Fazlodeks
Ukraina
Spis treści
INSTRUKCJA dotycz¹ca stosowania leczniczego leku Fazlodeks (FASLODEX®)
Sk³ad:
substancja czynna: 1 strzykawka wype³niona wstêpnie (5 ml) zawiera fulwestrant 250 mg;
substancje pomocnicze: etanol 96%, alkohol benzylowy, benzyl benzoan, olej rycynowy.
Postaæ farmaceutyczna. Roztwór do wstrzykiwañ.
G³ówne cechy fizykochemiczne: przejrzysta, od bezbarwnej do ¿ó³tej, lepka ciecz.
Grupa farmakoterapeutyczna. Antagoniści hormonów i œrodki analogiczne. rodki antyestrogenowe. Kod ATC L02B A03.
Właściwości farmakologiczne.
Farmakodynamika.
Mechanizm działania i efekty farmakodynamiczne
Fulwestrant jest konkurencyjnym antagonistą receptorów estrogenowych (ER), z powinowactwem porównywalnym do estradiolu. Fulwestrant blokuje działanie troficzne estrogenów, nie wykazując częściowej aktywności agonistycznej (podobnej do estrogenów). Mechanizm działania wiąże się z negatywną regulacją poziomu białek receptorów estrogenowych.
Badania kliniczne przeprowadzone u kobiet w okresie po menopauzie z pierwotnym rakiem piersi wykazały, że fulwestrant w porównaniu z placebo istotnie zmniejsza poziom białek ER w guzach ER- dodatnich. Zaobserwowano również istotne zmniejszenie ekspresji receptorów progesteronowych, co jest zgodne z brakiem efektów charakterystycznych dla agonistów estrogenów. Wykazano ponadto, że fulwestrant w dawce 500 mg w większym stopniu niż fulwestrant w dawce 250 mg hamuje poziom ER oraz marker proliferacji Ki67 w guzach piersi podczas leczenia neoadjuwantowego kobiet w okresie po menopauzie.
Bezpieczeństwo i skuteczność kliniczna leku w zaawansowanym raku piersi
Leczenie monoterapią
Przeprowadzono badanie kliniczne fazy 3 z udziałem 736 kobiet w okresie po menopauzie z przerostem raka piersi, u których doszło do nawrotu choroby podczas lub po adiuwantnej terapii endokrynnej albo do progresji choroby po terapii endokrynnej z powodu zaawansowanego schorzenia. W badaniu wzięło udział 423 pacjentki, u których choroba nawróciła lub postępowała podczas terapii antyestrogenowej (podgrupa AE), oraz 313 pacjentek, u których choroba nawróciła lub postępowała podczas leczenia inhibitorem aromatazy (podgrupa AI). Badanie to porównywało skuteczność i bezpieczeństwo stosowania leku Fazlodeks w dawce 500 mg (n = 362) i w dawce 250 mg (n = 374). Głównym punktem końcowym była przeżycie wolne od progresji (PFS); kluczowe wtórne punkty końcowe skuteczności obejmowały częstość odpowiedzi obiektywnej (ORR), częstość skuteczności klinicznej (CBR) oraz przeżycie ogólne (OS). Wyniki badania CONFIRM dotyczące skuteczności ujęto poniżej w tabeli 1.
Tabela 1. Podsumowanie wyników analizy głównego punktu końcowego skuteczności (PFS) i kluczowych wtórnych punktów końcowych skuteczności w badaniu CONFIRM
| Zmienna |
Typ oceny; porównanie leczenia |
Fazlodeks 500 mg (n = 362) |
Fazlodeks 250 mg (n = 374) |
Porównanie grup (Fazlodeks 500 mg/Fazlodeks 250 mg) |
||
| Stosunek ryzyka |
95 % CI |
p-wartość |
||||
| PZW |
Średnia K-M w miesiącach; stosunek ryzyka |
|||||
| Wszyscy pacjenci |
6,5 |
5,5 |
0,80 |
0,68; 0,94 |
0,006 |
|
|
8,6 |
5,8 |
0,76 |
0,62; 0,94 |
0,013 |
|
|
5,4 |
4,1 |
0,85 |
0,67; 1,08 |
0,195 |
|
| WZb |
Średnia K-M w miesiącach; stosunek ryzyka |
|||||
| Wszyscy pacjenci |
26,4 |
22,3 |
0,81 |
0,69; 0,96 |
0,016c |
|
|
30,6 |
23,9 |
0,79 |
0,63; 0,99 |
0,038c |
|
|
24,1 |
20,8 |
0,86 |
0,67; 1,11 |
0,241c |
|
| Zmienna |
Typ oceny; porównanie leczenia |
Fazlodeks 500 mg (n = 362) |
Fazlodeks 250 mg (n = 374) |
Porównanie między grupami (Fazlodeks 500 mg/Fazlodeks 250 mg) |
||
| Bezwzględna różnica w % |
95 % CI |
|||||
| ORZd |
% pacjentów z ORZ; bezwzględna różnica w % |
|||||
| Wszyscy pacjenci |
13,8 |
14,6 |
|
|
||
|
18,1 |
19,1 |
|
8,2; –9,3 |
||
|
7,3 |
8,3 |
|
|
||
| ORKe |
% pacjentów z KR; bezwzględna różnica w % |
|||||
| Wszyscy pacjenci |
45,6 |
39,6 |
6,0 |
|
||
|
52,4 |
45,1 |
7,3 |
|
||
|
36,2 |
32,3 |
3,9 |
|
||
a Fazlodeks jest wskazany u pacjentek, u których choroba nawróciła lub postępowała mimo terapii antyestrogenowej. Wyniki w podgrupie AI nie są ostateczne.
b Wartość OS przedstawiono dla ostatecznej analizy przeżycia przy 75 % opracowaniu danych.
c Nominalna wartość p bez żadnych korekt uwzględniających powtarzalność między pierwotnymi analizami przeżycia ogólnego przy 50 % opracowaniu danych a zaktualizowanymi analizami przeżycia przy 75 % opracowaniu danych.
d CBR analizowano u pacjentek, u których odpowiedź oceniano w stanе wyjściowym (tzn. miały chorobę ocenialną w stanе wyjściowym: 240 pacjentek w grupie Fazlodeksu w dawce 500 mg i 261 pacjentek w grupie Fazlodeksu w dawce 250 mg).
e Pacjenci z lepszą odpowiedzią obiektywną w postaci pełnej odpowiedzi, częściowej odpowiedzi lub stabilizacji choroby trwającej ≥ 24 tygodnie.
PFS – przeżycie wolne od postępu choroby; CBR – częstość odpowiedzi klinicznej; OR – odpowiedź obiektywna; CBR – częstość efektywności klinicznej; CE – efektywność kliniczna; OS – przeżycie ogólne; K-M – Kaplan-Meier; CI – przedział ufności; AI – inhibitor aromatazy; AE – antyestrogen.
Przeprowadzono randomizowane, podwójnie ślepe, wieloośrodkowe badanie kliniczne fazy 3 oceniające skuteczność leku Fazlodeks 500 mg w porównaniu z anastrozolem 1 mg u kobiet w okresie postmenopauzalnym z lokalnie zaawansowanym lub przerzutowym rakiem piersi z pozytywnymi receptorami estrogenowymi i/lub progesteronowymi, które wcześniej nie otrzymywały terapii hormonalnej. Łącznie 462 pacjentki zostało losowo przydzielonych w stosunku 1:1 do grupy fulwestrantu 500 mg lub anastrozolu 1 mg.
Randomizację przeprowadzono z uwzględnieniem warstwowania według stanu choroby (rak lokalnie zaawansowany lub przerzutowy), wcześniejszej chemioterapii w przypadku zaawansowanej choroby oraz klinicznych objawów choroby.
Pierwotnym punktem końcowym skuteczności była wolna od postępu choroby przeżycie (PFS) oceniane przez badacza zgodnie z kryteriami RECIST 1.1 (Response Evaluation Criteria in Solid Tumors). Kluczowymi wtórnymi punktami końcowymi skuteczności były przeżycie ogólne (OS) oraz częstość odpowiedzi obiektywnej (ORR).
