Droszpyfen® 30
Ukraina
Spis treści
INSTRUKCJA dot. stosowania leku Droszpyfen® 30
Skład:
substancje czynne: etyniloestradiol, drosperynon;
1 tabletka zawiera: 0,03 mg etyniloestradiolu i 3 mg drosperynonu;
substancje pomocnicze: laktoza monohydrat, skrobia kukurydziana, maltodekstryna, stearynian magnezu, Opadry 10A32290 żółty (hipromeloza, talk, dwutlenek tytanu (E 171), polisorbat 80, tlenek żelaza żółty (E 172)).
Postać farmaceutyczna. Tabletki powlekane.
Główne właściwości fizykochemiczne: okrągłe tabletki powlekane, żółte, bez wad powłoki.
Grupa farmakoterapeutyczna. Hormony gonadalne i leki stosowane w patologii układu rozrodczego. Hormonalne środki zapobiegające ciąży stosowane systemowo.
Gestageny i estrogeny, stałe kombinacje. Drosperynon i etyniloestradiol.
Kod ATX G03A A12.
Właściwości farmakologiczne.
Farmakodynamika.
Indeks Pearl niepowodzeń antykoncepcyjnych dla leku: 0,09 (dwustronny górny 95 % przedział ufności (PU): 0,32).
Ogólny indeks Pearl (niepowodzenia antykoncepcyjne + błędy po stronie pacjentek): 0,57 (dwustronny górny 95 % przedział ufności (PU): 0,90).
Droszpyfen® 30 to skojarzony doustny środek antykoncepcyjny zawierający etynylestradiol i drosperynon. W dawkach terapeutycznych drosperynon wykazuje działanie antyandrogenowe oraz umiarkowane działanie antymineralokortykoidowe. Nie posiada aktywności estrogenowej, glikokortykosteroidalnej ani antyglukokortykosteroidalnej. Drosperynon ma zatem profil farmakologiczny podobny do naturalnego progesteronu.
Mechanizm działania przeciwwapnowego opiera się na oddziaływaniu różnych czynników, z których najważniejsze to hamowanie owulacji i zmiany w wydzielinie szyjki macicy.
Zgodnie z danymi badań klinicznych umiarkowane działanie antymineralokortykoidowe leku Droszpyfen® 30 prowadzi do umiarkowanego wpływu antymineralokortykoidowego.
Farmakokinetyka.
Drosperynon
Wchłanianie. Po podaniu doustnym drosperynon jest szybko i niemal całkowicie wchłaniany. Maksymalna stężenie w surowicy – 38 ng/ml – osiągane jest około 1–2 godziny po jednorazowym podaniu. Bio dostępność wynosi 76–85 %. Jednoczesne spożycie pokarmu nie wpływa na biodostępność drosperynonu.
Rozkład. Po podaniu doustnym stężenie drosperynonu w surowicy maleje ze średniem końcowym okresem półtrwania około 31 godzin. Drosperynon wiąże się z albuminą surowicy, nie łącząc się z globuliną wiążącą sterydy płciowe (SHBG) ani z globuliną wiążącą kortykosteroidy (CBG). Tylko 3–5 % jego całkowitej ilości w surowicy krwi występuje w formie wolnej. Spowodowane etynylestradiolem zwiększenie poziomu SHBG nie wpływa na wiązanie drosperynonu z białkami surowicy krwi. Średni objętość rozkładu drosperynonu wynosi 3,7±1,2 l/kg.
Metabolizm. Drosperynon jest w znacznym stopniu metabolizowany po podaniu doustnym. Głównymi metabolitami w osoczu krwi są forma kwasowa drosperynonu, powstająca w wyniku otwarcia pierścienia laktonowego, oraz 4,5-dihydro-drosperynon-3-siarczan, powstający przez hydratację z późniejszym sulfowaniem. Drosperynon jest również poddawany metabolizmowi oksydacyjnemu katalizowanemu przez CYP3A4. In vitro drosperynon może słabo lub umiarkowanie hamować enzymy cytochromu P450: CYP1A1, CYP2C9, CYP2C19 oraz CYP3A4.
