Drospifem® 30
Ucraina
Indice
ISTRUZIONI PER L'USO MEDICINALE DEL MEDICINALE DROSPIFEM® 30
Composizione:
Principi attivi: etinilestradiolo, drospirenone;
1 compressa contiene: 0,03 mg di etinilestradiolo e 3 mg di drospirenone;
Eccipienti: lattosio monoidrato, amido di mais, maltodestrina, magnesio stearato, Opadry 10A32290 giallo (idrossipropilmetilcellulosa, talco, biossido di titanio (E 171), polisorbato 80, ossido di ferro giallo (E 172).
Forma farmaceutica. Compresse rivestite con film.
Principali caratteristiche fisico-chimiche: compresse rotonde rivestite con film di colore giallo, senza difetti del rivestimento.
Gruppo farmacoterapeutico. Ormoni delle ghiandole sessuali e farmaci utilizzati per patologie dell'apparato genitale. Contraccettivi ormonali per uso sistemico.
Progestinici ed estrogeni, combinazioni fisse. Drospirenone ed etinilestradiolo.
Codice ATC G03A A12.
Proprietà farmacologiche
Farmacodinamica
L’indice Pearl dei fallimenti contraccettivi per il medicinale: 0,09 (intervallo di confidenza bilaterale superiore al 95 % (IC): 0,32).
L’indice Pearl complessivo (fallimenti contraccettivi + errori da parte delle pazienti) per il medicinale: 0,57 (intervallo di confidenza bilaterale superiore al 95 % (IC): 0,90).
Drospifem® 30 è un contraccettivo orale combinato contenente etinilestradiolo e drospirenone. Alle dosi terapeutiche, il drospirenone manifesta proprietà antiandrogeniche e proprietà antimineralscorticoide moderate. Non presenta attività estrogenica, glucocorticoidica né antiglucocorticoidica. Pertanto, il profilo farmacologico del drospirenone è simile a quello del progesterone naturale.
L’azione contraccettiva del medicinale si basa sull’interazione di diversi fattori, tra cui i più importanti sono l’inibizione dell’ovulazione e le modifiche della secrezione cervicale.
Secondo i dati degli studi clinici, le proprietà antimineralscorticoide moderate del medicinale Drospifem® 30 determinano un effetto antimineralscorticoide moderato.
Farmacocinetica
Drospirenone
Assorbimento. Dopo somministrazione orale, il drospirenone viene assorbito rapidamente e quasi completamente. La concentrazione massima nel siero – 38 ng/ml – viene raggiunta circa 1-2 ore dopo una singola dose. La biodisponibilità è del 76-85 %. L’assunzione contemporanea di cibo non influenza la biodisponibilità del drospirenone.
Distribuzione. Dopo somministrazione orale, la concentrazione sierica di drospirenone diminuisce con un emivita terminale media di circa 31 ore. Il drospirenone si lega all’albumina sierica, senza legarsi al globulina legante gli steroidi sessuali (SHBG) né alla globulina legante i corticosteroidi (CBG). Solo il 3-5 % della sua concentrazione totale nel siero è presente in forma libera. L’aumento dell’SHBG indotto dall’etinilestradiolo non influenza il legame del drospirenone alle proteine sieriche. Il volume medio di distribuzione del drospirenone è di 3,7±1,2 l/kg.
Metabolismo. Il drospirenone viene ampiamente metabolizzato dopo somministrazione orale. I principali metaboliti nel plasma sono la forma acida del drospirenone, formata dall’apertura dell’anello lactonico, e il 4,5-diidro-drospirenone-3-solfato, formato tramite idratazione seguita da solfatazione. Il drospirenone è anche soggetto a metabolismo ossidativo catalizzato dal CYP3A4. In vitro, il drospirenone può inibire debolmente o moderatamente gli enzimi del citocromo P450: CYP1A1, CYP2C9, CYP2C19 e CYP3A4.
Eliminazione. La velocità di clearance metabolico del drospirenone dal siero è di circa 1,5±0,2 ml/min/kg. Solo una quantità trascurabile di drospirenone viene escreta in forma invariata. I metaboliti vengono eliminati con urina e feci in un rapporto di 1,2:1,4; l’emivita dei metaboliti è di circa 40 ore.
Condizione di equilibrio. Durante il ciclo di trattamento, la concentrazione sierica massima di equilibrio del drospirenone (circa 70 ng/ml) viene raggiunta dopo 8 giorni di assunzione. I livelli di drospirenone nel sangue aumentano di 3 volte a causa del rapporto tra l’emivita terminale e l’intervallo di dosaggio.
