Faslodex
Ukraine
Table of Contents
INSTRUCTIONS FOR MEDICAL USE OF THE MEDICINAL PRODUCT FASLODEX (FASLODEX®)
Composition:
Active substance: 1 pre-filled syringe (5 ml) contains fulvestrant 250 mg;
Excipients: ethanol 96%, benzyl alcohol, benzyl benzoate, ricinoleic oil.
Pharmaceutical form. Injection solution.
Main physicochemical properties: transparent, viscous liquid, ranging from colorless to yellow.
Pharmacotherapeutic group. Hormone antagonists and related agents. Anti-estrogenic agents. ATC code L02B A03.
Pharmacological Properties.
Pharmacodynamics.
Mechanism of Action and Pharmacodynamic Effects
Fulvestrant is a competitive antagonist of estrogen receptors (ER) with affinity comparable to that of estradiol. Fulvestrant blocks the trophic effects of estrogens without exhibiting any partial agonist (estrogen-like) activity. Its mechanism of action is associated with the downregulation of estrogen receptor protein levels.
Clinical studies in postmenopausal women with primary breast cancer have shown that fulvestrant significantly reduces levels of ER proteins in tumors with ER-positive status, compared to placebo. A significant reduction in progesterone receptor expression was also observed, consistent with the absence of typical estrogen agonist effects. Furthermore, it has been demonstrated that fulvestrant at a dose of 500 mg provides greater suppression of ER and the proliferation marker Ki67 in breast tumors than the 250 mg dose during neoadjuvant treatment of postmenopausal women.
Clinical Safety and Efficacy of the Medicinal Product in Advanced Breast Cancer
Monotherapy
A phase 3 clinical study was conducted in 736 postmenopausal women with advanced breast cancer who had disease recurrence during or after adjuvant endocrine therapy, or disease progression following endocrine therapy for advanced disease. The study included 423 patients whose disease recurred or progressed during antiestrogen therapy (AE subgroup) and 313 patients whose disease recurred or progressed during aromatase inhibitor therapy (AI subgroup). This study compared the efficacy and safety of the medicinal product Faslodex administered at a dose of 500 mg (n = 362) versus 250 mg (n = 374). The primary endpoint was progression-free survival (PFS); key secondary efficacy endpoints included objective response rate (ORR), clinical benefit rate (CBR), and overall survival (OS). The efficacy results from the CONFIRM study are summarized below in Table 1.
Table 1. Summary of results from the analysis of the primary efficacy endpoint (PFS) and key secondary efficacy endpoints in the CONFIRM study
| Variable |
Estimate type; treatment comparison |
Faslodex 500 mg (n = 362) |
Faslodex 250 mg (n = 374) |
Treatment comparison (Faslodex 500 mg/Faslodex 250 mg) |
||
| Hazard ratio |
95 % CI |
p-value |
||||
| PFS |
K-M median in months; hazard ratio |
|||||
| All patients |
6.5 |
5.5 |
0.80 |
0.68; 0.94 |
0.006 |
|
|
8.6 |
5.8 |
0.76 |
0.62; 0.94 |
0.013 |
|
|
5.4 |
4.1 |
0.85 |
0.67; 1.08 |
0.195 |
|
| OSb |
K-M median in months; hazard ratio |
|||||
| All patients |
26.4 |
22.3 |
0.81 |
0.69; 0.96 |
0.016c |
|
|
30.6 |
23.9 |
0.79 |
0.63; 0.99 |
0.038c |
|
|
24.1 |
20.8 |
0.86 |
0.67; 1.11 |
0.241c |
|
| Variable |
Estimate type; treatment comparison |
Faslodex 500 mg (n = 362) |
Faslodex 250 mg (n = 374) |
Comparison between groups (Faslodex 500 mg/Faslodex 250 mg) |
||
| Absolute difference in % |
95 % CI |
|||||
| ORRd |
% of patients with OR; absolute difference in % |
|||||
| All patients |
13.8 |
14.6 |
|
|
||
|
18.1 |
19.1 |
|
8.2; –9.3 |
||
|
7.3 |
8.3 |
|
|
||
| CBVe |
% of patients with CB; absolute difference in % |
|||||
| All patients |
45.6 |
39.6 |
6.0 |
|
||
|
52.4 |
45.1 |
7.3 |
|
||
|
36.2 |
32.3 |
3.9 |
|
||
and Faslodex is indicated for patients whose disease has recurred or progressed on antiestrogen therapy. Results in the AI subgroup are not mature.
b Hazard ratio value presented for final survival analysis at 75% data maturity.
c Nominal p-value with no adjustments made for multiplicity between the primary overall survival analysis at 50% data maturity and the updated survival analysis at 75% data maturity.
d ORR was analyzed in patients whose response was evaluable at baseline (i.e., they had disease measurable at baseline: 240 patients in the Faslodex 500 mg group and 261 patients in the Faslodex 250 mg group).
e Patients with best objective response of complete response, partial response, or stable disease lasting ≥24 weeks.
PFS – progression-free survival; ORR – objective response rate; OR – objective response; CBR – clinical benefit rate; CE – clinical efficacy; OS – overall survival; KM – Kaplan-Meier; CI – confidence interval; AI – aromatase inhibitor; AE – antiestrogen.
A randomized, double-blind, double-dummy, multicenter phase 3 study was conducted to evaluate the efficacy of Faslodex 500 mg compared to anastrozole 1 mg in postmenopausal women with estrogen and/or progesterone receptor-positive locally advanced or metastatic breast cancer who had not previously received hormonal therapy. A total of 462 patients were sequentially randomized 1:1 to receive fulvestrant 500 mg or anastrozole 1 mg.