Wydalanie. Prędkość klirensu metabolicznego drosperynonu z surowicy krwi wynosi około 1,5±0,2 ml/min/kg. Zaledwie niewielka ilość drosperynonu wydala się w postaci niezmienionej. Metabolity wydalane są z moczem i kałem w stosunku 1,2 : 1,4; okres półtrwania metabolitów wynosi około 40 godzin.
Stan równowagi. W trakcie cyklu stosowania maksymalne stężenie równowagowe drosperynonu w surowicy krwi (około 70 ng/ml) osiągane jest po 8 dniach przyjmowania. Poziomy drosperynonu we krwi wzrastały trzykrotnie w wyniku stosunku czasu trwania okresu końcowego półtrwania do odstępu dawkowania.
Osobne kategorie pacjentek:
- z zaburzeniem funkcji nerek: stężenie równowagowe drosperynonu w surowicy krwi u kobiet z niewydolnością nerek lekkiego stopnia nasilenia (klirens kreatyniny 50–80 ml/min) było porównywalne do tej wartości u kobiet z normalną funkcją nerek. Poziom drosperynonu w surowicy krwi był średnio o 37 % wyższy u kobiet z niewydolnością nerek średniego stopnia nasilenia (klirens kreatyniny 30–50 ml/min) w porównaniu do tej wartości u kobiet z normalną funkcją nerek. Terapia drosperynonem była dobrze tolerowana przez kobiety z niewydolnością nerek lekkiego i umiarkowanego stopnia. Wykazano, że przyjmowanie drosperynonu nie wywiera klinicznie istotnego wpływu na stężenie potasu w surowicy krwi;
- z zaburzeniem funkcji wątroby: w badaniu podania pojedynczej dawki klirens drosperynonu po podaniu doustnym obniżał się o około 50 % u osób z umiarkowaną niewydolnością wątroby w porównaniu z ochotnikami z normalną funkcją wątroby. Stwierdzone odchylenie klirensu nie spowodowało żadnej różnicy w stężeniach potasu we krwi między dwiema grupami ochotników. Nawet przy cukrzycy i współistniejącej terapii spironolaktonem (dwa czynniki mogące wywoływać hiperkaliemię) nie zaobserwowano wzrostu stężenia potasu w surowicy krwi powyżej górnej granicy normy. Można stwierdzić, że drosperynon jest dobrze tolerowany przez osoby z niewydolnością wątroby lekkiego lub średniego stopnia (klasa B wg klasyfikacji Childa-Pugha).
Etynylestradiol
Wchłanianie. Po podaniu doustnym etynylestradiol jest szybko i całkowicie wchłaniany. Po podaniu 30 µg maksymalne stężenie w surowicy 100 pg/ml osiągane jest w ciągu 1–2 godzin. Etynylestradiol podlega intensywnemu efektowi pierwszego przejścia, zależnemu od różnic indywidualnych.
Bezwzględna biodostępność wynosi około 45 %.
Rozkład. Oczekiwany objętość rozkładu etynylestradiolu wynosi około 5 l/kg, a wiązanie z białkami osocza krwi – około 98 %. Etynylestradiol indukuje syntezę w wątrobie SHBG oraz globulin wiążących hormony kortykosteroidowe. Po podaniu 30 µg etynylestradiolu stężenie SHBG w osoczu wzrasta od 70 do około 350 nmol/l.
Etynylestradiol w niewielkiej ilości przenika do mleka matki (0,02 % dawki).
Metabolizm. Etynylestradiol jest w znacznym stopniu metabolizowany w przewodzie pokarmowym i przy pierwszym przejściu przez wątrobę. Głównie zachodzi to poprzez hydroksylację pierścienia aromatycznego, z tworzeniem szerokiego spektrum hydroksylowanych i metylowanych metabolitów, występujących w formie wolnej oraz jako koniugaty z glukuronidami i siarczanami. Klirens metaboliczny etynylestradiolu wynosi około 5 ml/min/kg.
In vitro etynylestradiol jest odwracalnym inhibitorem CYP2C19, CYP1A1 i CYP1A2, a także inhibitorem CYP3A4/5, CYP2C8 i CYP2J2.
Wydalanie. Etynylestradiol praktycznie nie wydala się w niezmienionej formie. Metabolity etynylestradiolu wydalane są z moczem i żółcią w stosunku 4:6. Okres półtrwania metabolitów wynosi prawie 1 dobę. Okres półtrwania metabolitów wynosi 20 godzin.