Popolazioni speciali di pazienti:
- con alterazione della funzionalità renale: la concentrazione sierica di equilibrio del drospirenone in donne con insufficienza renale di grado lieve (clearance della creatinina 50-80 ml/min) è risultata comparabile a quella delle donne con funzionalità renale normale. I livelli di drospirenone nel siero sono stati mediamente del 37 % più elevati in donne con insufficienza renale di grado moderato (clearance della creatinina 30-50 ml/min) rispetto a quelle con funzionalità renale normale. La terapia con drospirenone è stata ben tollerata da donne con insufficienza renale di grado lieve o moderato. È stato dimostrato che l’assunzione di drospirenone non ha effetti clinicamente rilevanti sulla concentrazione di potassio nel siero;
- con alterazione della funzionalità epatica: in uno studio con dose singola, il clearance orale del drospirenone è risultato ridotto di circa il 50 % in soggetti con insufficienza epatica moderata rispetto a volontari con funzionalità epatica normale. Tale differenza nel clearance non ha determinato alcuna differenza significativa nei livelli di potassio nel sangue tra i due gruppi. Anche in presenza di diabete mellito e terapia concomitante con spironolattone (due fattori che possono favorire l’iperkaliemia), non è stato osservato un aumento della concentrazione di potassio nel siero oltre il limite superiore della norma. Si può concludere che il drospirenone è ben tollerato in soggetti con insufficienza epatica di grado lieve o moderato (classe B secondo la classificazione di Child-Pugh).
Etinilestradiolo
Assorbimento. Dopo somministrazione orale, l’etinilestradiolo viene assorbito rapidamente e completamente. Dopo l’assunzione di 30 µg, la concentrazione sierica di picco di 100 pg/ml viene raggiunta entro 1-2 ore. L’etinilestradiolo subisce un ampio effetto di primo passaggio epatico, variabile in base alle differenze individuali.
La biodisponibilità assoluta è di circa il 45 %.
Distribuzione. Il volume di distribuzione atteso dell’etinilestradiolo è di circa 5 l/kg e il legame alle proteine plasmatiche è di circa il 98 %. L’etinilestradiolo induce la sintesi epatica della SHBG e delle globuline leganti gli ormoni corticosteroidi. Con l’assunzione di 30 µg di etinilestradiolo, la concentrazione plasmatica di SHBG aumenta da 70 a circa 350 nmol/l.
L’etinilestradiolo passa in piccola quantità nel latte materno (0,02 % della dose).
Metabolismo. L’etinilestradiolo viene ampiamente metabolizzato nell’intestino e al primo passaggio epatico. Principalmente attraverso idrossilazione dell’anello aromatico, con formazione di una vasta gamma di metaboliti idrossilati e metilati, presenti sia in forma libera che come coniugati con glucuronidi e solfati. Il clearance metabolico dell’etinilestradiolo è di circa 5 ml/min/kg.
In vitro, l’etinilestradiolo è un inibitore reversibile di CYP2C19, CYP1A1 e CYP1A2, nonché un inibitore di CYP3A4/5, CYP2C8 e CYP2J2.
Eliminazione. L’etinilestradiolo viene praticamente eliminato solo in forma metabolizzata. I metaboliti dell’etinilestradiolo vengono eliminati con le urine e con la bile in un rapporto di 4:6. L’emivita dei metaboliti è di circa 1 giorno. L’emivita dei metaboliti è di 20 ore.
Condizione di equilibrio. La condizione di equilibrio viene raggiunta nella seconda metà del ciclo di assunzione, quando il livello sierico di etinilestradiolo aumenta da 1,4 a 2,1 volte.
Appartenenza etnica
Non sono state osservate differenze clinicamente significative nella farmacocinetica del drospirenone o dell’etinilestradiolo tra donne di nazionalità giapponese e donne di razza caucasica.
Dati preclinici di sicurezza
Negli animali da laboratorio, gli effetti del drospirenone e dell’etinilestradiolo si sono limitati a quelli associati al noto effetto farmacologico. In particolare, gli studi sulla tossicità riproduttiva negli animali hanno evidenziato effetti embriotossici e fetotossici specie-specifici. Con esposizioni superiori a quelle osservate negli utilizzatori di Drospifem® 30, in alcuni animali sono stati osservati effetti sulla differenziazione sessuale.