Randomization was stratified by disease extent (locally advanced or metastatic), prior chemotherapy for advanced disease, and disease sites.
The primary efficacy endpoint was progression-free survival (PFS) assessed by the investigator according to RECIST 1.1 (Response Evaluation Criteria in Solid Tumors). Key secondary efficacy endpoints were overall survival (OS) and objective response rate (ORR).
The median age of patients enrolled in this study was 63 years (range 39 to 90 years). The majority of patients (87.0%) had metastatic disease at study initiation. Fifty-five percent (55%) of patients had visceral metastases at study initiation. Overall, 17.1% of patients had previously received chemotherapy for advanced disease; 84.2% of patients had measurable disease.
Significant results were observed in most predefined patient subgroups. In the subgroup of patients with non-visceral metastases (n=208) receiving Faslodex, the HR (hazard ratio) was 0.592 (95% CI: 0.419; 0.837) compared to patients receiving anastrozole. In the subgroup of patients with visceral metastases (n=254) receiving Faslodex, the HR was 0.993 (95% CI: 0.740; 1.331), compared to patients receiving anastrozole. Efficacy results from the FALCON study are presented in Table 2 and Figure 1.
Table 2. Summary of results for the analysis of the primary efficacy endpoint (PFS) and key secondary efficacy endpoints in the FALCON study (investigator assessment, full analysis set "all randomized patients according to assigned treatment").
| Faslodex 500 mg (n = 230) |
Anastrozole 1 mg (n = 232) |
|
| Progression-free survival |
||
| Number of PFS events (%) |
143 (62.2%) |
166 (71.6%) |
| PFS hazard ratio (95% CI) and p-value |
HR 0.797 (0.637–0.999) p = 0.0486 |
|
| Median PFS [months (95% CI)] |
16.6 (13.8; 21.0) |
13.8 (12.0; 16.6) |
| Number of OS events* |
67 (29.1%) |
75 (32.3%) |
| Overall survival hazard ratio (95% CI) and p-value |
HR 0.875 (0.629–1.217) p = 0.4277 |
|
| TTP** |
89 (46.1%) |
88 (44.9%) |
| TTP odds ratio (95% CI) and p-value |
OR 1.074 (0.716–1.614) p = 0.7290 |
|
| Median duration of response (months) |
20.0 |
13.2 |
| CBR (clinical benefit rate) |
180 (78.3%) |
172 (74.1%) |
| CBR odds ratio (95% CI) and p-value |
OR 1.253 (0.815–1.932) p = 0.3045 |
|
*(31% processed) – analysis of OS is not final.
**(For patients with measurable disease.
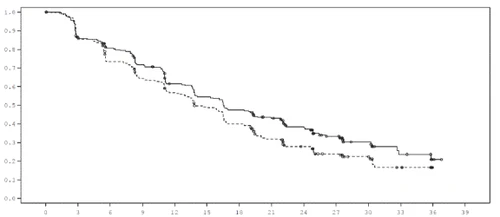
Figure 1. Kaplan-Meier curve for PFS (investigator assessment, "all randomized patients as per assigned treatment" population), FALCON study.
| Probability of BCFI |
Time from randomization (months) |
| Treatment: Fulvestrant 500 mg (n = 230) … Anastrozole 1 mg (n = 232) |
|
| Number of patients at risk Fulv. 500 230 187 171 150 124 110 96 81 63 44 24 11 2 0 Anast. 1 232 194 162 139 120 102 81 60 45 31 22 10 0 0 |
Two phase 3 clinical trials were conducted overall in 851 postmenopausal women with advanced breast cancer who had disease recurrence during or after adjuvant hormonal therapy or progression during hormonal therapy for advanced disease. Seventy-seven percent (77%) of the study population had estrogen receptor-positive breast cancer. These trials compared the safety and efficacy of monthly administration of 250 mg of the medicinal product Faslodex with daily administration of 1 mg of anastrozole (an aromatase inhibitor). Overall, Faslodex at a monthly dose of 250 mg was at least as effective as anastrozole in terms of progression-free survival, objective response rate, and time to death. There was no statistically significant difference between the two treatment groups for any of these endpoints. The primary endpoint was progression-free survival. A combined analysis of both studies showed that disease progression occurred in 83% of patients receiving Faslodex compared with 85% of patients receiving anastrozole. The combined analysis of both studies revealed a hazard ratio for Faslodex 250 mg versus anastrozole for progression-free survival of 0.95 (95% CI 0.82–1.10). The objective response rate for Faslodex 250 mg was 19.2% compared with 16.5% for anastrozole. Median time to death was 27.4 months for patients receiving Faslodex and 27.6 months for those receiving anastrozole. The hazard ratio for Faslodex 250 mg versus anastrozole for time to death was 1.01 (95% CI 0.86–1.19).
Combination therapy with palbociclib
An international, randomized, double-blind, multicenter, parallel-group phase 3 study was conducted to evaluate the use of Faslodex 500 mg with palbociclib 125 mg compared to Faslodex 500 mg with placebo in women with HR-positive, HER2-negative locally advanced breast cancer that was not amenable to surgical or radiation therapy with curative intent, regardless of menopausal status, whose disease had progressed after prior endocrine therapy in the (neo)adjuvant setting or for metastatic disease.
