Bretaris® Genueair®
UkrainaSpis treści
INSTRUKCJA dot. stosowania leku Bretaris® Genueair®
Skład:
substancja czynna: aclidinium bromide;
1 dawka wydzielona (dawka wydzielona z ustnika) zawiera 375 µg bromku akrydynii, co odpowiada 322 µg akrydynii. Odpowiada to dawce odmierzonej 400 µg bromku akrydynii, co odpowiada 343 µg akrydynii;
substancja pomocnicza: laktoza monohydrat.
Postać farmaceutyczna. Proszek do inhalacji.
Główne właściwości fizykochemiczne: biały lub prawie biały, drobnoziarnisty, sypki proszek bez widocznych agregatów lub zanieczyszczeń.
Grupa farmakoterapeutyczna.
Leki stosowane w obturacyjnych chorobach dróg oddechowych, leki antycholinergiczne. Kod ATC R03B B05.
Właściwości farmakologiczne.
Farmakodynamika.
Mechanizm działania.
Bromek akrydynii jest konkurencyjnym, selektywnym antagonistą receptorów muskarynowych (środek antycholinergiczny) o dłuższym czasie wiązania się z receptorami M3 niż z receptorami M2. Receptory M3 są pośrednikiem skurczu mięśni gładkich dróg oddechowych. Wdychany bromek akrydynii działa lokalnie w płucach jako antagonist receptorów M3 mięśni gładkich dróg oddechowych i powoduje rozszerzenie oskrzeli. Badania przedkliniczne in vitro i in vivo wykazały szybkie, zależne od dawki i długotrwałe hamowanie przez akrydynię skurczu oskrzeli wywołanego acetylocholiną. Bromek akrydynii szybko ulega rozkładowi w osoczu krwi, dlatego liczba systemowych reakcji niepożądanych o charakterze antycholinergicznym jest niska.
Działanie farmakodynamiczne.
Badania klinicznej skuteczności wykazały, że Bretaris® Genueair® zapewnia klinicznie istotne poprawienie funkcji płuc (mierzone objętością życiową wydechu [FEV1]) przez 12 godzin po porannej i wieczornej dawce, co objawiało się w ciągu 30 minut po podaniu pierwszej dawki (zwiększenie FEV1 w porównaniu z wartością wyjściową wyniosło 124–133 ml). Maksymalne rozszerzenie oskrzeli osiągane było w ciągu 1–3 godzin po podaniu dawki, przy średnim wzroście FEV1 w porównaniu z wartością wyjściową wynoszącym 227–268 ml w stanie równowagi.
Elektrofizjologia serca.
Nie zaobserwowano wpływu na interwał QT (skorygowany metodą Fridericia lub Bazetta lub indywidualnie skorygowany), gdy bromek akrydynii (200 μg lub 800 μg) podawano zdrowym osobom raz na dobę przez 3 dni.
Ponadto nie stwierdzono klinicznie istotnego wpływu leku Bretaris® Genueair® na rytm serca podczas 24-godzinnego monitorowania Holtera po 3 miesiącach leczenia u 336 pacjentów (164 z nich przyjmowało Bretaris® Genueair® dwa razy dziennie w dawce 322 μg).
Skuteczność i bezpieczeństwo kliniczne.
Program klinicznego rozwoju fazy III obejmował 269 pacjentów przyjmujących Bretaris® Genueair® w dawce 322 μg dwa razy dziennie w ramach jednego 6-miesięcznego, randomizowanego badania kontrolowanego placebo oraz 190 pacjentów przyjmujących Bretaris® Genueair® w dawce 322 μg dwa razy dziennie w ramach jednego 3-miesięcznego, randomizowanego badania kontrolowanego placebo. Skuteczność oceniano pod kątem funkcji płuc i skutków objawowych, takich jak duszność, charakterystyczny dla choroby stan zdrowia, stosowanie leków ratunkowych oraz występowanie zaostrzeń. W długotrwałych badaniach bezpieczeństwa Bretaris® Genueair® wykazał działanie rozszerzające oskrzela przez ponad 1 rok.
Rozszerzanie oskrzeli.
W 6-miesięcznym badaniu u pacjentów przyjmujących Bretaris® Genueair® w dawce 322 μg dwa razy dziennie zaobserwowano klinicznie istotne poprawienie funkcji płuc (mierzone za pomocą FEV1). Maksymalne działanie rozszerzające oskrzela objawiało się od pierwszego dnia i nasilało się w ciągu 6-miesięcznego okresu leczenia. Po 6 miesiącach terapii średnie poprawienie przed podaniem porannej dawki (minimum) FEV1 w porównaniu z placebo wyniosło 128 ml (95 % CI 85–170; p < 0,0001).
Podobne obserwacje dokonano dla Bretaris® Genueair® w trakcie 3-miesięcznych badań.