Caratteristiche cliniche.
Indicazioni.
Contraccezione orale.
La decisione di prescrivere Drospifem® 30 deve essere presa considerando i fattori di rischio individuali presenti della paziente, in particolare i fattori di rischio di tromboembolia venosa (TEV). È inoltre necessario confrontare il rischio di TEV durante il trattamento con Drospifem® 30 con quello durante il trattamento con altri contraccettivi ormonali combinati (COC) (per maggiori dettagli vedere le sezioni «Controindicazioni» e «Avvertenze speciali»).
Controindicazioni.
I COC non devono essere utilizzati in presenza di uno qualsiasi dei seguenti stati. Se uno qualsiasi di questi stati insorge per la prima volta durante l'assunzione di un COC, il farmaco deve essere immediatamente interrotto.
- Presenza o rischio di tromboembolia venosa (TEV):
- TEV in atto, ad esempio in corso di terapia anticoagulante, o anamnesi di TEV (ad esempio trombosi venosa profonda (TVP) o embolia polmonare (EP));
- predisposizione ereditaria o acquisita alla tromboembolia venosa, ad esempio resistenza alla proteina C attivata (inclusa la mutazione del fattore V di Leiden), deficit di antitrombina-III, deficit di proteina C, deficit di proteina S;
- interventi chirurgici maggiori con immobilizzazione prolungata (vedere la sezione «Avvertenze speciali»);
- alto rischio di tromboembolia venosa dovuto alla presenza di numerosi fattori di rischio (vedere la sezione «Avvertenze speciali»).
- Presenza o rischio di tromboembolia arteriosa (TEA):
- tromboembolia arteriosa in atto o anamnesi di tromboembolia arteriosa (ad esempio infarto miocardico) o presenza di sintomi prodromici (ad esempio angina pectoris);
- ictus in atto o anamnesi di ictus, o presenza di sintomi prodromici (ad esempio attacco ischemico transitorio (TIA));
- predisposizione ereditaria o acquisita alla tromboembolia arteriosa, ad esempio iperomocisteinemia e anticorpi antifosfolipidi (anticorpi anti-cardiolipina, lupus anticoagulant);
- emicrania con sintomi neurologici focali in anamnesi;
- alto rischio di tromboembolia arteriosa dovuto alla presenza di numerosi fattori di rischio (vedere la sezione «Avvertenze speciali») o alla presenza di un singolo fattore di rischio grave, come:
- diabete mellito con complicanze vascolari;
- ipertensione arteriosa grave;
- dislipoproteinemia grave.
- Presenza o anamnesi di grave malattia epatica finché i parametri di funzionalità epatica non tornano alla normalità.
- Insufficienza renale grave o insufficienza renale acuta.
- Presenza o anamnesi di tumori epatici (benigni o maligni).
- Presenza o anamnesi di carcinoma mammario ormono-sensibile (vedere la sezione «Avvertenze speciali», sottosezione «Tumori»).
- Emorragia vaginale di etiologia sconosciuta.
- Assunzione concomitante di medicinali contenenti ombitasvir/paritaprevir/ritonavir e dasabuvir, o medicinali contenenti glecaprevir/pibrentasvir o sofosbuvir/velpatasvir/voxilaprevir (vedere la sezione «Interazioni con altri medicinali ed altre forme di interazione»).
- Ipersensibilità alle sostanze attive o a uno qualsiasi degli eccipienti del medicinale.
Interazioni con altri medicinali ed altre forme di interazione.
In presenza di uno qualsiasi degli stati o fattori di rischio indicati di seguito, si deve discutere con la paziente l'opportunità di utilizzare il medicinale Drospifem® 30.
Effetto di altri medicinali su Drospifem® 30
Sono possibili interazioni con medicinali che inducono gli enzimi microsomiali. Ciò può portare ad un aumento della clearance degli ormoni sessuali, con conseguente comparsa di sanguinamenti da rottura e/o perdita dell'efficacia contraccettiva.
Terapia
L'induzione enzimatica può manifestarsi già dopo alcuni giorni di trattamento. L'induzione massima degli enzimi si verifica generalmente dopo alcune settimane. Dopo l'interruzione del trattamento, l'induzione enzimatica può persistere per circa 4 settimane.