Mediana wieku pacjentek włączonych do badania wynosiła 63 lata (zakres od 39 do 90 lat). U większości pacjentek (87,0 %) na początku badania stwierdzono postać przerzutową choroby. Pięćdziesiąt pięć procent (55 %) pacjentek miało przerzuty do narządów wewnętrznych na początku badania. Łącznie 17,1 % pacjentek wcześniej otrzymywało chemioterapię z powodu zaawansowanej choroby; u 84,2 % pacjentek stwierdzono zmiany chorobowe możliwe do pomiaru.
Istotne wyniki zaobserwowano u większości pacjentek z wcześniej określonych podgrup. W podgrupie pacjentek z przerzutami poza narządy wewnętrzne (n=208), które otrzymywały Fazlodeks, wskaźnik HR (hazard ratio) wyniósł 0,592 (95 % CI: 0,419; 0,837) w porównaniu z pacjentkami, które otrzymywały anastrozol. W podgrupie pacjentek z przerzutami do narządów wewnętrznych (n = 254), które otrzymywały Fazlodeks, wskaźnik HR wyniósł 0,993 (95 % CI: 0,740; 1,331) w porównaniu z pacjentkami, które otrzymywały anastrozol. Wyniki skuteczności badania FALCON przedstawiono w tabeli 2 oraz na wykresie 1.
Tabela 2. Podsumowanie wyników analizy pierwotnego punktu końcowego skuteczności (PFS) i kluczowych wtórnych punktów końcowych skuteczności w badaniu FALCON (ocena badacza, populacja „wszyscy randomizowani według zamiaru leczenia”).
| Fazlodeks 500 mg (n = 230) |
Anastrozol 1 mg (n = 232) |
|
| Przeżycie wolne od postępu choroby |
||
| Liczba przypadków PFS (%) |
143 (62,2 %) |
166 (71,6 %) |
| PFS stosunek ryzyka (95 % CI) i wartość p |
HR 0,797 (0,637–0,999) p = 0,0486 |
|
| Mediana PFS [miesiąc (95 % CI)] |
16,6 (13,8; 21,0) |
13,8 (12,0; 16,6) |
| Liczba przypadków OS* |
67 (29,1 %) |
75 (32,3 %) |
| Całkowite przeżycie stosunek ryzyka (95% CI) i wartość p |
HR 0,875 (0,629–1,217) p = 0,4277 |
|
| ORR** |
89 (46,1%) |
88 (44,9%) |
| ORR stosunek szans (95 % CI) i wartość p |
OR 1,074 (0,716–1,614) p = 0,7290 |
|
| Mediana czasu trwania odpowiedzi (miesiące) |
20,0 |
13,2 |
| CBR (częstość efektywności klinicznej) |
180 (78,3%) |
172 (74,1%) |
| CBR stosunek szans (95 % CI) i wartość p |
OR 1,253 (0,815–1,932) p = 0,3045 |
|
*(31% przetworzone) – analiza PFS nie jest ostateczna.
**Dla pacjentek z mierzalnymi objawami choroby.
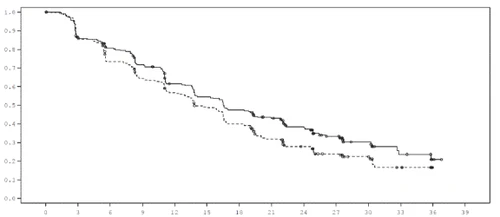
Wykres 1. Krzywa PFS według Kaplana-Meiera (ocena badacza, zbiorowość „wszyscy pacjenci zrandomizowani zgodnie z przypisanym leczeniem”), badanie FALCON.
| Prawdopodobieństwo WWP |
Czas od randomizacji (miesiące) |
| Leczenie: Fulvestrant 500 mg (n = 230) … Anastrozol 1 mg (n = 232) |
|
| Liczba pacjentów z ryzykiem Fulv. 500 230 187 171 150 124 110 96 81 63 44 24 11 2 0 Anast. 1 232 194 162 139 120 102 81 60 45 31 22 10 0 0 |
Dwa badania kliniczne fazy 3 przeprowadzono łącznie na 851 kobietach w okresie menopauzy z zaawansowanym rakiem piersi, u których doszło do nawrotu choroby podczas lub po adjuwantowej terapii hormonalnej lub do progresji po terapii hormonalnej z powodu zaawansowanego stadium choroby. Siedemdziesiąt siedem procent (77%) populacji badawczej miało raka piersi z pozytywnymi receptorami estrogenowymi. Badania te porównywały bezpieczeństwo i skuteczność miesięcznego podawania leku Fazlodeks w dawce 250 mg z codziennym podawaniem 1 mg anastrozolu (inhibitor aromatazy). Ogólnie rzecz biorąc, Fazlodeks w dawce 250 mg podawany raz miesięcznie był co najmniej tak samo skuteczny jak anastrozol pod względem przeżycia bez progresji choroby, odpowiedzi obiektywnej oraz czasu do śmierci. Nie stwierdzono statystycznie istotnej różnicy między dwiema grupami leczenia w odniesieniu do żadnego z tych punktów końcowych. Głównym punktem końcowym była przeżycie bez progresji choroby. Połączona analiza obu badań wykazała, że progresja wystąpiła u 83% pacjentek leczonych Fazlodeksem w porównaniu do 85% pacjentek leczonych anastrozolem. Połączona analiza obu badań wykazała, że stosunek ryzyka dla leku Fazlodeks w dawce 250 mg w porównaniu do anastrozolu w odniesieniu do przeżycia bez progresji choroby wynosił 0,95 (95% CI 0,82–1,10). Poziom odpowiedzi obiektywnej dla leku Fazlodeks w dawce 250 mg wynosił 19,2% w porównaniu do 16,5% dla anastrozolu. Mediana czasu do śmierci wynosiła 27,4 miesiąca dla pacjentek leczonych Fazlodeksem i 27,6 miesiąca dla pacjentek leczonych anastrozolem. Stosunek ryzyka dla leku Fazlodeks w dawce 250 mg w porównaniu do anastrozolu w odniesieniu do czasu do śmierci wynosił 1,01 (95% CI 0,86–1,19).
Terapia skojarzona z palbocyklibem
Międzynarodowe, randomizowane, podwójnie ślepe, wieloośrodkowe badanie fazy 3 z równoległymi grupami oceniało stosowanie leku Fazlodeks 500 mg w skojarzeniu z palbocyklibem 125 mg w porównaniu z Fazlodeksem 500 mg w skojarzeniu z placebo u kobiet z HR-pozytywnym, HER2-negatywnym lokalnie zaawansowanym rakiem piersi, nieuleczalnym chirurgicznie ani radioterapią z celem wyleczającym, niezależnie od statusu menopauzy, u których doszło do progresji choroby po wcześniejszej terapii endokrynnej w ramach leczenia adjuwantowego lub neoadjuwantowego lub w przebiegu choroby przerzutowej.
Łącznie 521 kobiet w okresie pre-/peri- i postmenopauzy z progresją choroby w ciągu 12 miesięcy lub po zakończeniu adjuwantowej terapii endokrynnej lub w ciągu 1 miesiąca lub po zakończeniu poprzedniej terapii endokrynnej w przebiegu choroby przerzutowej zostało zrandomizowanych w stosunku 2:1 do grupy leczenia Fazlodeksem w skojarzeniu z palbocyklibem lub Fazlodeksem w skojarzeniu z placebo i podzielonych według udokumentowanej wrażliwości na poprzednią terapię hormonalną, statusu menopauzy w momencie włączenia do badania (w okresie pre-/peri- lub postmenopauzy) oraz obecności przerzutów do narządów wewnętrznych. Kobiety w okresie pre-/perimenopauzy otrzymywały agonistę LH-RH gozerelina. Pacjenci z rozsianą/chorobą przerzutową, z objawami, z zaawansowanym zaangażowaniem narządów wewnętrznych i z ryzykiem powikłań zagrażających życiu w krótkim okresie (w tym pacjentki z dużym, niekontrolowanym wylewem [płucnym, osierdziowym, otrzewnowym], limfangiopatią płucną i zaangażowaniem wątroby powyżej 50%) nie mogli wziąć udziału w badaniu.