A total of 521 pre-/peri- and postmenopausal women with disease progression within 12 months or after completion of adjuvant endocrine therapy, or within 1 month or after completion of prior endocrine therapy for advanced disease, were randomized in a 2:1 ratio to receive either Faslodex plus palbociclib or Faslodex plus placebo, and stratified according to documented sensitivity to prior hormonal therapy, menopausal status at study entry (pre-/peri- or postmenopausal), and presence of visceral metastases. Women in the pre-/perimenopausal period received the LHRH agonist goserelin. Patients with advanced/metastatic, symptomatic, visceral disease at risk of life-threatening complications in the short term (including patients with massive uncontrolled effusions [pleural, pericardial, peritoneal], pulmonary lymphangitic carcinomatosis, and liver involvement exceeding 50%) were not eligible for the study.
Patients continued their assigned treatment until objective disease progression, worsening of symptoms, unacceptable toxicity, death, or withdrawal of consent. Cross-over between treatment groups was not permitted.
Patients were well balanced across baseline demographic and prognostic characteristics between the Faslodex plus palbociclib and Faslodex plus placebo groups. The median age of enrolled patients was 57 years (range 29–88). In each treatment group, the majority of study participants were of Caucasian race, had documented sensitivity to prior hormonal therapy, and were postmenopausal. Approximately 20% of patients were pre-/perimenopausal. All patients had received prior systemic therapy, and the majority in each treatment group had previously received chemotherapy for the primary diagnosis. More than half of the patients (62%) had an ECOG PS of 0, 60% had visceral metastases, and 60% had received more than one prior hormonal therapy for the primary diagnosis.
The primary endpoint of the study was progression-free survival (PFS), defined according to RECIST 1.1 criteria, as assessed by the investigator. Additional PFS analyses were determined based on independent central radiological review. Secondary endpoints included objective response rate (ORR), clinical benefit rate (CBR), overall survival (OS), safety, and time to deterioration (TTD) in the pain intensity endpoint.
The study achieved its primary endpoint—prolonged PFS as assessed by the investigator at an interim analysis of 82% of planned PFS events; results exceeded the prespecified Haybittle–Peto efficacy boundary (α=0.00135), demonstrating a statistically significant prolongation of PFS and a clinically meaningful treatment effect. The median progression-free survival was 11.2 months in the Faslodex plus palbociclib group versus 4.6 months in the Faslodex plus placebo group. Further details on efficacy are provided in Table 3.
After a median follow-up period of 45 months, a final OS analysis was performed based on 310 events (60% of randomized patients). A difference in median OS of 6.9 months was observed in the palbociclib plus fulvestrant group compared to the placebo plus fulvestrant group; however, this result was not statistically significant at the prespecified significance level of 0.0235 (one-sided). In the placebo plus fulvestrant group, 15.5% of randomized patients received palbociclib and other CDK4/6 inhibitors as subsequent lines of therapy after disease progression.
Results of PFS analyses and final OS data, as assessed by the investigator, from the PALOMA-3 study are presented in Table 3.
Table 3. Efficacy results, PALOMA-3 study (investigator assessment, patient population: "all randomized patients according to assigned treatment")
| Updated analysis (data cutoff date – October 23, 2015) |
||
| Faslodex plus palbociclib (N=347) |
Faslodex plus placebo (N=174) |
|
| Progression-free survival |
||
| Median [months (95% CI)] |
11.2 (9.5; 12.9) |
4.6 (3.5; 5.6) |
| Hazard ratio (95% CI) and p-value |
0.497 (0.398; 0.620), p<0.000001 |
|
| Secondary efficacy endpoints* |
||
| OR [% (95% CI)] |
26.2 (21.7; 31.2) |
13.8 (9.0; 19.8) |
| OR (measurable disease) [% (95% CI)] |
33.7 (28.1; 39.7) |
17.4 (11.5; 24.8) |
| CBR [% (95% CI)] |
68.0 (62.8; 72.9) |
39.7 (32.3; 47.3) |
| Final overall survival (OS) |
||
| Number of events (%) |
201 (57.9) |
109 (62.6) |
| Median [months (95% CI)] |
34.9 (28.8 – 40.0) |
28.0 (23.6 – 34.6) |
| Hazard ratio (95% CI) and p-value† |
0.814 (0.644 – 1.029) p=0.0429†* |
|
CRB – clinical benefit rate; CI – confidence interval; N – number of patients; OR – objective response.
Secondary endpoint results are based on confirmed and unconfirmed responses according to RECIST 1.1 criteria.
* Statistically not significant.
† One-sided p-values, stratified by log-rank test according to presence of visceral metastases and sensitivity to prior endocrine therapy at randomization.
A reduction in the risk of disease progression or death in favor of the fulvestrant plus palbociclib treatment group was observed across all prespecified patient subgroups defined by stratification factors and baseline characteristics. The effect was evident in pre-/perimenopausal women (HR 0.46 [95% CI: 0.28; 0.75]) and postmenopausal women (HR 0.52 [95% CI: 0.40; 0.66]), as well as in patients with visceral metastases (HR 0.50 [95% CI: 0.38; 0.65]) and those without visceral metastases (HR 0.48 [95% CI: 0.33; 0.71]). Benefit was also observed regardless of prior lines of therapy in the metastatic setting: 0 (HR 0.59 [95% CI: 0.37; 0.93]), 1 (HR 0.46 [95% CI: 0.32; 0.64]), 2 (HR 0.48 [95% CI: 0.30; 0.76]), or ≥3 prior lines (HR 0.59 [95% CI: 0.28; 1.22]). Additional efficacy measures (OR and PFS) assessed in subgroups of patients with or without visceral disease are presented in Table 4.
