Azytsytydyn Accord
UkrainaSpis treści
INSTRUKCJA dotyczaca stosowania leczniczego leku Azytsytydyn Accord (Azacitidine Accord)
Skład:
substancja czynna: azytsytydyn;
1 fiolka zawiera azytsytydynu 150 mg;
substancja pomocnicza: manitol (E 421).
Postać leku. Proszek do sporządzenia zawiesiny do wstrzykiwań.
Główne właściwości fizykochemiczne: biały proszek liofilizowany lub masa w przezroczystej szklanej fiolce.
Grupa farmakoterapeutyczna. Środki przeciwnowotworowe. Analogi pirymidyny.
Kod ATC L01B C07.
Właściwości farmakologiczne.
Farmakodynamika.
Mechanizm działania
Przyjmuje się, że azytsytydyn wywiera działanie przeciwnowotworowe poprzez wiele mechanizmów, w tym cytotoksyczność wobec nieprawidłowych komórek hematopoezy w szpiku kostnym oraz hipometylację DNA. Efekt cytotoksyczny azytsytydynu może wynikać z wielu mechanizmów, w tym hamowania syntezy DNA, RNA i białek, włączania do RNA i DNA oraz aktywacji szlaków uszkodzenia DNA. Komórki nieprzerastające są stosunkowo odporne na działanie azytsytydynu. Włączenie azytsytydynu do DNA prowadzi do inaktywacji DNA-metylotransferaz, co powoduje hipometylację DNA. Hipometylacja DNA nieprawidłowo metylowanych genów biorących udział w regulacji normalnego cyklu komórkowego, szlakach różnicowania i śmierci może prowadzić do ponownej ekspresji genów i przywrócenia funkcji supresji nowotworu w komórkach nowotworowych. Względna ważność hipometylacji DNA w porównaniu z cytotoksycznością lub inną aktywnością azytsytydynu w osiąganiu wyników klinicznych nie została ustalona.
Skuteczność i bezpieczeństwo kliniczne
Populacja dorosła (MDS, CMML i AML [20-30 % blastów szpiku kostnego])
Skuteczność i bezpieczeństwo azytsytydynu badano w międzynarodowym, wieloośrodkowym, kontrolowanym, otwartym, randomizowanym badaniu porównawczym fazy 3 z równoległymi grupami (AZA PH GL 2003 CL 001) u dorosłych pacjentów z: MDS o średnim i wysokim ryzyku wg Międzynarodowego Systemu Prognozowania (IPSS), oporną anemią z nadmiarem komórek promienistych (RAEB), oporną anemią z nadmiarem komórek promienistych w przebiegu transformacji (RAEB-T) oraz z modyfikowaną przewlekłą mielomonocytarną białaczką (mCMML) według francusko-amerykańsko-brytyjskiego (FAB) systemu klasyfikacji. Pacjenci z RAEB-T (21-30 % blastów) obecnie są uznawani za pacjentów z AML zgodnie z obecnym systemem klasyfikacji WHO. Schemat azytsytydyn + najlepsza terapia wspierająca (BSC) (n = 179) porównywano ze standardowymi schematami leczenia (CCR). Schematy CCR obejmowały wyłącznie BSC (n = 105), niską dawkę cytarabiny plus BSC (n = 49) lub standardową indukcyjną chemioterapię plus BSC (n = 25). Pacjenci byli wcześniej zakwalifikowani przez lekarza do jednego z trzech schematów CCR przed randomizacją. Pacjenci otrzymywali ten wcześniej wybrany schemat leczenia, jeśli nie zostali randomizowani do grupy otrzymującej azytsytydyn. Według kryteriów włączenia pacjenci musieli mieć status czynnościowy 0-2 według klasyfikacji Wschodniej Grupy Kooperacyjnej z Onkologii (ECOG). Pacjenci z wtórnym MDS zostali wykluczeni z badania. Głównym punktem końcowym badania było przeżycie ogólne. Azytsytydyn podawano podskórnie w dawce 75 mg/m² codziennie przez 7 dni, po czym następowała przerwa trwająca 21 dni (28-dniowy cykl leczenia); mediana liczby cykli wyniosła 9 (zakres = 1-39), a średnia liczba cykli – 10,2. W populacji poddanej leczeniu (ITT) średnia wieku wyniosła 69 lat (zakres od 38 do 88 lat).