Bretaris® Genueair® zapewniał klinicznie istotne zmniejszenie duszności (ocenianej za pomocą dynamicznego indeksu duszności [TDI]) z poprawą TDI po 6-miesięcznym leczeniu w porównaniu z 1,0 jednostki placebo (p < 0,001).
Odsetek pacjentów z klinicznie istotnym poprawieniem TDI (określony jako wzrost co najmniej o 1 jednostkę zmiany TDI) był wyższy w grupie leku Bretaris® Genueair® w porównaniu z placebo (56,9 % w porównaniu z 45,5 %; p = 0,004).
Bretaris® Genueair® wykazał klinicznie istotne złagodzenie objawów z poprawą ogólnych wyników w porównaniu z placebo – 4,6 jednostki (p < 0,0001). Odsetek pacjentów, którzy osiągnęli klinicznie istotne poprawienie w porównaniu z wartością wyjściową, był wyższy w grupie leku Bretaris® Genueair® w porównaniu z placebo (57,3 % w porównaniu z 41,0 % odpowiednio; p < 0,001).
Pacjentom przyjmującym Bretaris® Genueair® potrzebne było mniej leków ratunkowych niż pacjentom przyjmującym placebo (zmniejszenie o 0,95 wstrzyśnięć na dobę przez 6 miesięcy [p = 0,005]). Bretaris® Genueair® poprawiał również objawy przewlekłej obturacyjnej choroby płuc (POChP) w ciągu dnia (duszność, kaszel i wydzielanie się plwociny), jak również objawy nocne i poranne.
Analiza łączona skuteczności 6-miesięcznych i 3-miesięcznych badań kontrolowanych placebo wykazała istotne zmniejszenie częstości zaostrzeń umiarkowanych i ciężkich (wymagających leczenia antybiotykami lub kortykosteroidami lub kończących się hospitalizacją) przy stosowaniu 322 μg akrydynii dwa razy dziennie w porównaniu z placebo.
Długotrwałe badanie bezpieczeństwa i skuteczności trwające do 3 lat.
Wpływ bromku akrydynii na występowanie głównych niepożądanych zdarzeń sercowo-naczyniowych (major adverse cardiovascular events, MACE) oceniano w ramach randomizowanego, podwójnie ślepego, kontrolowanego placebo badania równoległego z udziałem 3630 dorosłych pacjentów w wieku od 40 do 91 lat, cierpiących na POChP od umiarkowanego do bardzo ciężkiego stopnia, leczonych przez okres do 36 miesięcy. Spośród nich 58,7 % stanowili mężczyźni, a 90,7 % należało do rasy europejskiej, ze średnią wartością FEV1 po podaniu leku rozszerzającego oskrzela wynoszącą 47,9 % wartości przewidywanej oraz średnią wartością CAT (skala oceny POChP) 20,7. Wszyscy pacjenci mieli w wywiadzie choroby układu sercowo-naczyniowego lub naczyń mózgowych i/lub istotne czynniki ryzyka chorób układu sercowo-naczyniowego. U 59,8 % pacjentów wystąpił przynajmniej jeden epizod zaostrzenia POChP w ciągu ostatnich 12 miesięcy po wizycie skriningowej. U około 48 % pacjentów biorących udział w badaniu w wywiadzie stwierdzono przynajmniej jedno udokumentowane zdarzenie ze strony układu sercowo-naczyniowego; choroby naczyń mózgowych (13,1 %), choroby tętnic wieńcowych (35,4 %), choroby naczyń obwodowych lub kuleństwo (13,6 %).
Badanie miało projekt kierowany zdarzeniami i zostało zakończone po osiągnięciu wystarczającej liczby zdarzeń MACE do przeprowadzenia pierwotnej analizy bezpieczeństwa. W przypadku wystąpienia MACE leczenie było przerywane, a pacjentów przenoszono do grupy obserwacji po leczeniu w trakcie badania. Według oceny badacza, 70,7 % pacjentów ukończyło udział w badaniu. Średni czas trwania leczenia w grupach pacjentów przyjmujących Bretaris® Genueair® i placebo wynosił odpowiednio 1,1 i 1 rok. Średni czas uczestnictwa w badaniu w grupach pacjentów przyjmujących Bretaris® Genueair® i placebo wynosił odpowiednio około 1,4 i 1,3 roku.
Pierwotnym punktem końcowym dotyczącym bezpieczeństwa był czas do pierwszego wystąpienia MACE, definiowanego jako jedno z następujących potwierdzonych zdarzeń: śmierć z przyczyn sercowo-naczyniowych, nieśmiertelny zawał mięśnia sercowego (ZM) lub nieśmiertelny udar niedokrwienny. Częstość wystąpienia przynajmniej jednego MACE u pacjentów wyniosła odpowiednio 3,85 % i 4,23 % w grupach stosujących akrydynię i placebo. Stosowanie leku Bretaris® Genueair® nie spowodowało zwiększenia ryzyka wystąpienia MACE u pacjentów z POChP w porównaniu z placebo przy dodaniu do aktualnej terapii podstawowej (stosunek ryzyka – hazard ratio (HR) [SR] 0,89; 95 % CI: 0,64, 1,23). Górna granica przedziału ufności wyklucza zdefiniowaną granicę ryzyka wynoszącą 1,8.