Stan równowagi. Stan równowagi osiągany jest w drugiej połowie cyklu przyjmowania, kiedy stężenie etynylestradiolu w surowicy wzrasta 1,4–2,1 razy.
Przynależność etniczna
Nie zaobserwowano klinicznie istotnych różnic w farmakokinetyce drosperynonu lub etynylestradiolu u kobiet japońskiego pochodzenia i kobiet rasy europejskiej.
Dane przedkliniczne dotyczące bezpieczeństwa.
U zwierząt laboratoryjnych efekty drosperynonu i etynylestradiolu ograniczały się do tych, które były związane z znanym działaniem farmakologicznym. W szczególności badania dotyczące wykrywania toksyczności rozrodczej u zwierząt wykazały obecność wpływu embrionotoksycznego i fetotoksycznego charakterystycznego dla gatunku. Przy ekspozycji przekraczającej ekspozycję użytkowników Droszpyfen® 30, u niektórych gatunków zwierząt obserwowano wpływ na różnicowanie płciowe.
Charakterystyka kliniczna.
Wskazania.
Antykoncepcja doustna.
Decyzję o przepisaniu Droszpyfen® 30 należy podjąć z uwzględnieniem indywidualnych czynników ryzyka pacjentki, w szczególności czynników ryzyka żylnej tromboembolii (ŻTE). Należy również porównać ryzyko ŻTE podczas leczenia Droszpyfen® 30 z ryzykiem podczas stosowania innych hormonalnych środków antykoncepcyjnych (HAK) (szczegóły w sekcjach „Przeciwwskazania” oraz „Szczególne ostrzeżenia i środki ostrożności podczas stosowania”).
Przeciwwskazania.
HAK nie należy stosować w przypadku występowania choćby jednego z poniższych stanów. Jeśli którykolwiek z tych stanów wystąpi po raz pierwszy podczas stosowania HAK, lek należy natychmiast odstawić.
- Występowanie lub ryzyko rozwoju żylnej tromboembolii (ŻTE):
- aktualna żylowa tromboembolia, np. wymagająca leczenia antykoagulantami, lub w wywiadzie (np. zakrzepica żył głębokich (ZŻG) lub zatorowość płucna (ZP));
- dziedziczna lub nabyta skłonność do żylnej tromboembolii, w szczególności oporność na aktywowany białko C (w tym mutacja czynnika V Leiden), niedobór antytrombiny-III, niedobór białka C, niedobór białka S;
- duże zabiegi chirurgiczne z długotrwałym unieruchomieniem (patrz sekcja „Szczególne ostrzeżenia i środki ostrożności podczas stosowania”);
- wysokie ryzyko żylnej tromboembolii ze względu na obecność wielu czynników ryzyka (patrz sekcja „Szczególne ostrzeżenia i środki ostrożności podczas stosowania”).
- Występowanie lub ryzyko rozwoju tętniczej tromboembolii (ATE):
- aktualna tętnicza tromboembolia lub w wywiadzie (np. zawał mięśnia sercowego) lub występowanie objawów prodromalnych (np. dławica piersiowa);
- aktualne zaburzenia krążenia mózgowego lub w wywiadzie, występowanie objawów prodromalnych (np. przejściowy napad niedokrwienny (TIA));
- dziedziczna lub nabyta skłonność do tętniczej tromboembolii, w szczególności hiperhomocysteinemia i przeciwciała przeciwko fosfolipidom (przeciwciała przeciwko kardiolipinie, antykoagulant wąsaczkowy);
- migrena z objawami ogniskowymi neurologicznymi w wywiadzie;
- wysokie ryzyko tętniczej tromboembolii ze względu na obecność wielu czynników ryzyka (patrz sekcja „Szczególne ostrzeżenia i środki ostrożności podczas stosowania”) lub ze względu na obecność jednego poważnego czynnika ryzyka, takiego jak:
- cukrzyca z powikłaniami naczyniowymi;
- ciężka nadciśnienie tętnicze;
- ciężka dyslipoproteynemia.
- Aktualne lub w wywiadzie ciężkie schorzenie wątroby, dopóki wskaźniki funkcji wątroby nie powrócą do normy.
- Niedostateczność nerek w ciężkim stopniu lub ostra niewydolność nerek.