W analizie populacji ITT z udziałem 358 pacjentów (179 w grupie azytsytydynu i 179 w grupie CCR) leczenie azytsytydynem wiązało się z medianą przeżycia 24,46 miesiąca w porównaniu do 15,02 miesiąca u pacjentów leczonych schematami CCR; różnica wyniosła 9,4 miesiąca, przy zastosowanym znormalizowanym logarytmicznym teście p=0,0001. Współczynnik ryzyka (HR) dla efektu leczenia wyniósł 0,58 (95 % CI: 0,43, 0,77). Wskaźniki przeżycia dwuletniego wyniosły 50,8 % u pacjentów leczonych azytsytydynem w porównaniu do 26,2 % u pacjentów leczonych CCR (p < 0,0001).
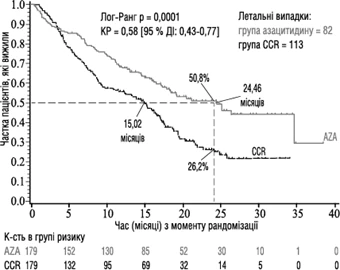
Skróty: AZA = azytsytydyn; CCR = standardowe schematy leczenia; CI = przedział ufności;
HR = współczynnik ryzyka.
Zwiększona długość przeżycia w grupie azytsytydynu była obserwowana konsekwentnie niezależnie od zastosowanego schematu CCR (wyłącznie BSC, niska dawka cytarabiny plus BSC lub standardowa intensywna chemioterapia plus BSC) w grupie kontrolnej.
W analizie podgrup cytogenetycznych IPSS podobne wyniki dotyczące mediany przeżycia ogólnego obserwowano we wszystkich grupach (dobry, pośredni, zły status cytogenetyczny, w tym monosomia 7).
W analizie podgrup wiekowych obserwowano wydłużenie średniego przeżycia ogólnego we wszystkich grupach (< 65 lat, ≥ 65 lat i ≥ 75 lat).
Leczenie azytsytydynem wiązało się również ze średnią czasem do zgonu lub transformacji w AML wynoszącym 13,0 miesiąca w porównaniu do 7,6 miesiąca u pacjentów leczonych CCR, co odpowiada poprawie o 5,4 miesiąca przy znormalizowanym logarytmicznym teście rangowym p = 0,0025.
Leczenie azytsytydynem wiązało się również ze zmniejszeniem cytopenii i związanych z nimi objawów. Leczenie azytsytydynem prowadziło do zmniejszenia potrzeby transfuzji erytrocytów (czerwonych krwinek) i płytek krwi. Wśród pacjentów w grupie azytsytydynu, którzy na początku byli uzależnieni od transfuzji erytrocytów, 45,0 % stało się niezależnych od transfuzji erytrocytów w okresie leczenia w porównaniu do 11,4 % pacjentów w połączonych grupach CCR (statystycznie istotna różnica (p < 0,0001) wyniosła 33,6 % (95 % CI: 22,4, 44,6). U pacjentów, którzy na początku byli uzależnieni od transfuzji erytrocytów i osiągnęli niezależność, średnia długość okresu niezależności od transfuzji erytrocytów wyniosła 13 miesięcy w grupie azytsytydynu.