Częstość występowania zaostrzeń POChP umiarkowanych lub ciężkich, obliczona na pacjenta w ciągu roku, w ciągu pierwszego roku leczenia była oceniana jako pierwotny punkt końcowy skuteczności badania. U pacjentów przyjmujących Bretaris® Genueair® zaobserwowano statystycznie istotne zmniejszenie o 22 % w porównaniu z placebo (stosunek częstości [SF] 0,78; 95 % CI od 0,68 do 0,89; p < 0,001). Ponadto stosowanie leku Bretaris® Genueair® spowodowało statystycznie istotne zmniejszenie częstości hospitalizacji o 35 % z powodu zaostrzeń POChP w trakcie leczenia w ciągu pierwszego roku w porównaniu z placebo (SF 0,65; 95 % CI od 0,48 do 0,89; p = 0,006).
W grupie pacjentów przyjmujących Bretaris® Genueair® opóźnienie czasu do pierwszego zaostrzenia choroby umiarkowanego lub ciężkiego stopnia w trakcie leczenia było statystycznie istotne w porównaniu z grupą pacjentów przyjmujących placebo. W grupie pacjentów przyjmujących bromek akrydynii zaobserwowano względne zmniejszenie ryzyka zaostrzenia choroby o 18 % (SR 0,82; 95 % CI [0,73, 0,92], p < 0,001).
Wytrzymałość na obciążenia fizyczne.
W 3-tygodniowym, krzyżowym, randomizowanym, kontrolowanym placebo klinicznym badaniu stosowanie leku Bretaris® Genueair® wiązało się ze statystycznie istotną poprawą wytrzymałości na obciążenia fizyczne w porównaniu z 58 sekundami przy stosowaniu placebo. Bretaris® Genueair® statystycznie istotnie zmniejszał hiperinflację płuc w stanie spoczynku, a także zwiększał objętość wdechu i zmniejszał duszność podczas ćwiczeń.
Farmakokinetyka.
Wchłanianie.
Bromek akrydynii szybko wchłania się z płuc, osiągając maksymalną stężenie w osoczu krwi w ciągu 5 minut po inhalacji u zdrowych osób i zazwyczaj w ciągu pierwszych 15 minut u pacjentów z POChP. Frakcja inhalowanej dawki, która osiągnęła obieg ogólny w postaci niezmienionego akrydynii, była bardzo niska, poniżej 5 %.
W stanie równowagi szczytowe stężenie w osoczu krwi osiągnięte u pacjentów z POChP po inhalacji proszku suchego w dawce 400 μg bromku akrydynii wynosiło około 224 pg/ml. Stan równowagi w osoczu krwi osiągany był w ciągu 7 dni przy stosowaniu dwa razy dziennie.
Rozkład.
Całkowita ilość bromku akrydynii inhalowanego przez inhalator Bretaris® Genueair®, która dociera do płuc, wynosiła około 30 % dawki zmierzonej.
Wiązanie bromku akrydynii z białkami osocza krwi in vitro najprawdopodobniej odnosi się do wiązania metabolitów z białkami z powodu szybkiego hydrolizy bromku akrydynii w osoczu krwi. Wiązanie z białkami osocza krwi wynosiło 87 % dla metabolitu kwasu karboksylowego i 15 % dla metabolitu alkoholowego. Głównym białkiem osocza krwi wiążącym bromek akrydynii jest albumina.
Biotransformacja.
Bromek akrydynii szybko i intensywnie ulega hydrolizie do swoich farmakologicznie nieaktywnych pochodnych alkoholowych i pochodnych kwasu karboksylowego. Zachodzi zarówno hydroliza chemiczna (nieenzymatyczna), jak i enzymatyczna z udziałem esteraz. Główną esterazą zaangażowaną w hydrolizę u człowieka jest butyrylocholinesteraza. Stężenie metabolitu kwasowego w osoczu krwi po inhalacji jest około 100 razy wyższe niż stężenie metabolitu alkoholowego i niezmienionej substancji czynnej. Niska absolutna biodostępność bromku akrydynii przy podaniu inhalacyjnym (< 5 %) wiąże się z tym, że bromek akrydynii podlega intensywnej systemowej i presystemowej hydrolizie zarówno w płucach, jak i po podaniu doustnym. Biotransformacja z udziałem enzymów CYP450 odgrywa niewielką rolę w ogólnym metabolizmie bromku akrydynii.
Badania in vitro wykazały, że bromek akrydynii w dawce terapeutycznej lub jego metabolity nie hamują ani nie indukują żadnych enzymów cytochromu P450 (CYP450) i nie hamują aktywności esteraz (karboksylaz, acetylocholinesterazy i butyrylocholinesterazy). Badania in vitro wykazały, że bromek akrydynii lub jego metabolity nie są substratami ani inhibitorami glikoproteiny P.