Table 4. Efficacy results from the PALOMA-3 study according to visceral and non-visceral disease (population: all randomized patients according to assigned treatment)
| Visceral disease |
Non-visceral disease |
|||
| Fulvestrant plus palbociclib (N=206) |
Fulvestrant plus placebo (N=105) |
Fulvestrant plus palbociclib (N=141) |
Fulvestrant plus placebo (N=69) |
|
| ORR [% (95% CI)] |
35.0 (28.5; 41.9) |
13.3 (7.5; 21.4) |
13.5 (8.3; 20.2) |
14.5 (7.2; 25.0) |
| PFS*, median [months (range)] |
3.8 (3.5; 16.7) |
5.4 (3.5; 16.7) |
3.7 (1.9; 13.7) |
3.6 (3.4; 3.7) |
*Results based on confirmed and unconfirmed responses.
N – number of patients; CI – confidence interval; OR – objective response; TTR – time to first tumor response.
Patient-reported symptoms and overall quality of life were assessed using the questionnaire developed by the European Organisation for Research and Treatment of Cancer (EORTC) QLQ-C30 and the breast cancer module (EORTC QLQ-BR23). Overall, 335 patients in the fulvestrant plus palbociclib treatment group and 166 patients in the fulvestrant plus placebo treatment group completed the questionnaire at baseline and at least once during the first post-baseline visit.
Time to deterioration was predefined as the time between baseline pain level and the first occurrence of an increase ≥10 points compared to baseline on the pain symptom scale. Adding palbociclib to fulvestrant treatment resulted in a significantly delayed time to pain symptom deterioration compared to fulvestrant with placebo (median 8.0 months vs. 2.8 months, HR 0.64 [95% CI: 0.49; 0.85]; p<0.001).
Endometrial effects in the postmenopausal period
Preclinical data indicate the absence of stimulatory effects of fulvestrant on the endometrium in the postmenopausal period. A two-week study in healthy postmenopausal women receiving ethinylestradiol 20 µg daily showed that pretreatment with fulvestrant 250 mg significantly reduced the stimulatory effect on the endometrium compared to pretreatment with placebo, as measured by ultrasound assessment of endometrial thickness.
Neoadjuvant treatment for up to 16 weeks in breast cancer patients receiving either fulvestrant 500 mg or fulvestrant 250 mg did not result in clinically significant changes in endometrial thickness, indicating the absence of agonistic effects. To date, there is no evidence of adverse effects on the endometrium in breast cancer patients treated with fulvestrant. Morphological data on endometrial structure are not available.
In two short-term studies (1 and 12 weeks) involving premenopausal women with benign gynecological conditions, no statistically significant differences in endometrial thickness were observed between fulvestrant and placebo treatment groups, as confirmed by ultrasound imaging.
Bone effects
Long-term data on the effects of fulvestrant on bone are lacking. Neoadjuvant treatment for up to 16 weeks in breast cancer patients receiving either fulvestrant 500 mg or fulvestrant 250 mg did not result in clinically significant changes in serum markers of bone remodeling.
Pediatric population
Faslodex is not indicated for the treatment of children. The European Medicines Agency has waived the obligation to submit results of studies with Faslodex in all pediatric subpopulations with breast cancer (see section "Posology and method of administration" for information on use in children).
In an open-label phase 2 study, the safety, efficacy, and pharmacokinetics of fulvestrant were evaluated in 30 girls aged 1 to 8 years with progressive precocious puberty associated with McCune-Albright syndrome (MAS). Children received monthly intramuscular injections of 4 mg/kg fulvestrant. This 12-month study evaluated a range of efficacy endpoints for fulvestrant use in MAS. Study results showed reduced frequency of vaginal bleeding and slowed progression of bone age advancement. Steady-state trough concentrations of fulvestrant in children in this study were comparable to those observed in adults (see section "Pharmacokinetics"). No new safety concerns emerged during this small study, although five-year data are not yet available.
Pharmacokinetics.
Absorption
After intramuscular injection of the prolonged-release formulation of Faslodex, fulvestrant is slowly absorbed, with peak plasma concentration (Cmax) reached approximately on day 5. With the 500 mg dosing regimen of Faslodex, steady-state or near-steady-state exposure levels are achieved within the first month of treatment (mean [coefficient of variation (CV)]: AUC 475 [33.4%] ng·day/mL, Cmax 25.1 [35.3%] ng/mL, Cmin 16.3 [25.9%] ng/mL, respectively). At steady state, plasma concentrations of fulvestrant remain within a relatively narrow range, with approximately a threefold difference between maximum and minimum concentrations. After intramuscular administration in the dose range of 50 to 500 mg, exposure is approximately dose-proportional.
Distribution
Fulvestrant is extensively and rapidly distributed. The large apparent volume of distribution at steady state (Vdss) of approximately 3 to 5 L/kg indicates predominantly extravascular distribution.
Fulvestrant is highly bound (99%) to plasma proteins. The main binding components are very low-density lipoprotein (VLDL), low-density lipoprotein (LDL), and high-density lipoprotein (HDL) fractions. Studies on competitive protein binding interactions have not been conducted. The role of sex hormone-binding globulin (SHBG) has not been established.
Biotransformation
The metabolism of fulvestrant is not fully characterized but involves a combination of numerous possible biotransformation pathways, similar to those of endogenous steroids. Identified metabolites (including 17-keto, sulphone, 3-sulphate, 3- and 17-glucuronide) are either less active or exhibit similar antiestrogenic activity to fulvestrant in antiestrogenic models. Studies using human liver preparations and recombinant human enzymes indicate that CYP3A4 is the only P450 isoenzyme involved in the oxidation of fulvestrant; however, non-P450 pathways are believed to predominate in vivo. In vitro data indicate that fulvestrant does not inhibit CYP450 isoenzymes.