Pacjentki kontynuowały otrzymywanie przypisanego leczenia aż do obiektywnej progresji choroby, nasilenia objawów, nieakceptowalnej toksyczności, śmierci lub odwołania zgody na udział w badaniu. Przejście z jednej grupy leczenia do drugiej nie było dozwolone.
Pacjentki zostały starannie zrównoważone pod względem początkowych cech demograficznych i prognostycznych w grupach Fazlodeks plus palbocyklib oraz Fazlodeks plus placebo. Mediana wieku pacjentek włączonych do badania wynosiła 57 lat (zakres 29–88). W każdej grupie leczenia większość uczestniczek należała do rasy europejskiej, miała udokumentowaną wrażliwość na poprzednią terapię hormonalną i była w okresie postmenopauzy. Około 20% pacjentek było w okresie pre-/perimenopauzy. Wszystkie pacjentki wcześniej otrzymywały leczenie systemowe, a większość pacjentek w każdej grupie leczenia wcześniej otrzymywała chemioterapię z powodu pierwotnego rozpoznania. Ponad połowa pacjentek (62%) miała funkcjonalny status ECOG PS 0, u 60% pacjentek stwierdzono przerzuty do narządów wewnętrznych, a 60% pacjentek otrzymało więcej niż jedną poprzednią terapię hormonalną z powodu pierwotnego rozpoznania.
Pierwotnym punktem końcowym badania była przeżycie bez progresji choroby zdefiniowanej według kryteriów RECIST 1.1, ocenianej przez badacza. Dodatkowe analizy PFS zostały przeprowadzone na podstawie oceny niezależnej centralnej ekspertyzy radiologicznej. Punkty końcowe wtórne obejmowały ORR, CBR, przeżycie ogólne (OS), bezpieczeństwo oraz czas do nasilenia objawów (TTD) jako punkt końcowy intensywności bólu.
Badanie osiągnęło swój główny punkt końcowy – istotne przedłużenie PFS według oceny badacza przy pośrednim analizie 82% zaplanowanych danych PFS; wyniki przekroczyły z góry ustaloną granicę skuteczności Haybitta–Petoe (α=0,00135), wykazując istotne statystycznie przedłużenie PFS oraz klinicznie istotny efekt leczenia. Mediana przeżycia bez progresji choroby przy stosowaniu kombinacji Fazlodeks plus palbocyklib wyniosła 11,2 miesiąca, a przy kombinacji Fazlodeks plus placebo – 4,6 miesiąca. Szczegółowe informacje dotyczące skuteczności przedstawiono w tabeli 3.
Po średnim czasie obserwacji wynoszącym 45 miesięcy przeprowadzono końcową analizę OS na podstawie 310 zdarzeń (60% zrandomizowanych pacjentów). Stwierdzono różnicę w średnim przeżyciu całkowitym wynoszącą 6,9 miesiąca na rzecz grupy leczonej palbocyklibem w połączeniu z fulwestrantem w porównaniu z grupą leczoną placebem w połączeniu z fulwestrantem; wynik ten nie był statystycznie istotny przy ustalonym poziomie istotności 0,0235 (jednostronnie). W grupie leczonej placebem w połączeniu z fulwestrantem 15,5% zrandomizowanych pacjentów otrzymało palbocyklib oraz inne inhibitory kinaz zależnych od cyklin jako kolejne linie leczenia po progresji choroby.
Wyniki analiz PFS oraz dane końcowe dotyczące OS, oceniane przez badacza, w badaniu PALOMA3 przedstawiono w tabeli 3.
Tabela 3. Wyniki dotyczące skuteczności, badanie PALOMA3 (według oceny badacza, populacja „wszyscy zrandomizowani pacjenci według przypisanego leczenia”)
| Zaktualizowana analiza (data zakończenia zbierania danych – 23 października 2015 r.) |
||
| Fazlodeks plus palbocyklib (N=347) |
Fazlodeks plus placebo (N=174) |
|
| Przeżycie wolne od postępu choroby |
||
| Mediana [miesiące (95 % CI)] |
11,2 (9,5; 12,9) |
4,6 (3,5; 5,6) |
| Stosunek ryzyka (95 % CI) i wartość p |
0,497 (0,398; 0,620), p<0,000001 |
|
| Pomocnicze punkty końcowe skuteczności* |
||
| OR [% (95% CI)] |
26,2 (21,7; 31,2) |
13,8 (9,0; 19,8) |
| OR (objawy choroby poddające się pomiarowi) [% (95% CI)] |
33,7 (28,1; 39,7) |
17,4 (11,5; 24,8) |
| COR [% (95% CI)] |
68,0 (62,8; 72,9) |
39,7 (32,3; 47,3) |
| Ostateczna całkowita przeżywalność (OS) |
||
| Liczba zdarzeń (%) |
201 (57,9) |
109 (62,6) |
| Mediana [miesiące (95 % CI)] |
34,9 (28,8 – 40,0) |
28,0 (23,6 – 34,6) |
| Stosunek ryzyka (95 % CI) i wartość p† |
0,814 (0,644 – 1,029) p=0,0429†* |
|
CER – częstotliwość skuteczności klinicznej; CI – przedział ufności; N – liczba pacjentów; OR – odpowiedź obiektywna.
Wyniki punktów końcowych wtórnych oparte są na odpowiedziach potwierdzonych i niepotwierdzonych zgodnie z kryteriami RECIST 1.1.
* Statystycznie nieistotne.
† Jednostronne wartości p, ustratyfikowane według testu log-rank z uwzględnieniem obecności przerzutów do narządów wewnętrznych oraz wrażliwości na wcześniejszą terapię hormonalną w momencie randomizacji.
Zmniejszenie ryzyka progresji choroby lub śmierci na korzyść grupy leczonej Fazlodeksem plus palbocyklibem obserwowano we wszystkich oddzielnych podgrupach pacjentek wyodrębnionych według kryteriów ustratyfikowania i wyjściowych cech. Efekt był wyraźny u kobiet w okresie przed-/w trakcie menopauzy (HR 0,46 [95% CI: 0,28; 0,75]) oraz u kobiet po menopauzie (HR 0,52 [95% CI: 0,40; 0,66]), a także u pacjentek z przerzutami do narządów wewnętrznych (HR 0,50 [95% CI: 0,38; 0,65]) i u pacjentek bez przerzutów do narządów wewnętrznych (HR 0,48 [95% CI: 0,33; 0,71]). Korzyść obserwowano również niezależnie od linii poprzedniego leczenia w przypadku przerzutów: przy 0 (HR 0,59 [95% CI: 0,37; 0,93]), 1 (HR 0,46 [95% CI: 0,32; 0,64]), 2 (HR 0,48 [95% CI: 0,30; 0,76]) lub ≥3 liniach (HR 0,59 [95% CI: 0,28; 1,22]). Dodatkowe wskaźniki skuteczności (OR i PFS), oceniane w podgrupach pacjentów z zaangażowaniem narządów wewnętrznych lub bez, przedstawiono w tabeli 4.
Tabela 4. Wyniki badania PALOMA3 dotyczące skuteczności w przypadku zaangażowania narządów wewnętrznych i bez takiego zaangażowania (populacja „wszyscy pacjenci zrandomizowani zgodnie z przypisanym leczeniem”)
| Choroba wewnętrzna |
Choroba pozawewnętrzną |
|||
| Fazlodeks plus palbocyklib (N=206) |
Fazlodeks plus placebo (N=105) |
Fazlodeks plus palbocyklib (N=141) |
Fazlodeks plus placebo (N=69) |
|
| OR [% (95% CI)] |
35,0 (28,5; 41,9) |
13,3 (7,5; 21,4) |
13,5 (8,3; 20,2) |
14,5 (7,2; 25,0) |
| PFS*, mediana [miesiące (zakres)] |
3,8 (3,5; 16,7) |
5,4 (3,5; 16,7) |
3,7 (1,9; 13,7) |
3,6 (3,4; 3,7) |
*Wyniki odpowiedzi na podstawie potwierdzonych i niepotwierdzonych odpowiedzi.