Terapia a breve termine
Le donne che assumono medicinali in grado di indurre enzimi devono temporaneamente utilizzare un metodo barriera o un altro metodo contraccettivo in aggiunta al contraccettivo orale combinato (COC). Il metodo barriera deve essere utilizzato per tutta la durata della terapia concomitante e per ulteriori 28 giorni dopo la sua interruzione.
Se la terapia concomitante prosegue oltre l'assunzione dell'ultima compressa del blister di COC, si deve iniziare immediatamente con le compresse del blister successivo, senza la consueta pausa tra i cicli.
Terapia a lungo termine
Alle donne in terapia a lungo termine con sostanze attive che inducono gli enzimi epatici si raccomanda di utilizzare un metodo contraccettivo barriera o un altro metodo non ormonale affidabile.
Le seguenti interazioni sono state segnalate sulla base di dati pubblicati
Sostanze attive che aumentano la clearance dei COC (riduzione dell'efficacia dei COC per induzione enzimatica), ad esempio:
barbiturici, bosentan, carbamazepina, fenitoina, primidone, rifampicina; medicinali utilizzati nel trattamento dell'infezione da HIV: ritonavir, nevirapina ed efavirenz; inoltre, possibilmente felbamato, griseofulvina, osscarbazepina, topiramato e prodotti vegetali contenenti estratto di erba di San Giovanni (Hypericum perforatum).
Sostanze attive con effetto variabile sulla clearance dei COC:
L'assunzione concomitante di COC con un gran numero di inibitori della proteasi HIV e inibitori non nucleosidici della trascrittasi inversa, comprese combinazioni con inibitori del virus dell'epatite C (VHC), può aumentare o ridurre le concentrazioni plasmatiche di estrogeni o progestinici. L'effetto complessivo di tali modifiche può essere clinicamente rilevante in alcuni casi.
Pertanto, per identificare potenziali interazioni e ulteriori raccomandazioni, si deve consultare l'informazione medica relativa al medicinale utilizzato per il trattamento di HIV/VHC assunto contemporaneamente. In caso di dubbi, le donne devono utilizzare un metodo barriera aggiuntivo durante la terapia con inibitori della proteasi o inibitori non nucleosidici della trascrittasi inversa.
Sostanze attive che riducono la clearance dei COC (inibitori enzimatici)
Il significato clinico delle potenziali interazioni con inibitori enzimatici rimane incerto.
L'assunzione concomitante di inibitori forti del CYP3A4 può aumentare le concentrazioni plasmatiche di estrogeno, di progestinico o di entrambi.
In uno studio con dosi multiple della combinazione drospirenone (3 mg/giorno)/etinilestradiolo (0,02 mg/giorno) con co-somministrazione di ketoconazolo, un inibitore forte del CYP3A4, per 10 giorni, i valori di AUC(0-24h) del drospirenone e dell'etinilestradiolo sono aumentati rispettivamente di 2,7 e 1,4 volte.
L'assunzione concomitante di etoricoxib alle dosi da 60 a 120 mg/giorno con un contraccettivo ormonale combinato contenente 0,035 mg di etinilestradiolo ha determinato un aumento delle concentrazioni plasmatiche di etinilestradiolo da 1,4 a 1,6 volte rispettivamente.
Effetto sui altri medicinali. I contraccettivi orali possono influenzare il metabolismo di altri farmaci. Possono modificare la concentrazione delle sostanze attive nel plasma e nei tessuti: aumentandola (ad esempio ciclosporina) o riducendola (ad esempio lamotrigina).
Sulla base di studi noti di inibizione e interazioni in vivo condotti su volontarie che assumevano omeprazolo, simvastatina e midazolam come substrati indicatori, è improbabile che drospirenone alla dose di 3 mg influenzi altri medicinali il cui metabolismo coinvolge il citocromo P450.
Dati clinici indicano che l'etinilestradiolo inibisce la clearance dei substrati del CYP1A2, causando un lieve (ad esempio teofillina) o moderato (ad esempio tizanidina) aumento delle loro concentrazioni plasmatiche.
Altre forme di interazione. In pazienti con insufficienza renale, l'assunzione concomitante di drospirenone con inibitori dell'ACE o farmaci antiinfiammatori non steroidei non ha mostrato un effetto significativo sui livelli di potassio nel siero. In questi casi è necessario effettuare un monitoraggio del potassio sierico durante il primo ciclo di trattamento (vedere anche la sezione «Avvertenze speciali»).