Elimination
Fulvestrant is primarily eliminated in metabolized form. The main route of elimination is fecal, with less than 1% excreted in urine. Fulvestrant has a high clearance of 11 ± 1.7 mL/min/kg, indicating a high hepatic extraction ratio. The terminal elimination half-life (t1/2) after intramuscular administration is determined by the absorption rate and is estimated at 50 days.
Special patient populations
Population pharmacokinetic analysis of phase 3 study data showed no differences in the pharmacokinetic profile of fulvestrant with regard to age (range 33 to 89 years), body weight (40 to 127 kg), or race.
Renal impairment
The impact of mild or moderate renal impairment on the pharmacokinetics of fulvestrant is not considered clinically significant.
Hepatic impairment
The pharmacokinetics of fulvestrant were evaluated in a clinical study using a single dose in women with mild and moderate hepatic impairment (Child-Pugh class A and B). A high dose for intramuscular injection was administered for a short duration. Compared to healthy subjects, women with hepatic impairment showed an almost 2.5-fold increase in AUC. An increase in exposure of this magnitude in patients receiving Faslodex is expected to be well tolerated. Women with severe hepatic impairment (Child-Pugh class C) were not studied.
Pediatric population
The pharmacokinetics of fulvestrant were evaluated in a clinical study involving 30 girls with progressive precocious puberty associated with McCune-Albright-Stevenson syndrome (see section "Pharmacodynamics"). Pediatric patients were aged 1 to 8 years and received intramuscular fulvestrant at a dose of 4 mg/kg monthly. The geometric mean (standard deviation) steady-state trough concentration (Cmin,ss) and AUCss were 4.2 (0.9) ng/mL and 3680 (1020) ng·h/mL, respectively. Although data are limited, steady-state trough concentrations of fulvestrant in children are likely comparable to those in adults.
Clinical characteristics.
Indications.
Faslodex is indicated:
- as monotherapy for the treatment of locally advanced or metastatic estrogen receptor-positive breast cancer in postmenopausal women:
- who have not previously received endocrine therapy, or
- in case of disease recurrence during or after adjuvant antiestrogen therapy or disease progression during antiestrogen therapy;
- in combination with palbociclib for the treatment of hormone receptor-positive (HR-positive), human epidermal growth factor receptor 2-negative (HER2-negative) locally advanced or metastatic breast cancer in women who have received prior endocrine therapy (see section "Pharmacodynamics").
In premenopausal or perimenopausal women, combination treatment with palbociclib should be administered in combination with a gonadotropin-releasing hormone (GnRH) agonist.
Contraindications.
Hypersensitivity to the active substance or to any of the excipients.
Pregnancy and lactation (see section "Use in pregnancy or breastfeeding").
Severe hepatic impairment (see sections "Special precautions" and "Pharmacokinetics").
Interaction with other medicinal products and other forms of interaction.
A clinical interaction study with midazolam (a CYP3A4 substrate) demonstrated that fulvestrant does not inhibit CYP3A4. Clinical interaction studies with rifampicin (a CYP3A4 inducer) and ketoconazole (a CYP3A4 inhibitor) showed no clinically significant changes in fulvestrant clearance. Therefore, dose adjustment is not required for patients receiving fulvestrant concomitantly with CYP3A4 inhibitors or inducers.
Special precautions for use
Faslodex should be used with caution in patients with mild to moderate hepatic impairment (see sections "Method of administration and dosage", "Contraindications", and "Pharmacokinetics").
Faslodex should be used with caution in patients with severe renal impairment (creatinine clearance less than 30 mL/min).
Due to the intramuscular route of administration, Faslodex should be used with caution in patients with hemorrhagic diathesis, thrombocytopenia, or those receiving anticoagulant therapy.
Thromboembolic events are commonly observed in women with advanced breast cancer and have been reported in clinical trials with Faslodex (see section "Adverse reactions"). This should be taken into account when prescribing Faslodex to patients at risk.
Injection site reactions, including sciatica, neuralgia, neuropathic pain, and peripheral neuropathy, have been reported following administration of Faslodex. Due to the proximity of the sciatic nerve, caution should be exercised when administering Faslodex into the upper outer quadrant of the gluteal region (see sections "Method of administration and dosage" and "Adverse reactions").
There are no long-term data on the effects of fulvestrant on bone. Due to the mechanism of action of fulvestrant, there is a potential risk of developing osteoporosis.
The safety and efficacy of Faslodex (as monotherapy or in combination with palbociclib) in patients with severe internal organ disease have not been studied.
For combination therapy with Faslodex and palbociclib, refer also to the summary of product characteristics for palbociclib.
Effect on estradiol assays using antibodies
Due to the structural similarity between fulvestrant and estradiol, fulvestrant may interfere with immunoassay-based estradiol measurements, leading to falsely elevated estradiol levels.
Ethanol
Faslodex contains 10% w/v ethanol (alcohol) as an excipient, i.e., up to 500 mg per injection, equivalent to 10 mL of beer or 4 mL of wine. This may be harmful to individuals suffering from alcoholism and should be considered in high-risk patients, particularly those with liver disease or epilepsy.
Benzyl alcohol
Faslodex contains benzyl alcohol as an excipient, which may cause allergic reactions.
Pediatric population
Faslodex is not recommended for use in children and adolescents, as its safety and efficacy in this age group have not been established (see section "Pharmacodynamics").