Odpowiedź na leczenie oceniano przez badacza lub przez Niezależny Komitet Oceny (IRC). Ogólna odpowiedź (pełna remisja [CR] + częściowa remisja [PR]), określona przez badacza, wyniosła 29 % w grupie azytsytydynu i 12 % w grupie połączonych CCR (p = 0,0001). Ogólna odpowiedź (CR + PR), określona przez komitet IRC w badaniu AZA PH GL 2003 CL 001, wyniosła 7 % (12/179) w grupie azytsytydynu w porównaniu do 1 % (2/179) w grupie połączonych CCR (p=0,0113). Różnice między ocenami odpowiedzi przez komitet IRC i badaczy wynikały z kryteriów Międzynarodowej Grupy Roboczej (IWG), które wymagały poprawy parametrów krwi obwodowej i utrzymania tej poprawy przez co najmniej 56 dni. Korzyść w przeżyciu została również wykazana u pacjentów, którzy nie osiągnęli pełnej/cząstkowej odpowiedzi po leczeniu azytsytydynem. Hematologiczna poprawa (duża lub niewielka), zgodnie z oceną IRC, została osiągnięta u 49 % pacjentów leczonych azytsytydynem w porównaniu do 29 % pacjentów leczonych połączonymi CCR (p < 0,0001).
U pacjentów z jedną lub więcej anomaliami cytogenetycznymi na początku odsetek pacjentów z istotną odpowiedzią cytogenetyczną był podobny w grupach azytsytydynu i połączonych CCR.
Niewielka odpowiedź cytogenetyczna była statystycznie istotnie (p=0,0015) wyższa w grupie azytsytydynu (34 %) w porównaniu do grupy połączonych CCR (10 %).
Populacja dorosła w wieku ≥ 65 lat z AML z > 30 % blastów szpiku kostnego
Wyniki podane poniżej dotyczą populacji pacjentów poddanych leczeniu badanej w badaniu AZA-AML-001 (zatwierdzone wskazania podano w sekcji „Sposób stosowania i dawki”).
Skuteczność i bezpieczeństwo azytsytydynu badano w międzynarodowym, wieloośrodkowym, kontrolowanym, otwartym badaniu fazy 3 z równoległymi grupami u pacjentów w wieku ≥ 65 lat z nowo zdiagnozowanym de novo lub wtórnym AML z > 30 % blastów szpiku kostnego zgodnie z klasyfikacją WHO, którzy nie spełniali kryteriów do przeszczepienia komórek macierzystych (HSCT). Azytsytydyn plus BSC (n = 241) porównywano ze schematem CCR. Schemat CCR obejmował wyłącznie BSC (n = 45), niskie dawki cytarabiny plus BSC (n = 158) lub standardową intensywną chemioterapię cytarabiną i antracykliną plus BSC (n = 44). Pacjenci byli wcześniej zakwalifikowani przez lekarza do jednego z trzech schematów leczenia CCR przed randomizacją. Pacjenci otrzymywali wcześniej wybrany schemat, jeśli nie zostali randomizowani do grupy azytsytydynu. Jako część kryteriów włączenia pacjenci musieli mieć status czynnościowy 0-2 według skali ECOG i anomalie cytogenetyczne średniego lub niskiego ryzyka. Głównym punktem końcowym badania było przeżycie ogólne.
Azytsytydyn podawano podskórnie w dawce 75 mg/m²/dobę przez 7 dni, z następującą przerwą trwającą 21 dni (28-dniowy cykl leczenia), z medianą 6 cykli leczenia (zakres: od 1 do 28): pacjenci otrzymujący wyłącznie BSC, z medianą 3 cykle (zakres: od 1 do 20); pacjenci otrzymujący niskie dawki cytarabiny, z medianą 4 cykle (zakres od 1 do 25); pacjenci otrzymujący standardową intensywną chemioterapię, z medianą 2 cykle (zakres: od 1 do 3, cykl indukcyjny plus 1 lub 2 cykle konsolidacji).
Początkowe parametry były porównywalne między grupami azytsytydynu i CCR. Mediana wieku uczestników wyniosła 75,0 roku (zakres: od 64 do 91 lat), 75,2 % to rasa kaukaska, 59,0 % to mężczyźni. Na początku 60,7 % pacjentów sklasyfikowano jako AML nieokreślonego typu, 32,4 % jako AML z cechami związanymi z dysplazją mieloidalną, 4,1 % miało nowotwory mieloidalne związane z terapią, a 2,9 % miało AML z powtarzającymi się anomaliami genetycznymi zgodnie z klasyfikacją WHO.