Eliminacja.
U pacjentów z POChP po inhalacji leku dwa razy dziennie w dawce 400 μg okres półtrwania końcowy i efektywny okres półtrwania bromku akrydynii wynoszą odpowiednio około 14 godzin i 10 godzin.
Po podaniu dożylnym zdrowym osobom badanym 400 μg radioaktywnie znakowanego bromku akrydynii około 1 % dawki wydalane było w niezmienionej postaci z moczem. Do 65 % dawki wydalane było w postaci metabolitów z moczem i do 33 % – w postaci metabolitów z kałem.
Po podaniu inhalacyjnym zdrowym osobom oraz pacjentom z POChP 200 μg i 400 μg bromku akrydynii bardzo mała ilość, około 0,1 % przyjętej dawki, wydalała się w niezmienionej postaci z moczem, co wskazuje, że klirens nerkowy odgrywa niewielką rolę w ogólnym klirensie akrydynii z osocza krwi.
Liniowość/nieliniowość.
Bromek akrydynii w zakresie terapeutycznym wykazał kinetykę liniową i niezależną od czasu farmakokinetykę.
Grupy specjalne pacjentów.
Pacjenci z zaburzeniem funkcji wątroby.
Badania z udziałem pacjentów z zaburzeniem funkcji wątroby nie były przeprowadzane. Ponieważ bromek akrydynii metabolizuje się głównie poprzez chemiczne i enzymatyczne rozszczepienie w osoczu krwi, bardzo mało prawdopodobne jest, że zaburzenie funkcji wątroby zmieni jego wpływ systemowy. Dla pacjentów z POChP i zaburzeniem funkcji wątroby nie jest wymagana korekta dawki.
Pacjenci w podeszłym wieku.
Właściwości farmakokinetyczne bromku akrydynii u pacjentów z POChP od umiarkowanego do ciężkiego stopnia są analogiczne do takich u pacjentów w wieku 40–59 lat i u pacjentów w wieku powyżej 70 lat. Dlatego dla pacjentów w podeszłym wieku z POChP nie jest wymagana korekta dawki.
Pacjenci z zaburzeniem funkcji nerek.
U pacjentów z normalną funkcją nerek i z jej zaburzeniem nie stwierdzono istotnych różnic w farmakokinetyce. Dlatego dla pacjentów z POChP i zaburzeniem funkcji nerek nie jest wymagana korekta dawki ani dodatkowa obserwacja.
Przynależność rasowa.
U mieszkańców Japonii i Europejczyków po powtarzanych inhalacjach obserwowano podobny wpływ systemowy bromku akrydynii.
Związek farmakokinetyka/farmakodynamika.
Ponieważ bromek akrydynii działa lokalnie w płucach i szybko ulega rozkładowi w osoczu krwi, nie ma bezpośredniego związku między farmakokinetyką a farmakodynamiką.
Dane przedkliniczne dotyczące bezpieczeństwa.
W danych przedklinicznych dotyczących bezpieczeństwa, opartych na tradycyjnych badaniach farmakologicznych bezpieczeństwa, toksyczności przy wielokrotnym podawaniu, genotoksyczności, działania rakotwórczego, toksyczności rozrodczej nie wykazano szczególnych ryzyk dla człowieka. W badaniach przedklinicznych efekty dotyczące parametrów układu sercowo-naczyniowego (zwiększenie częstości skurczów serca u psów), toksyczności rozrodczej (działanie fetotoksyczne) i płodności (niewielkie zmniejszenie częstości zapłodnień, liczby ciał żółtych oraz przed- i poimplantacyjna śmierć embrionów) obserwowano tylko przy wartościach ekspozycji znacznie przekraczających maksymalne działanie na człowieka, co wskazuje na małą istotność dla zastosowania klinicznego. Niska toksyczność stwierdzona w badaniach przedklinicznych toksyczności jest częściowo spowodowana szybkością metabolizmu bromku akrydynii w osoczu i brakiem istotnej aktywności farmakologicznej większości metabolitów. Zakres bezpieczeństwa dla człowieka przy ekspozycji systemowej wynoszącej 400 μg dwa razy dziennie w tych badaniach wahał się od 7- do 73-krotnego maksymalnego dawki leku, która nie prowadziła do pojawienia się obserwowanych reakcji niepożądanych.
Właściwości kliniczne.
Wskazania.
Leczenie wspomagające z rozszerzaniem oskrzeli w celu złagodzenia objawów przewlekłej obturacyjnej choroby płuc (POChP) u dorosłych pacjentów (patrz rozdział „Farmakodynamika”).
Przeciwwskazania.
Nadwrażliwość na bromek akrydynii lub na substancje pomocnicze.