Use during pregnancy or breastfeeding
Women of reproductive potential
Women of reproductive potential must use effective contraception during treatment with Faslodex and for 2 years after the last dose.
Pregnancy
Faslodex is contraindicated during pregnancy (see section "Contraindications"). Studies in rats and rabbits have shown that fulvestrant crosses the placental barrier after a single intramuscular dose. Animal studies have demonstrated reproductive toxicity, including increased frequency of fetal malformations and fetal death. If a patient becomes pregnant while receiving Faslodex, she should be informed of the potential risk to the fetus and the potential risk of pregnancy loss.
Breastfeeding
Breastfeeding should be discontinued during treatment with Faslodex. Fulvestrant is excreted in milk in lactating rats. It is unknown whether fulvestrant is excreted in human breast milk. Due to the potential for serious adverse reactions in breastfed infants caused by fulvestrant, use of this medicinal product is contraindicated during breastfeeding (see section "Contraindications").
Fertility
The effect of Faslodex on human fertility has not been studied.
Ability to affect reaction speed when driving or operating machinery
Faslodex has no or negligible influence on the ability to drive or operate machinery. However, since asthenia has been reported very commonly during treatment with Faslodex, patients who experience this adverse reaction while driving or operating machinery should exercise caution.
Method of Administration and Dosage
Dosage
Adult women (including elderly patients)
The recommended dose is 500 mg, administered at one-month intervals; an additional 500 mg dose should be given two weeks after the first injection.
When using the medicinal product Faslodex in combination with palbociclib, also refer to the summary of product characteristics for palbociclib.
Women of pre-/perimenopausal status should receive LHRH agonists (luteinizing hormone-releasing hormone agonists) before and during combination therapy with Faslodex and palbociclib, in accordance with local clinical practice guidelines.
Special patient populations
Renal impairment
No dose adjustment is required in patients with mild to moderate renal impairment (creatinine clearance ≥ 30 mL/min). The efficacy and safety of the medicinal product have not been evaluated in patients with severe renal impairment (creatinine clearance < 30 mL/min); therefore, the drug should be used with caution in such patients (see section "Special precautions for use").
Hepatic impairment
No dose adjustment is required in patients with mild to moderate hepatic impairment. However, Faslodex should be used with caution in these patients due to the potential for increased fulvestrant exposure. Data in patients with severe hepatic impairment are lacking (see sections "Contraindications", "Special precautions for use", and "Pharmacokinetics").
Method of administration
Faslodex should be administered as two consecutive slow (1–2 minutes per injection) intramuscular injections of 5 mL each, one into each buttock (gluteal area).
Due to the proximity of the sciatic nerve, caution should be exercised when administering Faslodex into the upper outer quadrant of the gluteal region.
Instructions for administration
The medicinal product should be administered according to local guidelines for administering large-volume intramuscular injections.
NOTE. Due to the proximity of the sciatic nerve, caution should be exercised when administering Faslodex into the upper outer quadrant of the gluteal region (see section "Special precautions for use").
Warning: Do not autoclave the safety needle (the capped subcutaneous needle “BDSafetyGlide™”) before use.
At all times during use and disposal, hands should remain behind the needle.
For each of the two syringes:
|
Figure 1
|
|
Figure 2
|
|
Figure 3
|
|
Figure 4
|
NOTE. When activating, keep the needle pointed away from yourself and others. Listen for the click and visually confirm that the needle tip is fully covered. |
Figure 5
|
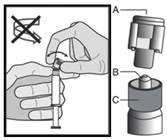
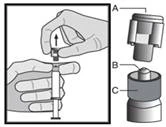
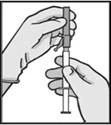
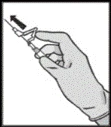
Disposal
Prefilled syringes are intended for single use only.
This medicinal product may be hazardous to the aquatic environment. Any unused medicinal product or waste material must be disposed of in accordance with local requirements.
Children.
The safety and efficacy of Faslodex in children (under 18 years of age) have not been established. The available data described in the sections “Pharmacokinetics” and “Pharmacodynamics” are insufficient to establish dosage recommendations in children.
Overdose.
There have been isolated reports of Faslodex overdose in humans. In the event of overdose, symptomatic and supportive treatment is recommended. Animal studies indicate no effects of higher doses of fulvestrant other than those directly or indirectly related to its antiestrogenic activity.
Adverse reactions.
Summary of safety profile
Monotherapy
This section provides information on all adverse reactions reported from clinical trials, post-marketing studies, or spontaneous reports. In the pooled monotherapy data set for fulvestrant, the most frequently reported adverse reactions are injection site reactions, asthenia, nausea, and increased levels of liver enzymes (ALT (alanine aminotransferase), AST (aspartate aminotransferase), ALP (alkaline phosphatase)).
The frequency categories of adverse reactions listed in Table 5 were derived from the pooled safety analysis of studies comparing Fulvestrant 500 mg versus Fulvestrant 250 mg [CONFIRM (study D6997C00002), FINDER 1 (study D6997C00004), FINDER 2 (study D6997C00006), and NEWEST (study D6997C00003)] or from the individual FALCON study (study D699BC00001), which compared Fulvestrant 500 mg with anastrozole 1 mg. Where the frequency of adverse reactions differed between the pooled safety analysis and the FALCON study, the higher frequency was used. The frequencies listed in Table 5 are based on data for all reported adverse reactions, regardless of the investigator's assessment of causal relationship. The median duration of treatment with fulvestrant 500 mg in the pooled data set (including the studies listed above and the FALCON study) was 6.5 months.