- Aktualne lub w wywiadzie guzy wątroby (łagodne lub złośliwe).
- Aktualne lub w wywiadzie raka piersi, który może być wrażliwy na hormony (patrz sekcja „Szczególne ostrzeżenia i środki ostrożności podczas stosowania”, podsekcja „Nowotwory”).
- Niepoddające się wyjaśnieniu pochodzenia krwawienie pochwowe.
- Jednoczesne stosowanie z lekami zawierającymi ombitaswir/paritaprewir/rytonawir i dazabuwir, z lekami zawierającymi glekaprewir/pibrentaswir lub sofosbuvir/velpataswir/woxilaprewir (patrz sekcja „Interakcje z innymi lekami i inne formy interakcji”).
- Nadwrażliwość na substancje czynne lub którykolwiek z dodatkowych składników leku.
Interakcje z innymi lekami i inne formy interakcji.
W przypadku występowania któregokolwiek z poniższych stanów lub czynników ryzyka należy omówić z pacjentką celowość stosowania leku Droszpyfen® 30.
Wpływ innych leków na Droszpyfen® 30
Możliwe są interakcje z lekami indukującymi enzymy mikrosomalne. Może to prowadzić do zwiększenia klirensu hormonów płciowych, co z kolei może spowodować wystąpienie krwawień „przerwowych” i/lub utratę skuteczności środka antykoncepcyjnego.
Leczenie
Indukcja enzymów może pojawić się już po kilku dniach leczenia. Maksymalna indukcja enzymów występuje zazwyczaj po kilku tygodniach. Po odstawieniu leczenia indukcja enzymów może trwać około 4 tygodni.
Leczenie krótkoterminowe
Kobiety przyjmujące leki indukujące enzymy powinny tymczasowo stosować metodę barierową lub inny środek antykoncepcyjny dodatkowo do hormonalnego środka antykoncepcyjnego doustnego (HAD). Metodę barierową należy stosować przez cały okres wspomocniczego leczenia i przez kolejne 28 dni po jego zakończeniu.
Jeśli wspomocnicze leczenie trwa po zakończeniu przyjmowania ostatnich tabletek HAD z opakowania, należy od razu rozpocząć przyjmowanie tabletek z następnego opakowania HAD bez przerwy w przyjmowaniu tabletek.
Leczenie długoterminowe
Kobietom poddawanym długoterminowemu leczeniu substancjami czynnymi indukującymi enzymy wątrobowe, zaleca się stosowanie metody barierowej lub innego niehormonalnego, niezawodnego środka antykoncepcyjnego.
<Poniżej wymienione interakcje zostały potwierdzone na podstawie opublikowanych danych
Substancje czynne zwiększające klirens HAD (obniżenie skuteczności HAD poprzez indukcję enzymów), np.:
barbiturany, bosen tan, karbamazepina, fenytoina, primidon, ryfampicyna; leki stosowane w HIV: rytonawir, nevirapyna i efawi rencja; a także, możliwe, felbamata, gryzeofulwina, okskarbazepina, topiramat i roślinne leki zawierające ekstrakt z zielca (Hypericum perforatum).
Substancje czynne o niestabilnym wpływie na klirens HAD:
Jednoczesne stosowanie z HAD duża liczba kombinacji inhibitorów HIV-proteazy i nie-nukleozydowych inhibitorów odwrotnej transkryptazy, w tym kombinacje z inhibitorami wirusa zapalenia wątroby C (WZW C), może zwiększać lub zmniejszać stężenia estrogenów lub progestagenów we krwi. Łączny wpływ takich zmian może być klinicznie istotny w niektórych przypadkach.
Dlatego należy zapoznać się z informacjami dotyczącymi stosowania leku leczącego HIV/WZW C, które są przyjmowane równocześnie, w celu wykrycia potencjalnych interakcji i wszelkich innych zaleceń. W przypadku jakichkolwiek wątpliwości kobiety powinny dodatkowo stosować metodę barierową podczas terapii inhibitorami proteazy lub nie-nukleozydowymi inhibitorami odwrotnej transkryptazy.
Substancje czynne obniżające klirens HAD (inhibitory enzymów)
Kliniczne znaczenie potencjalnej interakcji z inhibitorami enzymów pozostaje niejasne.