Analisi di laboratorio
L'uso di COC può influenzare i risultati di alcuni esami di laboratorio, come i parametri biochimici di funzionalità epatica, tiroidea, surrenale e renale, la concentrazione nel plasma di proteine leganti, come il globulina legante i corticosteroidi, la concentrazione nel plasma delle frazioni lipidiche/lipoproteiche, i parametri del metabolismo dei carboidrati, della coagulazione e della fibrinolisi. Di norma, tali modifiche rientrano nei limiti normali. Il drospirenone aumenta l'attività plasmatica della renina e dell'aldosterone, indotta dalla sua attività antimineralecorticoide moderata.
Interazioni farmacodinamiche
Durante studi clinici su pazienti trattati per infezione da epatite C (VHC) con medicinali contenenti ombitasvir/paritaprevir/ritonavir e dasabuvir con o senza ribavirina, si è osservato un aumento significativo delle transaminasi (ALT), superiore al limite superiore della norma (LSN) di 5 volte più frequente nelle donne che assumevano medicinali contenenti etinilestradiolo, come i contraccettivi ormonali combinati (COC). Inoltre, un aumento dei livelli di ALT è stato osservato anche con l'uso di medicinali antivirali contenenti glecaprevir/pibrentasvir o sofosbuvir/velpatasvir/voxilaprevir in donne che assumevano medicinali contenenti etinilestradiolo, come i COC (vedere le sezioni «Controindicazioni»). Pertanto, le pazienti che assumono Drospifem® 30 devono passare a un metodo contraccettivo alternativo (ad esempio contraccezione solo a base di progestinici o metodi contraccettivi non ormonali) prima di iniziare il trattamento con queste combinazioni di medicinali. Il trattamento con Drospifem® 30 può essere ripreso 2 settimane dopo la fine del trattamento con queste combinazioni di medicinali.
Caratteristiche d'uso.
In presenza di uno qualsiasi dei seguenti stati o fattori di rischio, si deve attentamente valutare il potenziale rischio e il beneficio atteso dall'uso dei COC in ogni singolo caso. In caso di peggioramento, intensificazione o insorgenza di uno qualsiasi dei seguenti stati o fattori di rischio, si raccomanda di consultare il medico, il quale potrà decidere di interrompere l'assunzione del medicinale.
In caso di sospetta o confermata TVP o ATE, l'uso del medicinale deve essere interrotto. Se è stata iniziata una terapia anticoagulante, si deve garantire un'adeguata contraccezione alternativa a causa dell'effetto teratogeno degli anticoagulanti (cumarine).
- Disturbi circolatori
L'incidenza di malattie trombotiche e tromboemboliche venose ed arteriose nelle donne senza fattori di rischio che assumono COC con basso dosaggio di estrogeni (inferiore a 50 mcg di etinilestradiolo), come Drospifem® 30, è compresa tra 20 e 40 casi ogni 100.000 donne all'anno, ma questo rischio varia in base al livello di progestinico. A titolo di confronto, nelle donne che non usano contraccettivi, l'incidenza di tali malattie è compresa tra 5 e 10 casi ogni 100.000.
Rischio di sviluppo di tromboembolia venosa (TVEM)
L'uso di qualsiasi CCO aumenta il rischio di sviluppare tromboembolia venosa (TVEM) nelle donne che li assumono, rispetto a quelle che non li assumono. I medicinali contenenti levonorgestrel, norgestimato o noretisterone sono associati a un rischio inferiore di TVEM. L'uso di altri medicinali, come Drospifem® 30, può raddoppiare tale rischio. La decisione di utilizzare medicinali diversi da quelli con il rischio più basso di TVEM deve essere presa solo dopo un'adeguata discussione con la donna. È necessario assicurarsi che la paziente comprenda il rischio di TVEM associato all'uso del medicinale Drospifem® 30, l'impatto dei fattori di rischio presenti e il fatto che il rischio di TVEM è massimo durante il primo anno di trattamento. Secondo alcuni dati, il rischio di TVEM può aumentare quando si riprende l'uso di CCO dopo un'interruzione di 4 settimane o più.
Il rischio di complicanze tromboemboliche venose nelle donne che assumono CCO può essere significativamente più elevato in presenza di ulteriori fattori di rischio, specialmente se multipli (vedi tabella).