Tabulated list of adverse reactions
The adverse reactions listed below are classified by frequency and by system organ class. Frequency groupings are defined as follows: very common (≥ 1/10), common (≥ 1/100 to < 1/10), uncommon (≥ 1/1000 to < 1/100). Within each frequency group, adverse reactions are listed in order of decreasing severity.
Table 5. Adverse reactions reported in patients during monotherapy with the medicinal product Faslodex.
| Adverse reactions classified by frequency and system organ class |
||
| Infections and infestations |
Common |
Urinary tract infections |
| Blood and lymphatic system disorders |
Common |
Decreased platelet count |
| Immune system disorders |
Very common |
Hypersensitivity reactions |
| Uncommon |
Anaphylactic reactions |
|
| Metabolism and nutrition disorders |
Common |
Anorexia |
| Nervous system disorders |
Common |
Headache |
| Vascular disorders |
Very common |
Hot flushes |
| Common |
Venous thromboembolism |
|
| Gastrointestinal disorders |
Very common |
Nausea |
| Common |
Vomiting, diarrhea |
|
| Hepatobiliary disorders |
Very common |
Elevated liver enzymes (ALT, AST, ALP) |
| Common |
Elevated bilirubin levels |
|
| Uncommon |
Hepatic failure, hepatitis, elevated GGT levels |
|
| Skin and subcutaneous tissue disorders |
Very common |
Rash |
| Musculoskeletal and connective tissue disorders |
Very common |
Joint and muscle pain |
| Common |
Back pain |
|
| Reproductive system and breast disorders |
Common |
Vaginal bleeding |
| Uncommon |
Vaginal candidiasis, leukorrhea |
|
| General disorders and administration site conditions |
Very common |
Asthenia, injection site reactions |
| Common |
Peripheral neuropathy, sciatica |
|
| Uncommon |
Hemorrhage at injection site, hematoma at injection site, neuralgia |
|
a Includes adverse drug reactions for which a relationship to Faslodex cannot be established due to the underlying disease.
b The term "injection site reactions" does not include the terms "injection site haemorrhage" and "injection site haematoma", "sciatica", "neuralgia", "peripheral neuropathy".
c The reaction was not observed in large clinical trials (CONFIRM, FINDER 1, FINDER 2, NEWEST). The frequency was calculated using the upper limit of the 95% confidence interval for the point estimate. It was calculated as 3/560 (where 560 is the number of patients in the large clinical trials), corresponding to the frequency category "uncommon".
d Includes arthralgia and less frequently musculoskeletal pain, myalgia, and limb pain.
e There are some differences in the frequency of adverse reactions between the corresponding categories based on pooled safety data and the FALCON study.
f Adverse reactions were not observed in the FALCON study.
Description of selected adverse reactions.
The following description is based on safety analysis of a group of 228 female patients who received at least one (1) dose of fulvestrant and a group of 232 female patients who received at least one (1) dose of anastrozole in the phase 3 FALCON study.
Joint pain and musculoskeletal pain
According to data from the FALCON study, the number of patients reporting joint pain and musculoskeletal pain was 65 (31.2%) and 48 (24.1%) in patients treated with fulvestrant and anastrozole, respectively. Of the 65 patients receiving Faslodex, 40% (26/65) experienced joint and musculoskeletal pain within the first month of treatment, and 66.2% (43/65) within the first 3 months of treatment. None of the patients reported events of grade ≥3 according to CTCAE, or cases requiring dose reduction, temporary interruption, or discontinuation of the drug due to these adverse reactions.
Combination therapy with palbociclib
The overall safety profile of fulvestrant when used in combination with palbociclib is based on data from 517 female patients with HR-positive, HER2-negative locally advanced or metastatic breast cancer in the randomized PALOMA3 trial (see section "Pharmacodynamics"). The most common (≥20%) adverse reactions of any grade reported in patients receiving fulvestrant in combination with palbociclib were neutropenia, leukopenia, infections, fatigue, nausea, anemia, stomatitis, diarrhea, thrombocytopenia, and vomiting. The most common (≥2%) adverse reactions of grade ≥3 were neutropenia, leukopenia, infections, anemia, increased AST levels, thrombocytopenia, and fatigue.
Table 6 presents data on adverse reactions observed in the PALOMA3 study.
The median duration of fulvestrant treatment was 11.2 months in the fulvestrant + palbociclib group and 4.8 months in the fulvestrant + placebo group. The mean duration of palbociclib treatment in the fulvestrant + palbociclib group was 10.8 months.