Interakcje z innymi lekami i inne rodzaje interakcji.
Nie zaleca się jednoczesnego stosowania bromku akrydynii z innymi lekami przeciwdziałającymi działaniu acetylocholiny, ponieważ nie przeprowadzono badań w tej dziedzinie. Choć formalnych badań interakcji lekowych in vivo nie przeprowadzono, bromek akrydynii w postaci inhalacyjnej stosowano razem z innymi lekami stosowanymi w terapii POChP, w tym z rozszerzaczami oskrzeli: sympatykomimetykami, metyloksantynami oraz steroidami – w postaci inhalacyjnej i doustnej, bez pojawienia się objawów klinicznych interakcji lekowych. Badania in vitro wykazały, że bromek akrydynii w dawce terapeutycznej lub jego metabolity nie oddziałują z substancjami czynnymi, które są substratami białka glikoproteiny P (P-gp), ani z substancjami czynnymi, które są metabolizowane przez enzymy cytochromu P450 (CYP450) i esterazy (patrz rozdział „Farmakokinetyka”).
Szczególne ostrzeżenia i środki ostrożności dotyczące stosowania.
Paradoksalny skurcz oskrzeli.
Zastosowanie leku Bretaris® Genueair® może powodować paradoksalny skurcz oskrzeli. W przypadku wystąpienia takiego zjawiska należy natychmiast przerwać leczenie lekiem Bretaris® Genueair® i rozważyć możliwość zastosowania terapii alternatywnej.
Wspływanie przebiegu choroby.
Aklidyniumu bromek jest lekiem podtrzymującym działanie rozszerzające oskrzela i nie powinien być stosowany do złagodzenia ostrych napadów skurczu oskrzeli, tj. jako lek do leczenia nagłego. Jeśli podczas leczenia aklidyniumu bromkiem u pacjenta zaobserwuje się nasilenie objawów POChP, które wymaga dodatkowego leczenia ratunkowego, należy ponownie ocenić stan pacjenta oraz jego schemat leczenia.
Wpływ na układ sercowo-naczyniowy
Artytmie serca, w tym migotanie przedsionków i tachykardia nadkomorowa przerywana, obserwowano po podaniu leku Bretaris® Genueair® (zob. rozdział „Działania niepożądane”). Dlatego lek Bretaris® Genueair® należy stosować z ostrożnością u pacjentów z arytmiami serca, z wywiadem arytmii serca lub z czynnikami ryzyka arytmii serca.
Doświadczenie z badań klinicznych obejmujących pacjentów z chorobami współistniejącymi układu sercowo-naczyniowego jest ograniczone (zob. rozdział „Farmakodynamika”). Te stany mogą być wpływowane przez antycholinergiczny mechanizm działania leku.
Działanie antycholinergiczne
Suchość w ustach, która może występować podczas leczenia lekami o działaniu antycholinergicznym, w przypadku długotrwałego stosowania może być związana z rozwojem próchnicy zębów.
Z uwagi na działanie antycholinergiczne aklidyniumu bromek należy stosować z ostrożnością u pacjentów z przewlekłym przerostem gruczołu krokowego, zwężeniem szyjki pęcherza moczowego lub z zamkniętym kątem jaskry (mimo że bezpośrednie narażenie oczu na lek jest mało prawdopodobne).
Substancje pomocnicze.
Ten lek zawiera laktozę. Każda dawka wyzwalana zawiera około 12 mg laktozy (w postaci monohydratu). Pacjenci z rzadkimi, dziedzicznymi formami nietolerancji galaktozy, pełnym niedoborem laktoazy lub zaburzeniami wchłaniania glukozy/galaktozy nie powinni przyjmować tego leku.
Stosowanie w czasie ciąży lub karmienia piersią.
Ciąża.
Brak danych dotyczących stosowania aklidyniumu bromku u kobiet w ciąży.
Badania na zwierzętach wykazały działanie toksyczne na płód tylko w dawkach znacznie przekraczających maksymalną stężenie aklidyniumu bromku u ludzi (zob. „Dane przedkliniczne dotyczące bezpieczeństwa”). Aklidyniumu bromek może być stosowany w czasie ciąży tylko wtedy, gdy oczekiwana korzyść przewyższa potencjalne ryzyko.
Karmienie piersią.
Nie wiadomo, czy aklidyniumu bromek/metabolity przenikają do mleka matki. Badania na zwierzętach wykazały, że aklidyniumu bromek i/lub jego metabolity przenikają do mleka w niewielkich ilościach. Nie można wykluczyć ryzyka dla noworodków/dzieci. Decyzję o zaprzestaniu karmienia piersią lub o zaprzestaniu/zachowaniu leczenia lekiem Bretaris® Genueair® należy podjąć, biorąc pod uwagę korzyści wynikające z karmienia piersią dla dziecka oraz korzyści z leczenia dla kobiety.
Plodność.