N – liczba pacjentów; CI – przedział ufności; OR – odpowiedź obiektywna; CTF – czas do pierwszej odpowiedzi guza.
Zebrano informacje o objawach zgłaszanych przez pacjentów oraz przeprowadzono ogólną ocenę jakości życia z wykorzystaniem kwestionariusza opracowanego przez Europejską Organizację ds. Badań i Leczenia Raka (EORTC) (QLQ)-C30 oraz modułu dla raka piersi (EORTC QLQ-BR23). Ogółem 335 pacjentów z grupy leczonej lekiem Fazlodeks z palbocyklibem oraz 166 pacjentów z grupy leczonej lekiem Fazlodeks z placebo odpowiedziało na kwestionariusz na początku badania i przynajmniej podczas 1. wizyty po rozpoczęciu.
Czas do pogorszenia został zdefiniowany a priori jako czas pomiędzy początkowym poziomem bólu a pierwszym wystąpieniem zwiększenia ≥10 punktów w porównaniu z poziomem początkowym według skali objawów bólu. Włączenie palbocyklibu do schematu leczenia lekiem Fazlodeks skutkowało istotnym opóźnieniem czasu do pogorszenia objawów bólu w porównaniu z leczeniem lekiem Fazlodeks z placebo (średnio 8,0 miesiąca w porównaniu z 2,8 miesiąca, HR 0,64 [95 % CI: 0,49; 0,85]; p<0,001).
Wpływ na endometrium w okresie menopauzalnym
Dane przedkliniczne wskazują na brak stymulującego wpływu fulwestrantu na endometrium w okresie menopauzalnym. Dwutygodniowe badanie z udziałem zdrowych ochotniczek w okresie menopauzalnym, które otrzymywały etyniloestradiol w dawce 20 μg na dobę, wykazało, że wcześniejsze leczenie lekiem Fazlodeks w dawce 250 mg w porównaniu z wcześniejszym leczeniem placebo znacząco zmniejszało stymulujący wpływ na endometrium w okresie menopauzalnym, co potwierdzono pomiarami ultrasonograficznymi grubości endometrium.
Leczenie neoadiuwantowe trwające do 16 tygodni u pacjentek z rakiem piersi, które otrzymywały leczenie lekiem Fazlodeks w dawce 500 mg lub Fazlodeks w dawce 250 mg, nie prowadziło do klinicznie istotnych zmian grubości endometrium, co wskazuje na brak wpływu agonistycznego. Obecnie nie ma dowodów na niepożądane działanie na endometrium podczas leczenia pacjentek z rakiem piersi. Brak dostępnych danych dotyczących morfologicznej struktury endometrium.
W dwóch krótkoterminowych badaniach (1 i 12 tygodni) z udziałem pacjentek w okresie przedmenopauzalnym z łagodnymi chorobami ginekologicznymi nie zaobserwowano żadnych istotnych statystycznie różnic w grubości endometrium między grupami leczonymi fulwestrantem a placebo, co potwierdzono danymi badania ultrasonograficznego.
Wpływ na kości
Brak długoterminowych danych dotyczących wpływu fulwestrantu na kości. Leczenie neoadiuwantowe trwające do 16 tygodni u pacjentek z rakiem piersi, które otrzymywały leczenie Fazlodeksem w dawce 500 mg lub Fazlodeksem w dawce 250 mg, nie prowadziło do klinicznie istotnych zmian poziomów surowiczych markerów remodelowania kości.
Populacja pediatryczna
Fazlodeks nie jest wskazany do leczenia dzieci. Europejska Agencja Leków wydała decyzję o zwolnieniu z obowiązku przedstawiania wyników badań z zastosowaniem leku Fazlodeks we wszystkich podgrupach pediatrycznych pacjentek z rakiem piersi (informacje dotyczące stosowania leku u dzieci znajdują się w punkcie „Sposób podania i dawki”).
W otwartym badaniu fazy 2 oceniano bezpieczeństwo, skuteczność i farmakokinetykę fulwestrantu u 30 dziewcząt w wieku od 1 do 8 lat z postępującym przedwczesnym dojrzewaniem płciowym związanym z zespołem Albrighta – McCune’a (MAS). Dzieci otrzymywały miesięcznie fulwestrant w dawce 4 mg/kg w formie wstrzyknięć do mięśnia. W trakcie tego 12-miesięcznego badania oceniano szereg punktów końcowych dotyczących skuteczności leku w przypadku MAS. Wyniki badania wykazały zmniejszenie częstości krwawień pochwy i spowolnienie tempa dojrzewania wieku kostnego. Stabilne minimalne stężenia fulwestrantu w stanie równowagi u dzieci w tym badaniu odpowiadały stężeniom u dorosłych (patrz punkt „Farmakokinetyka”). W trakcie tego małego badania nie pojawiły się nowe zagadnienia dotyczące bezpieczeństwa leku, jednak dane pięcioletnie nie są jeszcze dostępne.
Farmakokinetyka.
Wchłanianie
Po wstrzyknięciu leku Fazlodeks w formie przedłużonego działania wstrzykniętego do mięśnia fulwestrant jest wchłaniany powoli, a maksymalne stężenie w osoczu (Cmax) osiągane jest po około 5 dniach. Przy schemacie podawania leku Fazlodeks w dawce 500 mg stężenia ekspozycji w stanie równowagi lub bliskim równowadze osiągane są w ciągu pierwszego miesiąca leczenia (średnia wartość [współczynnik zmienności (CV)]: AUC 475 [33,4 %] ng·dzień/ml, Cmax 25,1 [35,3 %] ng/ml, Cmin 16,3 [25,9 %] ng/ml odpowiednio). W stanie równowagi stężenia fulwestrantu w osoczu utrzymują się w stosunkowo wąskim zakresie, z różnicą około trzykrotną między maksymalnym a minimalnym stężeniem. Po wstrzyknięciu do mięśnia w zakresie dawek od 50 do 500 mg ekspozycja jest w przybliżeniu proporcjonalna do dawki.
Rozkład
Fulwestrant jest szeroko i szybko rozprowadzany. Znaczny pozorny objętościowy rozkład w stanie równowagi (Vdss), wynoszący około 3–5 l/kg, wskazuje na przede wszystkim rozdział pozazastawny.
Fulwestrant w znacznym stopniu (99 %) wiąże się z białkami osocza. Głównymi frakcjami wiążącymi są lipoproteiny o bardzo niskiej gęstości (VLDL), lipoproteiny o niskiej gęstości (LDL) i lipoproteiny o wysokiej gęstości (HDL). Nie przeprowadzono badań dotyczących konkurencyjnego wiązania białek. Rola globuliny wiążącej hormony płciowe (SHBG) nie została ustalona.
Biotransformacja
Metabolizm fulwestrantu nie został w pełni poznany, ale obejmuje kombinację licznych możliwych szlaków biotransformacji, które są analogiczne do szlaków biotransformacji endogennych steroidów. Zidentyfikowane metabolity (w tym metabolity 17-ketonu, sulfonu, 3-sulfatu, 3- i 17-glukuronidu) w modelach antyestrogenowych są albo mniej aktywne, albo wykazują aktywność podobną do fulwestrantu. Badania z wykorzystaniem wycinków wątroby ludzkiej oraz rekombinowanych ludzkich enzymów wskazują, że CYP3A4 jest jedynym izoenzymem cytochromu P450 biorącym udział w utlenianiu fulwestrantu; jednak uważa się, że in vivo przeważają szlaki metabolizmu niezwiązane z P450. Dane in vitro wskazują, że fulwestrant nie hamuje izoenzymów CYP450.