Jednoczesne stosowanie silnych inhibitorów CYP3A4 może zwiększyć stężenia estrogenów lub progestagenów we krwi, lub obu składników.
W badaniu wielokrotnych dawek kombinacji drosperenonu (3 mg/dobę)/etyniloestradiolu (0,02 mg/dobę) z jednoczesnym stosowaniem silnego inhibitora CYP3A4 ketokonazolu przez 10 dni wartości AUC(0-24h) drosperenonu i etyniloestradiolu zwiększyły się odpowiednio 2,7 i 1,4 razy.
Podczas jednoczesnego przyjmowania etorykoksibu w dawkach od 60 do 120 mg/dobę z hormonalnym środkiem antykoncepcyjnym zawierającym 0,035 mg etyniloestradiolu stwierdzono wzrost stężeń etyniloestradiolu we krwi odpowiednio 1,4–1,6 razy.
Wpływ na inne leki. Środki antykoncepcyjne doustne mogą wpływać na metabolizm innych leków. Mogą one zmieniać stężenia substancji czynnych we krwi i tkankach: zwiększać (np. cyklosporyna) lub zmniejszać (np. lamotrygina).
Na podstawie dostępnych badań in vivo i badań interakcji in vitro przeprowadzonych na kobietach ochotniczkach przyjmujących omeprazol, symwastatynę i midazolam jako substancje wskaźnikowe, wpływ drosperenonu w dawce 3 mg na inne leki, których metabolizm odbywa się przy udziale cytochromu P450, jest mało prawdopodobny.
Dane kliniczne wskazują, że etyniloestradiol hamuje klirens substratów CYP1A2, co prowadzi do słabego (np. teofiline) lub umiarkowanego (np. tizanidyna) wzrostu ich stężeń we krwi.
Inne formy interakcji. U pacjentek z niewydolnością nerek jednoczesne stosowanie drosperenonu i inhibitorów ACE lub niesteroidowych leków przeciwzapalnych nie wykazuje istotnego wpływu na poziom potasu w surowicy. W takim przypadku konieczne jest badanie poziomu potasu w surowicy w pierwszym cyklu przyjmowania leku (patrz również sekcja „Szczególne ostrzeżenia i środki ostrożności podczas stosowania”).
Badania laboratoryjne
Stosowanie HAD może wpływać na wyniki niektórych badań laboratoryjnych, takich jak biochemiczne wskaźniki funkcji wątroby, tarczycy, nadnerczy i nerek, na stężenie we krwi białek transportowych, takich jak globulina wiążąca kortykosteroidy, na stężenie we krwi frakcji lipidów/lipoproteinów, na wskaźniki gospodarki węglowodanowej, krzepnięcia i fibrynolizy. Zazwyczaj takie zmiany mieszczą się w granicach normy. Drosperenon zwiększa aktywność reniny i aldosteronu we krwi, co jest spowodowane jego umiarkowaną aktywnością antymineralokortykoidową.
Interakcje farmakodynamiczne
Podczas badań klinicznych u pacjentów leczonych z powodu zakażenia WZW C za pomocą leków zawierających ombitaswir/paritaprewir/rytonawir i dazabuwir z lub bez rybawiryny, znacznie częściej występowało podwyższenie stężenia aminotransferaz (ALT) przekraczające górny limit normy (GLN) 5-krotnie u kobiet stosujących leki zawierające etyniloestradiol, takie jak hormonalne środki antykoncepcyjne (HAK). Ponadto podwyższenie stężenia ALT obserwowano również podczas stosowania leków przeciwwirusowych zawierających glekaprewir/pibrentaswir lub sofosbuvir/velpataswir/woxilaprewir u kobiet przyjmujących leki zawierające etyniloestradiol, takie jak HAK (patrz sekcja „Przeciwwskazania”). W związku z tym pacjentki przyjmujące Droszpyfen® 0 powinny przejść na alternatywną metodę antykoncepcji (np. tylko progestagenową antykoncepcję lub niehormonalne metody antykoncepcji) przed rozpoczęciem leczenia tymi kombinowanymi schematami leczenia. Leczenie Droszpyfen® 30 można wznowić po 2 tygodniach od zakończenia leczenia tymi kombinowanymi schematami leczenia.
Szczególne zagadnienia dotyczące stosowania.