Badania na zwierzętach wykazały niewielkie zmniejszenie płodności tylko przy zastosowaniu dawek znacznie przekraczających maksymalne stężenie aklidyniumu bromku u ludzi (zob. „Dane przedkliniczne dotyczące bezpieczeństwa”). Uważa się za mało prawdopodobne, aby aklidyniumu bromek stosowany w zalecanej dawce wpływał na płodność u ludzi.
Wpływ na zdolność prowadzenia pojazdów i obsługi mechanizmów.
Aklidyniumu bromek może mieć nieznaczny wpływ na szybkość reakcji podczas prowadzenia samochodu lub obsługiwanie innych maszyn. Pojawienie się bólu głowy, zawrotów głowy lub zamazanego widzenia po podaniu aklidyniumu bromku (zob. rozdział „Działania niepożądane”) może wpływać na szybkość reakcji podczas prowadzenia pojazdów lub obsługi innych mechanizmów.
Sposób stosowania i dawki.
Do inhalacji.
Pacjenci powinni zostać poinstruowane o właściwym stosowaniu leku, ponieważ inhalator Genueair® może działać inaczej niż inhalatory, z których pacjenci mogli wcześniej korzystać. Ważne jest, aby pacjent dokładnie przeczytał instrukcję stosowania.
Zalecana dawka to 1 inhalacja 322 μg akrydynium 2 razy dziennie.
W przypadku pominięcia dawki, następną dawkę należy zastosować tak szybko jak to możliwe. Jednak jeśli czas podania następnej dawki jest już bliski, pominiętej dawki nie należy stosować.
Pacjenci w podeszłym wieku.
U pacjentów w podeszłym wieku nie jest wymagana korekta dawki (patrz sekcja „Farmakokinetyka”).
Upośledzenie funkcji nerek.
U pacjentów z zaburzeniami funkcji nerek nie jest wymagana korekta dawki (patrz sekcja „Farmakokinetyka”).
Upośledzenie funkcji wątroby.
U pacjentów z zaburzeniami funkcji wątroby nie jest wymagana korekta dawki (patrz sekcja „Farmakokinetyka”).
Instrukcja stosowania.
Rozpoczęcie stosowania.
Przed rozpoczęciem stosowania leku należy przeczytać niniejszą instrukcję.
Inhalator białego koloru z wbudowanym wskaźnikiem dawek i zielonym przyciskiem dawkującym. Maska ustna jest pokryta zdejmowanym ochronnym kapturkiem. Inhalator znajduje się w plastikowym foliowanym opakowaniu, umieszczonym w tekturowej puszce.
Zapoznaj się z elementami składającymi się na inhalator Genueair®.
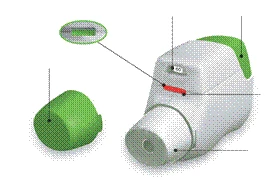
Rysunek A
Przed użyciem:
- Przed pierwszym użyciem należy rozciąć opakowanie i wyjąć inhalator. Opakowanie wyrzuć.
- Nie naciskaj zielonego przycisku, dopóki nie będziesz gotowy(-wa) do przyjęcia dawki.
- Zdejmij kaptur ochronny, delikatnie naciskając na strzałki oznaczone z każdej strony (rysunek B).

Rysunek B
-
- Trzymaj inhalator poziomo, maską ustną skierowaną w swoją stronę, zielony przycisk powinien być skierowany do góry (rysunek C).
**
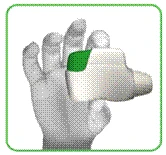
**
Rysunek C
-
- Naciśnij zielony przycisk w dół do oporu, aby przygotować dawkę leku (rysunek D).
Gdy naciskasz przycisk całkowicie w dół, okienko kontrolne zmienia kolor z czerwonego na zielony.
Upewnij się, że zielony przycisk znajduje się u góry. Nie nachylaj inhalatora.
-
- Puść zielony przycisk (rysunek E).
Upewnij się, że puściłeś(-aś) przycisk, aby inhalator mógł poprawnie działać.

Rysunek E
Dawka leku nie została przygotowana. Wróć do sekcji „KROK 1: Przygotuj dawkę leku” i powtórz punkty od 1.1 do 1.6.
KROK 2: Wykonaj inhalację leku
Przeczytaj dokładnie punkty od 2.1 do 2.7 przed zastosowaniem leku. Nie nachylaj inhalatora.
-
- Trzymając inhalator w odległości od ust, wypchnij całkowicie powietrze z płuc. Nigdy nie wydychaj powietrza do inhalatora (rysunek F).
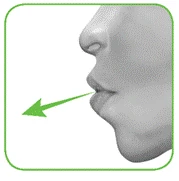
Rysunek F
-
- Trzymaj głowę w pozycji pionowej, umieść maskę ustną między wargami i ciasno zacisknij wargi wokół niej (rysunek G).
Nie naciskaj zielonego przycisku podczas wdechu.