Wydalanie
Fulwestrant jest wydalany głównie w formie zmetabolizowanej. Główną drogą wydalania jest kał, przy czym mniej niż 1 % wydala się z moczem. Fulwestrant charakteryzuje się wysokim kliremsem, wynoszącym 11 ± 1,7 ml/min/kg, co wskazuje na wysoki wątrobowy współczynnik ekstrakcji. Okres półtrwania końcowego (t1/2) po wstrzyknięciu do mięśnia jest określany przez szybkość wchłaniania i szacowany na 50 dni.
Specjalne grupy pacjentów
W analizie populacyjnej farmakokinetyki danych z badań fazy 3 nie stwierdzono różnic w profilu farmakokinetycznym fulwestrantu ze względu na wiek (zakres od 33 do 89 lat), masę ciała (od 40 do 127 kg) ani przynależność rasową.
Zaburzenia funkcji nerek
Stopień wpływu łagodnego lub umiarkowanego zaburzenia funkcji nerek na farmakokinetykę fulwestrantu nie osiąga istotności klinicznej.
Zaburzenia funkcji wątroby
Farmakokinetykę fulwestrantu oceniano w badaniu klinicznym z zastosowaniem pojedynczej dawki leku, przeprowadzonym u kobiet z łagodnym i umiarkowanym zaburzeniem funkcji wątroby (klasa A i B według Childa-Pugh). Stosowano krótkotrwałe podawanie wysokiej dawki leku w formie wstrzyknięć do mięśnia. W porównaniu ze zdrowymi osobami u kobiet z zaburzeniem funkcji wątroby zaobserwowano zwiększenie AUC o prawie 2,5 raza. Oczekuje się, że zwiększenie ekspozycji do takiego stopnia u pacjentów otrzymujących Fazlodeks będzie dobrze tolerowane. Kobiety z ciężkim zaburzeniem funkcji wątroby (klasa C według Childa-Pugh) nie były oceniane.
Populacja pediatryczna
Farmakokinetykę fulwestrantu oceniano w badaniu klinicznym przeprowadzonym u 30 dziewcząt z postępującym przedwczesnym dojrzewaniem płciowym związanym z zespołem Albrighta – McCune’a – Sterneberga (patrz punkt „Farmakodynamika”). Pacjentki pediatryczne miały wiek od 1 do 8 lat i otrzymywały fulwestrant w dawce 4 mg/kg wstrzykiwany do mięśnia co miesiąc. Geometryczna średnia (odchylenie standardowe) minimalne stężenie w stanie równowagi (Cmin,ss) i AUCss wynosiły odpowiednio 4,2 (0,9) ng/ml i 3680 (1020) ng*godz/ml. Chociaż dane są ograniczone, stężenia fulwestrantu w stanie równowagi u dzieci prawdopodobnie są zgodne ze stężeniami u dorosłych.
Charakterystyki kliniczne.
Wskazania.
Fazlodeks jest wskazany:
- jako monoterapia w leczeniu lokalnie zaawansowanego lub przerzutowego raka piersi z receptorami pozytywnymi wobec estrogenów u kobiet w okresie poklimakterycznym:
- które wcześniej nie otrzymywały terapii endokrynnej, lub
- u których doszło do nawrotu choroby podczas lub po adiuwantowej terapii antyestrogenowej lub progresji choroby podczas terapii antyestrogenowej;
- w połączeniu z palbocyklibem w leczeniu raka piersi z dodatnimi receptorami hormonów (HR-dodatni), negatywnym wobec ludzkiego receptora czynnika wzrostu epidermy 2 (HER2-negatywny), lokalnie zaawansowanego lub przerzutowego u kobiet, które wcześniej otrzymywały terapię endokrynną (patrz rozdział „Farmakodynamika”).
U kobiet w okresie przedklimakterycznym lub w klimakterium leczenie skojarzone z palbocyklibem należy prowadzić w połączeniu z agonistą hormonu uwalniającego hormon gonadotropinowy (LHRH).
Przeciwwskazania.
Nadwrażliwość na substancję czynną lub na którąkolwiek z substancji pomocniczych.
Ciąża i laktacja (patrz rozdział „Stosowanie w czasie ciąży lub karmienia piersią”).
Ciężka niewydolność wątroby (patrz rozdziały „Szczególne ostrzeżenia i środki ostrożności dotyczące stosowania” i „Farmakokinetyka”).
Interakcje z innymi lekami i inne rodzaje interakcji.
Badania kliniczne interakcji z midazolamem (substratem CYP3A4) wykazały, że fulwestrant nie hamuje CYP3A4. Badania kliniczne interakcji z ryfampicyną (induktorem CYP3A4) i ketokonazolem (inhibitorem CYP3A4) nie wykazały klinicznie istotnych zmian klirensu fulwestrantu. W związku z tym u pacjentów otrzymujących jednocześnie fulwestrant oraz inhibitory lub induktory CYP3A4 nie jest wymagana korekta dawki.
Szczególne ostrzeżenia i środki ostrożności dotyczące stosowania
Fazlodeks należy stosować z ostrożnością u pacjentów z zaburzeniami funkcji wątroby o lekkim i umiarkowanym nasileniu (patrz punkty „Sposób stosowania i dawki”, „Przeciwwskazania” oraz „Farmakokinetyka”).
Fazlodeks należy stosować z ostrożnością u pacjentów z ciężkim zaburzeniem funkcji nerek (klirens kreatyniny < 30 ml/min).
Z uwagi na sposób podania do wewnątrzmięśniowego, Fazlodeks należy stosować z ostrożnością u pacjentów z diatezą krwotoczną, trombocytopenią oraz u tych przyjmujących leki przeciwwątrociowe.
Zwykle u kobiet z zaawansowanym rakiem piersi obserwowano reakcje zakrzepowo-zatorowe, które odnotowano również w badaniach klinicznych leku Fazlodeks (patrz punkt „Efekty niepożądane”). Należy to uwzględnić przy przepisywaniu leku Fazlodeks pacjentkom z grupy wysokiego ryzyka.
Podczas stosowania leku Fazlodeks zgłaszano reakcje związane z miejscem wstrzyknięcia, w tym ischias, neuralgię, ból neuropatyczny i neuropatię obwodową. Z uwagi na bliskość nerwu siadłowego należy zachować ostrożność podczas wstrzykiwania leku Fazlodeks do górnego zewnętrznego kwadrantu pośladka (patrz punkty „Sposób stosowania i dawki” oraz „Efekty niepożądane”).
Nie ma danych z długoterminowych badań dotyczących wpływu fulwestrantu na tkankę kostną. Z uwagi na mechanizm działania fulwestrantu istnieje potencjalne ryzyko rozwoju osteoporozy.
Bezpieczeństwo i skuteczność stosowania leku Fazlodeks (jako monoterapii lub w połączeniu z palbocyklibem) u pacjentów z ciężkimi chorobami narządów wewnętrznych nie były badane.
W przypadku stosowania leku Fazlodeks w połączeniu z palbocyklibem należy również zapoznać się z krótkim opisem produktu leczniczego palbocyklibu.
Wpływ na oznaczanie estradiolu metodą opartą na przeciwciałach
Z uwagi na podobieństwo strukturalne fulwestrantu i estradiolu, fulwestrant może wpływać na wyniki oznaczeń poziomu estradiolu metodą opartą na przeciwciałach, powodując fałszywie podwyższone wartości.
Etyloalkohol
Fazlodeks zawiera 10 % mas./obj. etanolu (alkoholu) jako substancję pomocniczą, co odpowiada do 500 mg w jednym wstrzyknięciu, co jest równoważne 10 ml piwa lub 4 ml wina. Może to szkodzić osobom cierpiącym na alkoholizm, a także należy to wziąć pod uwagę u pacjentów z grupy wysokiego ryzyka, w szczególności u pacjentów z uszkodzeniem wątroby i padaczką.