W przypadku występowania któregokolwiek z poniżej wymienionych stanów lub czynników ryzyka należy dokładnie przeanalizować potencjalne ryzyko i oczekiwaną korzyść z zastosowania COC w każdym przypadku indywidualnym. W przypadku nasilenia się, pogorszenia lub pojawienia się któregokolwiek z poniżej wymienionych stanów lub czynników ryzyka zaleca się skonsultować się z lekarzem, który może podjąć decyzję o zaprzestaniu przyjmowania leku.
W przypadku podejrzenia lub potwierdzenia VTE lub ATE należy zaprzestać stosowania leku. Jeśli rozpoczęto terapię lekami przeciwkrzepicznymi, należy zapewnić alternatywną skuteczną metodę antykoncepcji ze względu na działanie teratogenne leków przeciwkrzepiczych (kumaryny).
- Zaburzenia krążenia
Częstość występowania chorób zakrzepowo-zatorowych żylnych i tętniczych u kobiet bez czynników ryzyka przyjmujących COC o niskiej dawce estrogenów (poniżej 50 µg etynylowej estradiolu), takich jak Droszpyfen® 30, wynosi od 20 do 40 przypadków na 100 000 kobiet na rok, jednak ryzyko to waha się w zależności od poziomu progestyn. Dla porównania: u kobiet nie stosujących środków antykoncepcyjnych częstość tych chorób wynosi od 5 do 10 przypadków na 100 000.
Ryzyko wystąpienia żylnej tromboembolii (VTE)
Stosowanie dowolnych COC zwiększa ryzyko wystąpienia żylnej tromboembolii (VTE) u kobiet je przyjmujących, w porównaniu z kobietami, które ich nie stosują. Leki zawierające lewonorzegestrel, norgestymat lub norogetrion są związane z niższym ryzykiem VTE. Stosowanie innych leków, takich jak Droszpyfen® 30, może zwiększać ryzyko dwukrotnie. Decyzję o stosowaniu leków innych niż te o najniższym ryzyku VTE należy podejmować wyłącznie po konsultacji z kobietą. Należy upewnić się, że kobieta rozumie ryzyko VTE związane ze stosowaniem leku Droszpyfen® 30, wpływ posiadanych przez nią czynników ryzyka oraz fakt, że ryzyko VTE jest najwyższe w pierwszym roku stosowania. Według niektórych danych ryzyko VTE może wzrastać po wznowieniu stosowania COC po przerwie trwającej 4 tygodnie lub dłużej.
Ryzyko wystąpienia żylnych powikłań zakrzepowo-zatorowych u kobiet stosujących COC może być znacznie wyższe przy obecności dodatkowych czynników ryzyka, szczególnie wielu (patrz tabela).
Droszpyfen® 30 jest przeciwwskazany u kobiet z kombinacją czynników ryzyka, które tworzą bardzo wysokie ryzyko żylnego zakrzepicy (patrz sekcja „Przeciwwskazania”). Jeśli kobieta ma więcej niż jeden czynnik ryzyka, wzrost ryzyka może być większy niż suma ryzyk związanych z każdym pojedynczym czynnikiem, dlatego należy wziąć pod uwagę ogólne ryzyko wystąpienia VTE. Jeśli stosunek korzyści do ryzyka uznaje się za negatywny, nie należy przepisywać COC (patrz sekcja „Przeciwwskazania”).
U około dwóch na 10 000 kobiet, które nie stosują COC i nie są w ciąży, rozwija się VTE w ciągu jednego roku obserwacji. Jednak dla każdej konkretnej kobiety ryzyko może być znacznie wyższe, w zależności od czynników ryzyka (patrz poniżej).
Ustalono, że u 9–12 kobiet spośród 10 000 kobiet stosujących COC zawierające droszpirenon rozwinie się VTE w ciągu jednego roku. Dane te oparte są na wszystkich dostępnych danych epidemiologicznych z uwzględnieniem ryzyka względnego związanego ze stosowaniem różnych COC w porównaniu ze stosowaniem COC zawierających lewonorzegestrel.
Porównując, wskaźnik wynosi 6 u kobiet stosujących COC zawierające lewonorzegestrel. Średnio 5–7 przypadków na 10 000 kobiet-roku, obliczonych na podstawie ryzyka względnego stosowania COC zawierających lewonorzegestrel w porównaniu z kobietami nieprzyjmującymi COC (około 2,3–3,6 przypadków).