Drospifem® 30 è controindicato nelle donne con una combinazione di fattori di rischio che determinano un elevatissimo rischio di trombosi venosa (vedi sezione «Controindicazioni»). Se una donna presenta più di un fattore di rischio, l'aumento del rischio può essere maggiore della somma dei rischi associati a ciascun fattore singolarmente; pertanto, si deve considerare il rischio complessivo di sviluppare TVEM. Se il rapporto rischio/beneficio è considerato sfavorevole, i CCO non devono essere prescritti (vedi sezione «Controindicazioni»).
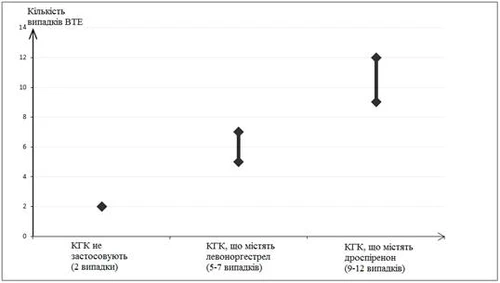
Circa 2 su 10.000 donne che non assumono CCO e non sono in stato di gravidanza sviluppano TVEM durante un periodo di osservazione di 1 anno. Tuttavia, per una singola donna, il rischio può essere molto più elevato, a seconda dei fattori di rischio (vedi sotto).
È stato stabilito che, su 10.000 donne che assumono CCO contenenti drospirenone, 9-12 svilupperanno TVEM entro un anno. Questi dati si basano su tutte le evidenze epidemiologiche disponibili, tenendo conto dei rischi relativi associati all'assunzione di diversi tipi di CCO, rispetto all'uso di CCO contenenti levonorgestrel.
Questo valore si confronta con un tasso di 6 nelle donne che assumono CCO contenenti levonorgestrel. In media, 5-7 casi ogni 10.000 donne-anno, calcolati in base al rischio relativo dell'uso di CCO contenenti levonorgestrel rispetto alle donne che non assumono CCO (circa 2,3-3,6 casi).
In entrambi i casi, il numero di episodi di TVEM all'anno è stato inferiore rispetto a quanto generalmente atteso durante la gravidanza o nel periodo post-partum.
La TVEM è letale nell'1-2% dei casi. Il rischio aggiuntivo di TVEM è massimo durante il primo anno di uso di un contraccettivo combinato.
Numero di casi di TVEM per 10.000 donne in un anno
Fattori di rischio per lo sviluppo di TVEM
Il rischio di sviluppare complicanze tromboemboliche venose nelle donne che assumono CCO può essere significativamente più elevato in presenza di ulteriori fattori di rischio, specialmente se multipli (vedi tabella).
L'uso del medicinale Drospifem® 30 è controindicato nelle donne con multipli fattori di rischio che possono aumentare il rischio di trombosi venosa (vedi sezione «Controindicazioni»). Se una donna presenta più di un fattore di rischio, l'aumento del rischio può essere maggiore della somma dei rischi associati a ciascun fattore singolarmente; pertanto, si deve considerare il rischio complessivo di sviluppo di TVEM. Se il rapporto rischio/beneficio è sfavorevole, non si devono prescrivere CCO (vedi sezione «Controindicazioni»).
Fattori di rischio per lo sviluppo di TVEM
| Fattore di rischio |
Nota |
| Obesità (indice di massa corporea superiore a 30 kg/m²). |
Il rischio aumenta significativamente con l'aumentare dell'indice di massa corporea. Richiede particolare attenzione soprattutto in presenza di altri fattori di rischio. |
| Immobilità prolungata, interventi chirurgici di grande entità, interventi agli arti inferiori o agli organi pelvici, interventi neurochirurgici o traumi gravi. Nota: l'immobilità temporanea, inclusi voli di durata superiore a 4 ore, può rappresentare un fattore di rischio per lo sviluppo di TVP, specialmente nelle donne con altri fattori di rischio. |
Si raccomanda di interrompere il trattamento (almeno 4 settimane prima di un intervento chirurgico programmato) e di non riprenderlo prima di 2 settimane dal completo recupero della mobilità. Per evitare una gravidanza indesiderata, si devono utilizzare altri metodi contraccettivi. Si deve valutare l'opportunità di una terapia antitrombotica qualora il trattamento non fosse stato sospeso preventivamente. |
| Anamnesi familiare (tromboembolia venosa in un familiare o in uno dei genitori, specialmente in età relativamente giovane, ad esempio prima dei 50 anni). |
In caso di predisposizione ereditaria, alle donne si raccomanda di consultare uno specialista prima di iniziare qualsiasi CCO. |
| Altri stati associati a TVP. |
Neoplasie, lupus eritematoso sistemico, sindrome emolitico-uremica, malattia infiammatoria cronica intestinale (morbo di Crohn o colite ulcerosa) e anemia falciforme. |
| Età. |
Particolarmente dopo i 35 anni. |
Non esiste un consenso univoco riguardo al possibile impatto della varicosi e della tromboflebite superficiale sullo sviluppo e il progresso del tromboembolismo venoso.