Alkohol benzylowy
Fazlodeks zawiera alkohol benzylowy jako substancję pomocniczą, który może powodować reakcje alergiczne.
Populacja pediatryczna
Fazlodeks nie jest zalecany do stosowania u dzieci i młodzieży, ponieważ bezpieczeństwo i skuteczność w tej grupie wiekowej nie zostały ustalone (patrz punkt „Farmakodynamika”).
Stosowanie w okresie ciąży lub karmienia piersią
Kobiety w wieku rozrodczym
Kobiety w wieku rozrodczym powinny stosować skuteczne metody antykoncepcji podczas leczenia lekiem Fazlodeks oraz przez 2 lata po podaniu ostatniej dawki.
Ciąża
Fazlodeks jest przeciwwskazany w czasie ciąży (patrz punkt „Przeciwwskazania”). Wykazano, że fulwestrant przenika przez barierę łożyskową po jednorazowym wstrzyknięciu do mięśnia u szczurów i królików. Badania na zwierzętach wykazały toksyczność reprodukcyjną, w tym zwiększenie częstości występowania wad rozwojowych i śmierci płodu. Jeżeli pacjentka zajdzie w ciążę w trakcie leczenia lekiem Fazlodeks, należy ją poinformować o potencjalnym niebezpieczeństwie dla płodu oraz o potencjalnym ryzyku przerwania ciąży.
Karmienie piersią
Karmienie piersią należy przerwać w trakcie leczenia lekiem Fazlodeks. Fulwestrant wydzielany jest z mlekiem u ssących szczurów. Nie wiadomo, czy fulwestrant wydzielany jest z mlekiem matki. Ze względu na możliwość wystąpienia poważnych niepożądanych reakcji u niemowląt karmionych piersią, stosowanie tego leku w okresie karmienia piersią jest przeciwwskazane (patrz punkt „Przeciwwskazania”).
Plodność
Wpływ leku Fazlodeks na płodność u ludzi nie był badany.
Wpływ na zdolność prowadzenia pojazdów i obsługiwanie maszyn
Fazlodeks nie wpływa lub ma nieznaczny wpływ na zdolność prowadzenia pojazdów i obsługiwanie maszyn. Jednakże, ponieważ podczas leczenia lekiem Fazlodeks bardzo często zgłaszano astenię, pacjenci, u których podczas prowadzenia pojazdów lub pracy z innymi mechanizmami wystąpi ten efekt niepożądany, powinni zachować ostrożność.
Sposób stosowania i dawki.
Dawkowanie
Dorosłe kobiety (w tym osoby starsze)
Zalecana dawka wynosi 500 mg, stosowaną w odstępie jednego miesiąca; dodatkową dawkę 500 mg podaje się po dwóch tygodniach od pierwszego wstrzyknięcia.
W przypadku stosowania leku Fazlodeks w połączeniu z palbocyklibem należy również zapoznać się z krótką charakterystyką produktu palbocyklibu.
Kobietom w okresie przedmenopauzalnym/w trakcie menopauzy, przed rozpoczęciem i w trakcie leczenia skojarzonego lekiem Fazlodeks i palbocyklibem, należy stosować agonisty LH-RH (hormonu uwalniającego gonadotropinę) zgodnie z lokalnymi standardami praktyki klinicznej.
Osobliwe grupy pacjentów
Zaburzenia funkcji nerek
U pacjentów z zaburzeniami funkcji nerek o lekkim i umiarkowanym nasileniu (klirens kreatyniny ≥ 30 ml/min) nie jest wymagana korekta dawki. Skuteczność i bezpieczeństwo leku nie były oceniane u pacjentów z ciężkimi zaburzeniami funkcji nerek (klirens kreatyniny < 30 ml/min), dlatego u tych pacjentów lek należy stosować z ostrożnością (patrz sekcja „Szczególne ostrzeżenia i środki ostrożności dotyczące stosowania”).
Zaburzenia funkcji wątroby
U pacjentów z zaburzeniami funkcji wątroby o lekkim i umiarkowanym nasileniu nie jest wymagana korekta dawki. Jednakże u tych pacjentów lek Fazlodeks należy stosować z ostrożnością ze względu na możliwy wzrost ekspozycji na fulwestrant. Brak danych dotyczących pacjentów z ciężkimi zaburzeniami funkcji wątroby (patrz sekcje „Przeciwwskazania”, „Szczególne ostrzeżenia i środki ostrożności dotyczące stosowania” oraz „Farmakokinetyka”).
Sposób stosowania
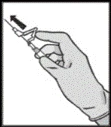
Fazlodeks należy podawać w postaci dwóch kolejnych, powolnych (1-2 minuty na wstrzyknięcie) wstrzyknięć do mięśnia po 5 ml, po jednym w każdą pośladę (w okolice pośladkowe).
Ze względu na bliskie położenie nerwu siadłowego należy zachować ostrożność podczas wstrzykiwania leku Fazlodeks w górny zewnętrzny kwadrant okolicy pośladkowej.
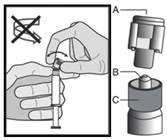
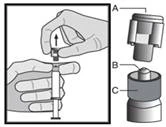
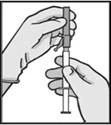
Instrukcja dotycząca wstrzykiwania
Lek należy podawać zgodnie z lokalnymi zasadami stosowania dużych objętościowo wstrzyknięć do mięśnia.
UWAGA. Ze względu na bliskie położenie nerwu siadłowego należy zachować ostrożność podczas wstrzykiwania leku Fazlodeks w górny zewnętrzny kwadrant okolicy pośladkowej (patrz sekcja „Szczególne ostrzeżenia i środki ostrożności dotyczące stosowania”).
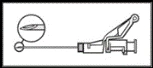
Ostrzeżenie: Nie sterylizować igły bezpiecznej (igły podskórnej pokrytej osłonką „BDSafetyGlide™”) w autoklawie przed użyciem.
W trakcie całego okresu użytkowania i usuwania ręce muszą znajdować się z tyłu igły.
Dla każdego z dwóch strzykawek:
|
Rysunek 1
|
|
Rysunek 2
|
|
Rysunek 3
|
|
Rysunek 4
|
UWAGA. Podczas aktywacji trzymać igłę skierowaną w stronę przeciwną do siebie i innych osób. Uważnie nasłuchiwać dźwięku kliknięcia i wizualnie upewnić się, że koniec igły został całkowicie zamknięty. |
Rysunek 5
|
Unikalizacja
Wypełnione strzykawki przeznaczone są wyłącznie do jednorazowego użytku.
Ten lek może stanowić zagrożenie dla środowiska wodnego. Wszelkie niewykorzystane leki lub ich odpady należy unieszkodliwiać zgodnie z lokalnymi przepisami.
Dzieci.
Bezpieczeństwo i skuteczność stosowania leku Fazlodeks u dzieci (do 18. roku życia) nie zostały ustalone. Obecnie dostępne dane opisane w sekcjach „Farmakokinetyka” i „Farmakodynamika” są niewystarczające, aby określić zalecenia dotyczące dawkowania u dzieci.
Przedawkowanie.
Istnieją pojedyncze doniesienia o przypadkach przedawkowania leku Fazlodeks u ludzi. W przypadku przedawkowania zaleca się leczenie objawowe wspierające. Badania na zwierzętach wskazują na brak efektów wyższych dawek fulwestrantu poza tymi, które są bezpośrednio lub pośrednio związane z działaniem antyestrogenowym.
Działania niepożądane
Podsumowanie profilu bezpieczeństwa
Monoterapia
W tej sekcji przedstawiono informacje dotyczące wszystkich działań niepożądanych zgłoszonych podczas badań klinicznych, badań po rejestracji lub zgłoszeń spontanicznych. W połączonym zbiorze danych dotyczących monoterapii fulwestrantem najczęściej zgłaszane działania niepożądane to reakcje w miejscu wstrzyknięcia, osłabienie (astenia), nudności oraz podwyższenie poziomu enzymów wątrobowych (ALT – aminotransferaza alaninowa, AST – aminotransferaza asparaginianowa, ALP – fosfataza alkaliczna).