W analizie populacji ITT z udziałem 488 pacjentów (241 w grupie azytsytydynu i 247 w grupie CCR) leczenie azytsytydynem wiązało się z medianą przeżycia 10,4 miesiąca w porównaniu do 6,5 miesiąca u pacjentów leczonych CCR – różnica wyniosła 3,8 miesiąca, przy znormalizowanym logarytmicznym teście rangowym p = 0,1009 (dwustronny). Współczynnik ryzyka dla efektu leczenia wyniósł 0,85 (95 % CI = 0,69, 1,03). Przeżycie roczne wyniosło 46,5 % u pacjentów leczonych azytsytydynem w porównaniu do 34,3 % u pacjentów leczonych schematem CCR.
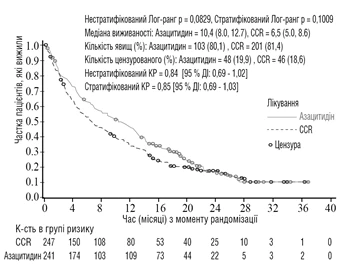
Skróty: CCR = standardowe schematy leczenia; CI = przedział ufności; HR = współczynnik ryzyka.
Zgodnie z modelem proporcjonalnych ryzyk Coxa, skorygowanym o wcześniej określone czynniki prognostyczne, HR dla azytsytydynu w porównaniu do schematu CCR wyniósł 0,80 (95 % CI = 0,66, 0,99; p = 0,0355).
Ponadto, choć badanie nie miało możliwości wykazania statystycznie istotnej różnicy przy porównaniu azytsytydynu z wcześniej wybranymi grupami CCR, przeżycie pacjentów leczonych azytsytydynem było dłuższe w porównaniu do tych opcji leczenia CCR, takich jak wyłącznie BSC, niska dawka cytarabiny plus BSC, i było podobne do standardowej intensywnej chemioterapii plus BSC.
We wszystkich wcześniej określonych podgrupach wieku [(< 75 lat i ≥ 75 lat), płeć, rasa, status czynnościowy wg skali ECOG (0, lub 1 i 2), początkowe ryzyko cytogenetyczne (średnie i niskie), region geograficzny, klasyfikacja AML WHO (w tym AML z cechami związanymi z dysplazją mieloidalną), początkowa liczba leukocytów (≤ 5 x 10⁹/l i > 5 x 10⁹/l), początkowa liczba blastów szpiku kostnego (≤ 50 % i > 50 %) oraz wcześniejsza historia choroby MDS], obserwowano tendencję do korzyści OS na korzyść azytsytydynu. W kilku wcześniej określonych podgrupach HR OS osiągnął istotność statystyczną, w tym u pacjentów z niskim ryzykiem cytogenetycznym, pacjentów z AML z cechami związanymi z dysplazją mieloidalną, pacjentów w wieku < 75 lat, pacjentek oraz pacjentów rasy kaukaskiej.
Odpowiedzi hematologiczne i cytogenetyczne oceniano przez badacza i komitet ekspertów instytucji (IRC) w porównaniu z wynikami analogicznymi. Ogólna częstość odpowiedzi (pełna remisja [CR] + pełna remisja z niepełnym przywróceniem analizy krwi [CRi]), jak określono przez IRC, wyniosła 27,8 % w grupie azytsytydynu i 25,1 % w grupie połączonej terapii CCR (p = 0,5384). U pacjentów, którzy osiągnęli CR lub CRi, średnia długość remisji wyniosła 10,4 miesiąca (95 % CI = 7,2, 15,2) u pacjentów leczonych azytsytydynem i 12,3 miesiąca (95 % CI = 9,0, 17,0) u pacjentów leczonych CCR. Poprawa przeżycia została również wykazana u pacjentów, którzy nie osiągnęli pełnej odpowiedzi na azytsytydyn w porównaniu do schematu CCR.