W obu przypadkach liczba wystąpień VTE w ciągu roku była mniejsza niż zwykle oczekuje się w okresie ciąży lub po porodzie.
VTE prowadzi do zgonu w 1–2% przypadków. Dodatkowe ryzyko wystąpienia VTE jest najwyższe w pierwszym roku stosowania kombinowanego środka antykoncepcyjnego.
Liczba przypadków VTE na 10 000 kobiet w ciągu 1 roku
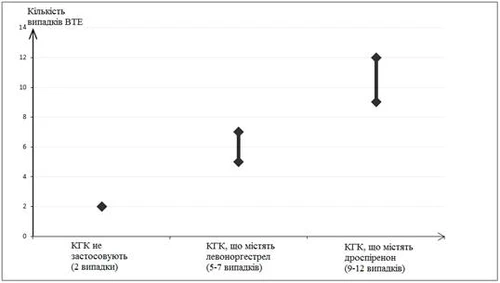
Czynniki ryzyka rozwoju VTE
Ryzyko wystąpienia żylnych powikłań zakrzepowo-zatorowych u kobiet stosujących COC może być znacznie wyższe przy obecności dodatkowych czynników ryzyka, szczególnie wielu (patrz tabela).
Droszpyfen® 30 jest przeciwwskazany u kobiet z wieloma czynnikami ryzyka, które mogą zwiększyć ryzyko żylnego zakrzepicy (patrz sekcja „Przeciwwskazania”). Jeśli kobieta ma więcej niż jeden czynnik ryzyka, wzrost ryzyka może być większy niż suma ryzyk związanych z każdym pojedynczym czynnikiem, dlatego należy wziąć pod uwagę ogólne ryzyko wystąpienia VTE. Jeśli stosunek korzyści do ryzyka jest niekorzystny, nie należy przepisywać COC (patrz sekcja „Przeciwwskazania”).
Czynniki ryzyka rozwoju VTE
| Czynnik ryzyka |
Uwaga |
| Otyłość (wskaźnik masy ciała przekracza 30 kg/m²). |
Ryzyko znacznie wzrasta wraz ze wzrostem wskaźnika masy ciała. Wymaga szczególnej uwagi, zwłaszcza przy obecności innych czynników ryzyka. |
| Długotrwała immobilizacja, duża interwencja chirurgiczna, operacja kończyn dolnych lub narządów miednicy, zabieg neurochirurgiczny lub poważny uraz. Uwaga: tymczasowa immobilizacja, w tym loty > 4 godziny, może również stanowić czynnik ryzyka wystąpienia ZTŻ, szczególnie u kobiet z innymi czynnikami ryzyka. |
Zaleca się przerwanie stosowania leku (w przypadku planowanego zabiegu chirurgicznego – co najmniej 4 tygodnie wcześniej) oraz nie wznowienie stosowania wcześniej niż po 2 tygodniach od pełnego przywrócenia aktywności ruchowej. W celu uniknięcia niepożądanej ciąży należy stosować inne metody antykoncepcji. Należy rozważyć celowość terapii przeciwcewnotrombotycznej, jeśli stosowanie leku nie zostało wcześniej przerwane. |
| Anamneza rodzinna (tromboembolia żylna u krewnych pierwszego stopnia lub rodziców, szczególnie w stosunkowo młodym wieku, np. poniżej 50 roku życia). |
W przypadku dziedzicznej predyspozycji przed zastosowaniem dowolnych CHC kobiety powinny skonsultować się ze specjalistą. |
| Inne stany związane z ZTŻ. |
Nowotwór, toczeń układowy, zespół hemolityczno-mocznicowy, przewlekłe zapalenie jelita (choroba Leśniowskiego-Crohna lub wrzodziejące zapalenie jelita grubego) oraz anemia sierpowata. |
| Wiek. |
Szczególnie powyżej 35 roku życia. |
Nie ma jednolitego stanowiska w kwestii możliwego wpływu żylaków oraz zakrzepowego zapalenia żył powierzchownych na rozwój i postęp zakrzepicy żylnej.