W tabeli 5 kategorie częstości występowania działań niepożądanych obliczono na podstawie danych z grupy leczonych lekiem Fazlodeks 500 mg z połączonej analizy bezpieczeństwa badań, w których porównywano Fazlodeks 500 mg i Fazlodeks 250 mg [CONFIRM (badanie D6997C00002), FINDER 1 (badanie D6997C00004), FINDER 2 (badanie D6997C00006) oraz NEWEST (badanie D6997C00003)], albo z osobnego badania FALCON (badanie D699BC00001), w którym porównywano Fazlodeks 500 mg i anastrozol 1 mg. W przypadku różnicy w częstości działań niepożądanych między połączoną analizą bezpieczeństwa a badaniem FALCON brano wyższą wartość częstości. Częstość podana w poniższej tabeli 5 została określona na podstawie danych dotyczących wszystkich zgłoszonych działań niepożądanych, niezależnie od oceny związku przyczynowego przez badacza. Mediana czasu trwania leczenia fulwestrantem 500 mg w połączonym zbiorze danych (w szczególności w powyższych badaniach oraz badaniu FALCON) wynosiła 6,5 miesiąca.
Wykaz działań niepożądanych w formie tabeli
Poniższe działania niepożądane sklasyfikowano według częstości występowania i układów narządów. Grupy częstości zdefiniowano zgodnie z następującymi kryteriami: bardzo często (≥ 1/10), często (≥ 1/100 do < 1/10), rzadko (≥ 1/1000 do < 1/100). W ramach grupy według częstości działania niepożądane wymieniono w kolejności malejącej według ich nasilenia.
Tabela 5. Działania niepożądane zgłaszane u pacjentów podczas monoterapii lekiem Fazlodeks.
| Reakcje niepożądane sklasyfikowane według częstości i układu narządów |
||
| Infekcje i inwazje |
Często |
Infekcje dróg moczowych |
| Z boku krwi i układu limfatycznego |
Często |
Obniżenie liczby płytek krwi |
| Z boku układu odpornościowego |
Bardzo często |
Reakcje nadwrażliwości |
| Niekonie |
Reakcje anafilaktyczne |
|
| Z boku metabolizmu i odżywiania |
Często |
Anoreksja |
| Z boku układu nerwowego |
Często |
Ból głowy |
| Z boku naczyń |
Bardzo często |
Zawroty |
| Często |
Żylna tromboembolia |
|
| Z boku przewodu pokarmowego |
Bardzo często |
Światłot |
| Często |
Wymioty, biegunka |
|
| Z boku wątroby i dróg żółciowych |
Bardzo często |
Podwyższenie poziomu enzymów wątrobowych (ALT, AST, ALP) |
| Często |
Podwyższenie poziomu bilirubiny |
|
| Niekonie |
Niewydolność wątroby, zapalenie wątroby, podwyższenie poziomu GGTP |
|
| Z boku skóry i tkanki podskórnej |
Bardzo często |
Wysypka |
| Z boku układu mięśniowo-szkieletowego i tkanki łącznej |
Bardzo często |
Ból stawów i mięśni szkieletowych |
| Często |
Ból pleców |
|
| Z boku układu rozrodczego i gruczołów mlekowych |
Często |
Krwawienie z pochwy |
| Niekonie |
Kandydoza pochwy, białe |
|
| Zaburzenia ogólne i stan w miejscu podania leku |
Bardzo często |
Astenia, reakcje w miejscu wstrzyknięcia |
| Często |
Obwodowa neuropatia, ischias |
|
| Niekonie |
Krwawienia w miejscu wstrzyknięcia, siniaki w miejscu wstrzyknięcia, neuralgia |
|
a Włączone są działania niepożądane związane z lekiem, dla których związek z Fazlodeksem nie może być ustalony z powodu choroby podstawowej.
b Termin „reakcje w miejscu wstrzyknięcia” nie obejmuje pojęć „krwawienie w miejscu wstrzyknięcia” i „siniak w miejscu wstrzyknięcia”, „iskiada”, „neuralgia”, „neuropatia obwodowa”.
c Reakcja nie była obserwowana w dużych badaniach klinicznych (CONFIRM, FINDER 1, FINDER 2, NEWEST). Częstość została obliczona przy użyciu górnego limitu 95% przedziału ufności dla szacunku punktowego. Obliczono ją jako 3/560 (gdzie 560 to liczba pacjentów w dużych badaniach klinicznych), co odpowiada częstości kategorii „nieczęsto”.
d Obejmuje artralgię oraz rzadziej – ból kostno-mięśniowy, mialgię i ból kończyn.
e Istnieją pewne różnice w częstości działań niepożądanych w odpowiednich kategoriach między połączonymi danymi dotyczącymi bezpieczeństwa a badaniem FALCON.
f Działania niepożądane nie były obserwowane w badaniu FALCON.
Opis wybranych działań niepożądanych.
Poniższy opis opiera się na analizie bezpieczeństwa grupy 228 pacjentek, które otrzymały co najmniej jedną dawkę fulwestrantu, oraz grupy 232 pacjentek, które otrzymały co najmniej jedną dawkę anastrozolu w badaniu III fazy FALCON.
Ból stawów i ból układu kostno-mięśniowego
Zgodnie z danymi z badania FALCON, liczba pacjentek zgłaszających ból stawów i ból układu kostno-mięśniowego wynosiła odpowiednio 65 (31,2%) i 48 (24,1%) przy stosowaniu fulwestrantu i anastrozolu. Spośród 65 pacjentek otrzymujących Fazlodeks, 40% (26/65) zgłosiło ból stawów i układu kostno-mięśniowego w pierwszym miesiącu leczenia, 66,2% (43/65) pacjentek – w pierwszych trzech miesiącach leczenia. Żaden z pacjentów nie zgłosił przypadków o nasileniu ≥3 wg CTCAE ani przypadków wymagających zmniejszenia dawki leku, tymczasowego przerwania leczenia lub odstawienia leku z powodu tych działań niepożądanych.
Leczenie skojarzone z palbocyklibem
Ogólne dane dotyczące bezpieczeństwa fulwestrantu w skojarzeniu z palbocyklibem oparte są na danych z badania randomizowanego PALOMA3 u 517 pacjentek z HR-dodatnim, HER2-ujemnym rakiem piersi lokalnie zaawansowanym lub przerzutowym (zobacz punkt „Farmakodynamika”). Najczęstsze (≥20%) działania niepożądane dowolnego stopnia zgłaszane u pacjentek otrzymujących fulwestrant w skojarzeniu z palbocyklibem to: neutropenia, leukopenia, infekcje, zmęczenie, nudności, anemia, stomatyt, biegunka, trombocytopenia i wymioty. Najczęstsze (≥2%) działania niepożądane o nasileniu ≥3 stopnia to: neutropenia, leukopenia, infekcje, anemia, podwyższenie poziomu AST, trombocytopenia i zmęczenie.
W tabeli 6 przedstawiono dane dotyczące działań niepożądanych obserwowanych w badaniu PALOMA3.
Średnia długość leczenia fulwestrantem wynosiła 11,2 miesiąca w grupie fulwestrant + palbocyklib oraz 4,8 miesiąca w grupie fulwestrant + placebo. Średnia długość leczenia palbocyklibem w grupie fulwestrant + palbocyklib wyniosła 10,8 miesiąca.