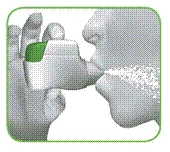
Rysunek G
-
- Wykonaj silny, głęboki wdech ustami. Kontynuuj wdech tak długo, jak to możliwe.
Jeśli inhalacja została wykonana poprawnie, rozlegnie się kliknięcie. Po usłyszeniu kliknięcia kontynuuj wdech tak długo, jak to możliwe. Niektórzy pacjenci mogą nie usłyszeć kliknięcia. Skorzystaj z okienka kontrolnego, aby upewnić się, że inhalacja została wykonana poprawnie.
-
- Wyciągnij inhalator z ust.
-
- Zatrzymaj oddech tak długo, jak to możliwe.
-
- Powoli wydechnij, ale nie do inhalatora.
Niektórzy pacjenci mogą odczuwać uczucie drobnych ziarenek w ustach lub słabe słodkie lub gorzkie posmakowanie. Nie wykonuj dodatkowego wdychania leku, nawet jeśli nie odczuwasz żadnego posmaku lub nie odczuwasz nic po inhalacji.
Zatrzymaj się i sprawdź:
-
- Upewnij się, że okienko kontrolne jest teraz czerwone (rysunek H). Oznacza to, że inhalacja została wykonana poprawnie.

Rysunek H
Oznacza to, że inhalacja została wykonana niepoprawnie. Wróć do sekcji „KROK 2: Wykonaj inhalację leku” i powtórz punkty od 2.1 do 2.7.
Jeśli okienko kontrolne nadal nie zmieniło koloru na czerwony, możliwe, że zapomniałeś(-łaś) puścić zielonego przycisku przed inhalacją lub wdech podczas inhalacji był niewystarczająco silny. W takim przypadku spróbuj ponownie. Upewnij się, że puściłeś(-aś) zielony przycisk i całkowicie wypchnąłeś(-aś) powietrze. Następnie wykonaj silny, głęboki wdech przez maskę ustną.
Skonsultuj się z lekarzem, jeśli po ponownych próbach okienko kontrolne pozostaje zielone.
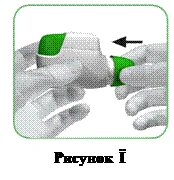
Po każdym użyciu załóż na maskę ustną ochronny kaptur (rysunek I), aby zapobiec zabrudzeniu inhalatora pyłem lub innymi substancjami. W przypadku utraty kapturka należy wyrzucić inhalator.
Dodatkowe informacje.
Co zrobić, jeśli przypadkowo przygotowałeś(-aś) dawkę leku przed jej zastosowaniem?
Przechowuj inhalator z założonym ochronnym kapturkiem do momentu zaplanowanego przyjęcia leku, a następnie zdejmij kaptur i przejdź do punktu 1.6.
Jak działa wskaźnik dawek?
Wskaźnik dawek pokazuje całkowitą liczbę pozostałych dawek leku w inhalatorze (rysunek J).
Przy pierwszym użyciu każdy inhalator zawiera co najmniej 60 dawek lub co najmniej 30 dawek – w zależności od wielkości opakowania.
Za każdym razem przy przygotowaniu dawki leku poprzez naciśnięcie zielonego przycisku wskaźnik dawek przesuwa się nieco w kierunku kolejnej liczby (50, 40, 30, 20, 10 lub 0).
Kiedy potrzebujesz nowego inhalatora?
Nowy inhalator jest potrzebny:
Jeśli Twój inhalator jest uszkodzony lub jeśli zgubiłeś(-aś) kaptur, lub
Gdy na wskaźniku dawek pojawią się czerwone paski, co oznacza zbliżanie się do ostatniej dawki leku (rysunek J), lub
Gdy Twój inhalator jest pusty (rysunek K).
Wskaźnik dawek powoli przesuwa się w kierunku od 60 do 0: 60, 50, 40, 30, 20, 10, 0.

Jak utrzymać inhalator w czystości?
NIGDY nie używaj wody do czyszczenia inhalatora, ponieważ może to uszkodzić lek.
Jeśli chcesz go wyczyścić, po prostu przetrzyj maskę ustną zewnętrznie suchą tkaniną lub chusteczką papierową.
Dzieci.
Lek Bretaris® Genueair® nie jest zalecany dzieciom (do 18 roku życia) ze względu na brak wystarczającego doświadczenia klinicznego.
Przedawkowanie.
Wysokie dawki bromku akrydynium mogą powodować objawy i objawy działania antycholinergicznego. Jednak jednorazowa dawka inhalacyjna bromku akrydynium do 6000 μg u zdrowych osób nie powodowała ogólnoustrojowych niepożądanych reakcji antycholinergicznych. Ponadto nie zaobserwowano klinicznie istotnych działań niepożądanych po 7-dniowym stosowaniu bromku akrydynium do 800 μg dwa razy dziennie u zdrowych osób.