Należy zwrócić uwagę na zwiększony ryzyko wystąpienia zakrzepowo-zatorowej choroby płucnej (ZTE) w okresie ciąży, szczególnie w ciągu 6 tygodni po porodzie (informacje dotyczące okresu ciąży lub karmienia piersią znajdują się w punkcie „Stosowanie w okresie ciąży lub karmienia piersią”).
Objawy ZTE (zakrzepica żył głębokich i zatorowość płucna)
Należy zalecić kobietom, aby w przypadku pojawienia się poniższych objawów niezwłocznie skontaktować się z lekarzem i poinformować o przyjmowaniu COC.
Objawami zakrzepicy żył głębokich (ZŽG) mogą być:
- jednostronne obrzęki nogi i/lub stopy lub obszaru wzdłuż żyły na nodze;
- ból lub zwiększona wrażliwość w nodze, które mogą być odczuwane tylko w pozycji stojącej lub podczas chodzenia;
- uczucie gorąca w dotkniętej nodze; zaczerwienienie lub zmiana koloru skóry na nodze.
Objawami zatorowości płucnej (ZP) mogą być:
- nagłe duszności o nieustalonej etiologii lub przyśpieszone oddychanie;
- nagłe kaszle, które mogą towarzyszyć plwocina z krwią;
- ostry ból w klatce piersiowej;
- silne zawroty głowy lub uczucie zawrotów;
- przyśpieszone lub nieregularne bicie serca.
Niektóre z powyższych objawów (np. duszność, kaszel) są niestosunkowe i mogą być błędnie interpretowane jako objawy częstszych i mniej poważnych stanów (np. jako objawy infekcji dróg oddechowych).
Innymi objawami okluzji naczyniowej mogą być: nagły ból kończyny, obrzęk, ostry ból brzucha oraz niewielki cianoz skóry kończyn.
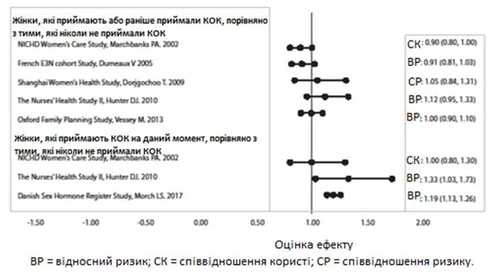
Dotyczące badania ryzyka raka piersi przy stosowaniu doustnych środków antykoncepcyjnych skojarzonych
W trzech badaniach porównywano ryzyko wystąpienia raka piersi u osób, które aktualnie przyjmowały COC lub przyjmowały je niedawno (< 6 miesięcy od ostatniego zażycia), z osobami, które nigdy nie przyjmowały COC. W jednym z tych badań nie odnotowano związku między ryzykiem wystąpienia raka piersi a stosowaniem COC. Dwa inne badania wykazały zwiększone ryzyko względne w granicach 1,19–1,33 przy aktualnym lub niedawnym stosowaniu COC. Oba badania wykazały zwiększone ryzyko wystąpienia raka piersi przy długotrwałym stosowaniu w momencie badania, przy czym ryzyko względne wahalo się od 1,03 przy stosowaniu COC krócej niż 1 rok do około 1,4 przy stosowaniu COC dłużej niż 8–10 lat.
Zgłaszanie podejrzewanych działań niepożądanych
Zgłaszanie podejrzewanych działań niepożądanych w okresie obserwacji pozarynkowej jest bardzo ważne. Pozwala to na ciągłe monitorowanie stosunku korzyści do ryzyka w stosowaniu leków. Osobom pracującym w ochronie zdrowia zaleca się zgłaszanie wszelkich podejrzewanych działań niepożądanych za pomocą krajowego systemu raportowania.
Okres ważności. 4 lata.
Warunki przechowywania. Przechowywać w miejscu niedostępnym dla dzieci, w oryginalnym opakowaniu, w temperaturze nie przekraczającej 30 °C.
Opakowanie. Po 21 tabletek w blaszance; po 1, 3 lub 6 blaszeczek w opakowaniu tekturowym.
Kategoria wydawania. Na receptę.
Producent. mibe GmbH Arzneimittel.
Miejsce produkcji oraz adres siedziby przedsiębiorstwa.
Münchenerstrasse 15, Brehna, Saksonia-Anhalt, 06796, Niemcy.