Tabela 6. Działania niepożądane według danych z badania PALOMA3 (N=517)
| Klasa układów narządów |
Fazlodeks + Palbocyklib |
Fazlodeks + placebo (N=172) |
||
| Wszystkie stopnie |
Stopień ≥ 3 |
Wszystkie stopnie n (%) |
Stopień ≥ 3 |
|
| Choroby zakaźne i pasożytnicze |
||||
| Bardzo często |
||||
| Zakażeniab |
188 (54,5) |
19 (5,5) |
60 (34,9) |
6 (3,5) |
| Zaburzenia układu krwi i chłonnego |
||||
| Bardzo często |
||||
| Neutropeniaс |
290 (84,1) |
240 (69,6) |
6 (3,5) |
0 |
| Leukopenia d |
207 (6,0) |
132 (38,3) |
9 (5,2) |
1 (0,6) |
| Anemia e |
109 (31,6) |
15 (4,3) |
24 (14,0) |
4 (2,3) |
| Trombocytopenia f |
88 (25,5) |
10 (2,9) |
0 |
0 |
| Nieczęsto |
||||
| Neutropenia febrilna |
3 (0,9) |
3 (0,9) |
0 |
0 |
| Zaburzenia przemiany materii i odżywiania |
||||
| Bardzo często |
||||
| Współczucie apetytu |
60 (17,4) |
4 (1,2) |
18 (10,5) |
1 (0,6) |
| Zaburzenia układu nerwowego |
||||
| Często |
||||
| Dysgezja |
27 (7,8) |
0 |
6 (3,5) |
0 |
| Zaburzenia oka |
||||
| Często |
||||
| Zwiększone łzawienie |
25 (7,2) |
0 |
2 (1,2) |
0 |
| Podwójne widzenie |
24 (7,0) |
0 |
3 (1,7) |
0 |
| Susza oczu |
15 (4,3) |
0 |
3 (1,7) |
0 |
| Zaburzenia układu oddechowego, klatki piersiowej i śródpiersia |
||||
| Często |
||||
| Krwawienie z nosa |
25 (7,2) |
0 |
4 (2,3) |
0 |
| Zaburzenia ze strony przewodu pokarmowego |
||||
| Bardzo często |
||||
| Światłowstręt |
124 (35,9) |
2 (0,6) |
53 (30,8) |
1 (0,6) |
| Stomatytg |
104 (30,1) |
3 (0,9) |
24 (14,0) |
0 |
| Biegunka |
94 (27,2) |
0 |
35 (20,3) |
2 (1,2) |
| Wymioty |
75 (21,7) |
2 (0,6) |
28 (16,3) |
1 (0,6) |
| Zaburzenia skóry i tkanki podskórnej |
||||
| Bardzo często |
||||
| Alopeacja |
67 (19,4) |
Nie dotyczy |
11 (6,4) |
Nie dotyczy |
| Wysypkh |
63 (18,3) |
3 (0,9) |
10 (5,8) |
0 |
| Często |
||||
| Susza skóry |
28 (8,1) |
0 |
3 (1,7) |
0 |
| Ogólne zaburzenia i reakcje w miejscu podania |
||||
| Bardzo często |
||||
| Zmęczenie |
152 (44,1) |
9 (2,6) |
54 (31,4) |
2 (1,2) |
| Gorączka |
47 (13,6) |
1 (0,3) |
10 (5,8) |
0 |
| Często |
||||
| Astenia |
27 (7,8) |
1 (0,3) |
13 (7,6) |
2 (1,2) |
| Wyniki badań laboratoryjnych |
||||
| Bardzo często |
||||
| Zwiększenie poziomu AST |
40 (11,6) |
11 (3,2) |
13 (7,6) |
4 (2,3) |
| Często |
||||
| Zwiększenie poziomu ALT |
30 (8,7) |
7 (2,0) |
10 (5,8) |
1 (0,6) |
ALT – alanina aminotransferaza; AST – asparaginian aminotransferaza; N/n – liczba pacjentów.
a Podano dominujące terminy (PT) dla reakcji zgodnie z MedDRA 17.1.
b Wszystkie PT należące do klasy „Zakażenia i pasożyty”.
c Neutropenia obejmuje następujące PT: neutropenia, zmniejszona liczba neutrofili.
d Leukopenia obejmuje następujące PT: leukopenia, zmniejszona liczba leukocytów.
e Anemia obejmuje następujące PT: anemia, obniżony poziom hemoglobiny, obniżony hematokryt.
f Trombocytopenia obejmuje następujące PT: trombocytopenia, zmniejszona liczba płytek krwi.
g Zapalenie jamy ustnej obejmuje następujące PT: aftowa stomatyt, zapalenie warg, zapalenie języka, glosodynia, owrzodzenie jamy ustnej, zapalenie błony śluzowej, ból jamy ustnej, dyskomfort w obrębie ust i gardła, ból w obrębie ust i gardła, stomatyt.
h Wysypka obejmuje następujące PT: wysypka, wysypka makularna i plamniczo-grudkowa, wysypka z świądem, wysypka rumieniowa, wysypka grudkowa, zapalenie skóry, zapalenie skóry typu trądzikowatego, toksyczne wysypki skórne.
Opis wybranych niepożądanych działań
Neutropenia
W badaniu PALOMA3, w którym stosowano fulwestrant w połączeniu z palbocyklibem, o neutropenii stopnia dowolnego donoszono u 290 (84,1%) pacjentów, o neutropenii stopnia 3 – u 200 (58,0%) pacjentów, a o neutropenii stopnia 4 – u 40 (11,6%) pacjentów. W grupie stosującej fulwestrant + placebo (n = 172) o neutropenii stopnia dowolnego donoszono u 6 (3,5%) pacjentów. O przypadkach neutropenii stopnia 3 i 4 w grupie stosującej fulwestrant + placebo nie donoszono.
U pacjentów otrzymujących fulwestrant w połączeniu z palbocyklibem mediana czasu do pierwszego epizodu neutropenii wynosiła 15 dni (zakres: 13–512 dni), a mediana trwania neutropenii stopnia ≥3 wynosiła 16 dni. U 3 (0,9%) pacjentów otrzymujących fulwestrant w połączeniu z palbocyklibem donoszono o przypadkach neutropenii febrylnej.
Wykazywanie podejrzewanych niepożądanych działań
Istotne jest wykazywanie podejrzewanych niepożądanych działań w okresie pogwarancyjnym stosowania leku. Pozwala to na ciągłe monitorowanie stosunku korzyści do ryzyka związanego ze stosowaniem leku. Osoby pracujące w ochronie zdrowia są zobowiązane do zgłaszania wszelkich przypadków podejrzewanych niepożądanych działań poprzez krajowy system raportowania.
Okres ważności.
4 lata.
Warunki przechowywania.
Przechowywać w temperaturze od 2 do 8 °C.
Przechowywać w miejscu niedostępnym dla dzieci.
Przechowywać wstępnie załadowane strzykawki w oryginalnym opakowaniu w celu ochrony przed światłem.
Fluktuacje temperatury poza zakresem od 2 do 8 °C powinny być ograniczone. Należy unikać przechowywania w temperaturach powyżej 30 °C i nie przekraczać 28-dniowego okresu, w którym średnia temperatura przechowywania leku jest poniżej 25 °C (ale powyżej 2–8 °C). Po wystąpieniu fluktuacji temperatury lek należy natychmiast ponownie umieścić w zalecanych warunkach przechowywania (przechowywanie i transport w lodówce w temperaturze od 2 do 8 °C). Fluktuacje temperatury mają skumulowany wpływ na jakość leku i 28-dniowy okres nie powinien być przekroczony w ciągu 4-letniego okresu ważności leku Fazlodeks. Oddziaływanie temperatur poniżej 2 °C nie powoduje uszkodzenia leku pod warunkiem, że lek nie jest przechowywany w temperaturze poniżej –20 °C.
Niezgodność.
Ponieważ brak badań zgodności, tego leku nie należy mieszać z innymi lekami.
Opakowanie.
1 kartonowa puszka zawiera opakowanie blisterowe zawierające 2 wstępnie załadowane szklane strzykawki z kontrolą pierwszego otwarcia, z których każda zawiera 5 ml roztworu, oraz 2 bezpieczne igły „BD SafetyGlide™”.
Kategoria wydawania.
Na receptę.
Producent.
AstraZeneca UK Limited.
Miejsce położenia producenta oraz jego adres siedziby.
Silk Road Business Park, Macclesfield, SK10 2NA, Wielka Brytania.