Rozwój ostrej toksyczności po przypadkowym zażyciu bromku akrydynium jest mało prawdopodobny ze względu na niską biodostępność po podaniu doustnym oraz mechanizm dawkowania inhalacyjnego inhalatora Genueair®.
Niepożądane działania.
Najczęściej występującymi niepożądanymi działaniami podczas stosowania leku Bretaris® Genueair® są ból głowy (6,6%) i zapalenia nosa i gardła (5,5%).
Częstość wymienionych poniżej niepożądanych działań ustalono na podstawie ogólnych współczynników występowania działań niepożądanych (tj. działań występujących w związku ze stosowaniem leku Bretaris® Genueair®). Obserwowano je podczas stosowania leku Bretaris® Genueair® w dawce 322 μg (636 pacjentów) w jednym sześciomiesięcznym oraz dwóch trzymiesięcznych randomizowanych badaniach klinicznych z kontrolą placebo lub w wynikach badań pozwoleniowych.
W badaniu z kontrolą placebo, w którym wzięło udział 1791 pacjentów z POChP o umiarkowanym do bardzo ciężkiego stopniu zaawansowania, przy długości leczenia lekiem Bretaris® Genueair® do 36 miesięcy, nie stwierdzono innych niepożądanych działań.
Częstość niepożądanych działań określono następująco: bardzo często (≥ 1/10), często (od ≥ 1/100 do < 1/10), nieczęsto (od ≥ 1/1000 do < 1/100), rzadko (od 1/10 000 do < 1/1000), bardzo rzadko (< 1/10 000); częstotliwość nieznana (nie można oszacować częstości na podstawie dostępnych danych).
| Klasy układu narządów |
Reakcje niepożądane |
Częstotliwość |
| Infekcje i choroby pasożytnicze |
Zapalenie zatok |
Często |
| Zapalenie nosa i gardła |
Często |
|
| Ze strony układu odpornościowego |
Reakcje nadwrażliwości |
Rzadko |
| Angioświedź |
Częstotliwość nieznana |
|
| Reakcja anafilaktyczna |
Częstotliwość nieznana |
|
| Ze strony układu nerwowego |
Ból głowy |
Często |
| Zawroty głowy |
Nieczęsto |
|
| Ze strony narządów wzroku |
Rozmazane widzenie |
Nieczęsto |
| Ze strony układu sercowo-naczyniowego |
Arystmie serca, w tym migotanie przedsionków i tachyarytmia paroksystyczna |
Nieczęsto |
| Tachykardia |
Nieczęsto |
|
| Przyspieszone bicie serca |
Nieczęsto |
|
| Ze strony układu oddechowego, klatki piersiowej i śródpiersia |
Kaszel |
Często |
| Dysfonia |
Nieczęsto |
|
| Ze strony układu pokarmowego |
Diaree |
Często |
| Świędzenie |
Często |
|
| Susza w ustach |
Nieczęsto |
|
| Stomatyt |
Nieczęsto |
|
| Ze strony skóry i tkanek podskórnych |
Ostrze |
Nieczęsto |
| Świąd |
Nieczęsto |
|
| Ze strony nerek i dróg moczowych |
Zatrzymanie moczu |
Nieczęsto |
Zgłaszanie podejrzanych działań niepożądanych.
Zgłaszanie działań niepożądanych po rejestracji produktu leczniczego ma istotne znaczenie. Umożliwia to prowadzenie monitorowania stosunku korzyści do ryzyka w przypadku stosowania tego produktu leczniczego. Pracownicy medyczni i farmaceutyczni, a także pacjenci lub ich upoważnieni przedstawiciele, powinni zgłaszać wszystkie przypadki podejrzewanych działań niepożądanych oraz braku skuteczności produktu leczniczego poprzez Automatyczny System Informacyjny do Nadzoru Farmakologicznego pod adresem: https://aisf.dec.gov.ua.
Okres ważności.
3 lata. Użyć w ciągu 90 dni od momentu otwarcia.
Warunki przechowywania.
Nie wymagane są specjalne warunki przechowywania. Inhalator Genueair® należy przechowywać w oryginalnym opakowaniu do rozpoczęcia użytkowania. Przechowywać w miejscu niedostępnym dla dzieci.
Opakowanie.
30 dawek proszku w inhalatorze; 1 inhalator w folii plastikowej w pudełku z tektury;
60 dawek proszku w inhalatorze; 1 lub 3 inhalatory, każdy w folii plastikowej, w pudełku z tektury.
Kategoria wydawania.
Na receptę.
Producent.
Industrias Farmacéuticas Almirall S.A.
Siedziba producenta oraz adres miejsca wykonywania działalności.
Ctra. de Martorell 41-61, 08740 Sant Andreu de la Barca (Barcelona), Hiszpania.
Wnioskodawca.
Berlin-Chemi AG.
Siedziba wnioskodawcy.
Glinkiker Weg 125, 12489 Berlin, Niemcy.