Leczenie azytsytydynem poprawiło parametry krwi obwodowej i prowadziło do zmniejszenia potrzeby transfuzji erytrocytów i płytek krwi. Pacjent był uznawany za uzależnionego od transfuzji erytrocytów lub płytek krwi na początku, jeśli miał jedną lub więcej transfuzji erytrocytów lub płytek krwi w ciągu 56 dni (8 tygodni) przed rozpoczęciem lub randomizacją. Pacjent był uznawany za niezależnego od transfuzji erytrocytów lub płytek krwi w okresie leczenia, jeśli nie miał transfuzji erytrocytów lub płytek krwi w ciągu dowolnych kolejnych 56 dni w okresie raportowania.
Wśród pacjentów w grupie azytsytydynu, którzy na początku byli uzależnieni od transfuzji erytrocytów, 38,5 % (95 % CI = 31,1, 46,2) tych pacjentów stało się niezależnych od transfuzji erytrocytów w okresie leczenia w porównaniu do 27,6 % (95 % CI = 20,9, 35,1) pacjentów w połączonych grupach CCR. U pacjentów, którzy na początku byli uzależnieni od transfuzji erytrocytów i osiągnęli niezależność od transfuzji krwi w trakcie leczenia, średnia długość okresu niezależności od transfuzji erytrocytów wyniosła 13,9 miesiąca w grupie azytsytydynu; w grupie CCR nie osiągnięto takiej niezależności.
Wśród pacjentów w grupie azytsytydynu, którzy na początku byli uzależnieni od transfuzji płytek krwi, 40,6 % (95 % CI = 30,9, 50,8) pacjentów stało się niezależnych od transfuzji płytek krwi w okresie leczenia w porównaniu do 29,3 % (95 % CI = 19,7, 40,4) pacjentów w połączonych grupach CCR. U pacjentów, którzy na początku byli uzależnieni od transfuzji płytek krwi i osiągnęli niezależność od transfuzji krwi po leczeniu, średnia długość okresu niezależności od transfuzji płytek krwi wyniosła 10,8 miesiąca w grupie azytsytydynu i 19,2 miesiąca w grupie CCR.
Jakość życia związaną ze zdrowiem (HRQoL) oceniano za pomocą Podstawowego Kwestionariusza Jakości Życia Europejskiej Organizacji ds. Badania i Leczenia Raka (EORTC QLQ-C30). Dane HRQoL mogą być analizowane dla podgrupy pełnej populacji badanej. Pomimo ograniczeń analizy, uzyskane dane wskazują, że pacjenci nie odczuwają istotnego pogorszenia jakości życia podczas leczenia azytsytydynem.
Dzieci
Badanie AZA-JMML-001 było międzynarodowym, wieloośrodkowym, otwartym badaniem fazy 2 oceniającym farmakokinetykę, farmakodynamikę, bezpieczeństwo i aktywność azytsytydynu przed przeszczepieniem HSCT u dzieci z niedawno zdiagnozowanym postępującym MDS lub JMML. Głównym celem badania klinicznego było ocenienie wpływu azytsytydynu na poziom odpowiedzi na leczenie w 3. cyklu, w dniu 28.
Pacjenci (z MDS: n = 10; z JMML: n = 18, w wieku od 3 miesięcy do 15 lat; 71 % mężczyzn) otrzymywali leczenie azytsytydynem dożylnie w dawce 75 mg/m² codziennie od dnia 1 do 7, 28-dniowego cyklu przez co najmniej trzy cykle i maksymalnie sześć cykli.
Włączenie pacjentów z MDS do grupy badawczej zostało zakończone po zarekrutowaniu 10 pacjentów z MDS z powodu niewystarczającej skuteczności leczenia: u tych 10 pacjentów nie odnotowano potwierdzonych odpowiedzi.