Table 6. Adverse reactions from the PALOMA3 study (N=517)
| System organ class |
Faslodex + Palbociclib |
Faslodex + placebo (N=172) |
||
| All grades |
Grade ≥ 3 |
All grades n (%) |
Grade ≥ 3 |
|
| Infections and infestations |
||||
| Very common |
||||
| Infectionsb |
188 (54.5) |
19 (5.5) |
60 (34.9) |
6 (3.5) |
| Blood and lymphatic system disorders |
||||
| Very common |
||||
| Neutropeniac |
290 (84.1) |
240 (69.6) |
6 (3.5) |
0 |
| Leukopeniad |
207 (60.0) |
132 (38.3) |
9 (5.2) |
1 (0.6) |
| Anemiae |
109 (31.6) |
15 (4.3) |
24 (14.0) |
4 (2.3) |
| Thrombocytopeniaf |
88 (25.5) |
10 (2.9) |
0 |
0 |
| Uncommon |
||||
| Febrile neutropenia |
3 (0.9) |
3 (0.9) |
0 |
0 |
| Metabolism and nutrition disorders |
||||
| Very common |
||||
| Decreased appetite |
60 (17.4) |
4 (1.2) |
18 (10.5) |
1 (0.6) |
| Nervous system disorders |
||||
| Common |
||||
| Dysgeusia |
27 (7.8) |
0 |
6 (3.5) |
0 |
| Eye disorders |
||||
| Common |
||||
| Lacrimation increased |
25 (7.2) |
0 |
2 (1.2) |
0 |
| Blurred vision |
24 (7.0) |
0 |
3 (1.7) |
0 |
| Dry eye |
15 (4.3) |
0 |
3 (1.7) |
0 |
| Respiratory, thoracic and mediastinal disorders |
||||
| Common |
||||
| Nasopharyngitis |
25 (7.2) |
0 |
4 (2.3) |
0 |
| Gastrointestinal disorders |
||||
| Very common |
||||
| Nausea |
124 (35.9) |
2 (0.6) |
53 (30.8) |
1 (0.6) |
| Stomatitisg |
104 (30.1) |
3 (0.9) |
24 (14.0) |
0 |
| Diarrhea |
94 (27.2) |
0 |
35 (20.3) |
2 (1.2) |
| Vomiting |
75 (21.7) |
2 (0.6) |
28 (16.3) |
1 (0.6) |
| Skin and subcutaneous tissue disorders |
||||
| Very common |
||||
| Alopecia |
67 (19.4) |
Not applicable |
11 (6.4) |
Not applicable |
| Rashh |
63 (18.3) |
3 (0.9) |
10 (5.8) |
0 |
| Common |
||||
| Dry skin |
28 (8.1) |
0 |
3 (1.7) |
0 |
| General disorders and administration site conditions |
||||
| Very common |
||||
| Fatigue |
152 (44.1) |
9 (2.6) |
54 (31.4) |
2 (1.2) |
| Pyrexia |
47 (13.6) |
1 (0.3) |
10 (5.8) |
0 |
| Common |
||||
| Asthenia |
27 (7.8) |
1 (0.3) |
13 (7.6) |
2 (1.2) |
| Investigations |
||||
| Very common |
||||
| Increased AST |
40 (11.6) |
11 (3.2) |
13 (7.6) |
4 (2.3) |
| Common |
||||
| Increased ALT |
30 (8.7) |
7 (2.0) |
10 (5.8) |
1 (0.6) |
ALT – alanine aminotransferase; AST – aspartate aminotransferase; N/n – number of patients.
a Preferred terms (PT) according to MedDRA 17.1 are indicated.
b All PTs belonging to the class "Infections and infestations".
c Neutropenia includes the following PTs: neutropenia, decreased neutrophil count.
d Leukopenia includes the following PTs: leukopenia, decreased white blood cell count.
e Anemia includes the following PTs: anemia, decreased hemoglobin level, decreased hematocrit level.
f Thrombocytopenia includes the following PTs: thrombocytopenia, decreased platelet count.
g Stomatitis includes the following PTs: aphthous stomatitis, cheilitis, glossitis, glossodynia, oral ulceration, mucosal inflammation, oral pain, oropharyngeal discomfort, oropharyngeal pain, stomatitis.
h Rash includes the following PTs: rash, maculopapular rash, rash with pruritus, erythematous rash, papular rash, dermatitis, acneiform dermatitis, toxic skin eruption.
Description of selected adverse reactions
Neutropenia
In the PALOMA3 study, in which fulvestrant was administered in combination with palbociclib, neutropenia of any grade was reported in 290 (84.1%) patients, grade 3 neutropenia in 200 (58.0%) patients, and grade 4 neutropenia in 40 (11.6%) patients. In the fulvestrant + placebo group (n = 172), neutropenia of any grade was reported in 6 (3.5%) patients. No cases of grade 3 or 4 neutropenia were reported in the fulvestrant + placebo group.
In patients receiving fulvestrant in combination with palbociclib, the median time to first episode of neutropenia was 15 days (range: 13–512 days), and the median duration of grade ≥3 neutropenia was 16 days. Febrile neutropenia was reported in 3 (0.9%) patients receiving fulvestrant in combination with palbociclib.
Reporting of suspected adverse reactions
It is important to report suspected adverse reactions during the post-marketing period of the medicinal product. This allows for continuous monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are obliged to report any suspected adverse reactions through the national reporting system.
Shelf life.
4 years.
Storage conditions.
Store at 2 to 8 °C.
Keep out of the reach of children.
Store pre-filled syringes in the original packaging to protect from light.
Temperature excursions outside the range of 2 to 8 °C should be limited. Exposure to temperatures above 30 °C should be avoided, and the cumulative time at which the average storage temperature of the medicinal product is below 25 °C (but above 2–8 °C) must not exceed 28 days. After temperature excursions, the medicinal product should be returned immediately to the recommended storage conditions (refrigerated storage and transport at 2 to 8 °C). Temperature excursions have a cumulative effect on the quality of the medicinal product, and the 28-day period must not be exceeded during the 4-year shelf life of Faslodex. Exposure to temperatures below 2 °C does not damage the medicinal product provided it is not stored below –20 °C.
Incompatibilities.
Since compatibility studies are lacking, this medicinal product must not be mixed with other medicinal products.
Packaging.
1 cardboard box contains a blister pack containing 2 pre-filled glass syringes with tamper-evident device, each containing 5 ml of solution, with two safety needles “BD SafetyGlide™”.
Prescription category.
Prescription only.
Manufacturer.
AstraZeneca UK Limited.
Manufacturer’s name and address.
Silk Road Business Park, Macclesfield, SK10 2NA, United Kingdom.