È necessario prestare attenzione al rischio aumentato di tromboembolia durante il periodo di gravidanza, specialmente nei 6 settimane successive al parto (per informazioni riguardanti il periodo di gravidanza o allattamento al seno, vedere la sezione «Uso durante la gravidanza o l’allattamento al seno»).
Sintomi di TVP (trombosi venosa profonda e embolia polmonare)
Alle donne deve essere raccomandato di rivolgersi immediatamente al medico e di informarlo che stanno assumendo un contraccettivo ormonale combinato (COC), in caso di comparsa dei seguenti sintomi.
I sintomi della trombosi venosa profonda (TVP) possono essere:
- gonfiore unilaterale della gamba e/o del piede o di una zona lungo la vena della gamba;
- dolore o ipersensibilità nella gamba, che può manifestarsi solo in posizione eretta o durante la deambulazione;
- sensazione di calore nella gamba colpita; arrossamento o cambiamento del colore della pelle della gamba.
I sintomi dell’embolia polmonare (EP) possono essere:
- comparsa improvvisa di dispnea di origine sconosciuta o respirazione accelerata;
- tosse improvvisa, che può essere accompagnata da emottisi;
- dolore acuto al torace;
- grave capogiro o vertigini;
- battito cardiaco accelerato o irregolare.
Alcuni di questi sintomi (ad esempio dispnea, tosse) sono aspecifici e possono essere interpretati erroneamente come manifestazioni di condizioni più comuni e meno gravi (ad esempio infezioni respiratorie).
Altri segni di occlusione vascolare possono includere: dolore improvviso in un arto, gonfiore, addome acuto e lieve cianosi della pelle degli arti.
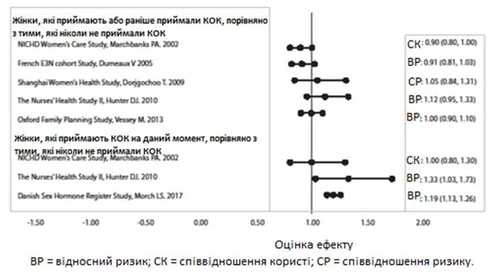
Studi pertinenti sul rischio di cancro al seno con l’uso di contraccettivi orali combinati
Tre studi hanno confrontato il rischio di cancro al seno tra donne che assumevano COC al momento dello studio o che avevano interrotto di recente l’assunzione (< 6 mesi dall’ultima assunzione) e donne che non avevano mai assunto COC. In uno di questi studi non è stato riportato alcun legame tra il rischio di cancro al seno e l’uso di COC. Gli altri due studi hanno evidenziato un aumento del rischio relativo compreso tra 1,19 e 1,33 nei soggetti che assumevano attualmente o di recente COC. Entrambi gli studi hanno rilevato un aumento del rischio di cancro al seno con un uso prolungato al momento dello studio, con un rischio relativo che variava da 1,03 per un uso inferiore a 1 anno a circa 1,4 per un uso superiore a 8–10 anni.
Segnalazione delle reazioni avverse sospette
La segnalazione delle reazioni avverse sospette dopo l’immissione in commercio è di fondamentale importanza. Permette un monitoraggio continuo del rapporto beneficio/rischio del medicinale. Si raccomanda agli operatori sanitari di segnalare qualsiasi reazione avversa sospetta attraverso il sistema nazionale di segnalazione.
Periodo di validità. 4 anni.
Condizioni di conservazione. Conservare in un luogo inaccessibile ai bambini, nell’imballaggio originale, a una temperatura non superiore a 30 °C.
Confezionamento. 21 compresse in un blister; 1, 3 o 6 blister in un imballaggio di cartone.
Categoria di prescrizione. Medicinale soggetto a prescrizione medica.
Produttore. mibe GmbH Arzneimittel.
Indirizzo del produttore e sede operativa.
Muenchenstrasse 15, Brehna, Sassonia-Anhalt, 06796, Germania.