienie wewnątrzczaszkowe.
Zgłaszanie podejrzewanych działań niepożądanych.
Zgłaszanie podejrzewanych działań niepożądanych po rejestracji leku ma istotne znaczenie. Umożliwia to ciągłe monitorowanie stosunku korzyści do ryzyka związanego z zastosowaniem leku.
Okres ważności. 3 lata.
Warunki przechowywania. Przechowywać w temperaturze 2–8 °C w oryginalnym opakowaniu w celu ochrony przed światłem.
Odtworzony roztwór może być przechowywany w temperaturze 2–8 °C przez 28 dni w oryginalnym opakowaniu w celu ochrony przed światłem.
Przechowywać w miejscu niedostępnym dla dzieci.
Niezgodność. Ponieważ nie przeprowadzono badań niezgodności, zabrania się mieszania tego leku z innymi lekami.
Opakowanie. 5,3 mg: 1 wstępnie napełnione pióro zawierające 1 dwukomorowy kartusz (przednia komora z proszkiem, tylna komora z rozpuszczalnikiem) w pudełku kartonowym.
12 mg: 1 lub 5 wstępnie napełnionych piór, z których każde zawiera 1 dwukomorowy kartusz (przednia komora z proszkiem, tylna komora z rozpuszczalnikiem), w pudełku kartonowym.
Kategoria wydania. Na receptę.
Producent.
Pfizer Manufacturing Belgium NV.
Adres producenta i miejsce prowadzenia działalności.
Rijksweg 12, Puurs-Sint-Amands, 2870, Belgia.