Do grupy badawczej JMML włączono 18 pacjentów (13 z somatycznymi mutacjami PTPN11, 3 z mutacjami NRAS, 1 z mutacją KRAS i 1 z klinicznym rozpoznaniem neurofibromatozy typu 1 [NF-1]). 16 pacjentów ukończyło trzy cykle terapii, a 5 z nich ukończyło 6 cykli. Łącznie 11 pacjentów z JMML miało odpowiedź kliniczną w cyklu 3, w dniu 28; z tych 11 uczestników badania 9 (50 %) miało potwierdzoną odpowiedź kliniczną (3 uczestników z cCR i 6 uczestników z cPR). Wśród kohorty pacjentów z JMML otrzymujących azytsytydyn, 7 (43,8 %) pacjentów miało trwałą odpowiedź na poziomie płytek krwi (liczba ≥ 100 x 10⁹/l), a 7 (43,8 %) pacjentów wymagało transfuzji krwi podczas HSCT. 17 z 18 pacjentów przeszło do procedury HSCT.
Ze względu na projekt badania (mała liczba pacjentów i różne czynniki utrudniające analizę), na podstawie tego badania klinicznego niemożliwe jest stwierdzenie, czy zastosowanie azytsytydynu przed procedurą HSCT poprawia wyniki przeżycia u pacjentów z JMML.
Badanie AZA-AML-004 było wieloośrodkowym, otwartym badaniem fazy 2 oceniającym bezpieczeństwo, farmakodynamikę i skuteczność azytsytydynu w porównaniu z brakiem leczenia przeciwnowotworowego u dzieci i młodych dorosłych z AML w stadium molekularnego nawrotu po CR1.
Siedem pacjentów (mediana wieku 6,7 roku [zakres od 2 do 12 lat]; 71,4 % mężczyzn) otrzymywało dożylne podanie azytsytydynu w dawce 100 mg/m² codziennie od dnia 1 do 7 każdego 28-dniowego cyklu przez maksymalnie 3 cykle.
Pięciu pacjentów zgodnie z oceną wykazywało objawy minimalnej choroby resztkowej (MRD) w dniu 84, przy czym 4 pacjentów osiągnęło albo molekularną stabilizację (n = 3), albo molekularną poprawę (n = 1), a u jednego pacjenta zaobserwowano nawrót kliniczny. Sześć z siedmiu pacjentów (90 % [95 % CI = 0,4, 1,0]), którzy otrzymywali azytsytydyn, przeszło do HSCT.
Ze względu na małą liczbę pacjentów niemożliwe jest ustalenie skuteczności azytsytydynu u dzieci z AML. Informacje o bezpieczeństwie podano w sekcji „Działania niepożądane”.
Farmakokinetyka.
Wchłanianie
Po podskórnym podaniu pojedynczej dawki 75 mg/m² azytsytydyn szybko wchłaniał się, osiągając maksymalną stężenie w osoczu krwi 750 ± 403 ng/ml, obserwowane po 0,5 godziny po podaniu dawki (pierwszy punkt poboru).
Bezwzględna biodostępność azytsytydynu po podaniu podskórnym w porównaniu z dożylnym (pojedyncza dawka 75 mg/m²) wynosiła około 89 %, na podstawie wartości pola pod krzywą farmakokinetyczną (AUC).
Pole pod krzywą i maksymalne stężenie w osoczu krwi (Cmax) po podaniu podskórnym azytsytydynu były w przybliżeniu proporcjonalne w zakresie dawek od 25 do 100 mg/m².
Rozkład
Po dożylnej podaniu średnia objętość rozkładu wyniosła 76 ± 26 l, a klirens systemowy – 147 ± 47 l/godz.
Biotransformacja
Zgodnie z danymi in vitro, metabolizm azytsytydynu nie jest pośredniczony przez izoenzymy cytochromu P450 (CYP), UDP-glukuronozylotransferazy (UGT), sulfotransferazy (SULT) i glutatyonotransferazy (GST).
Azytsytydyn ulega samorzutnemu hydrolizie i deaminacji z udziałem cytydynadezaminazy. W frakcjach S9 wątroby ludzkiej powstawanie metabolitów nie zależało od zredukowanego nikotynamidoadeninodinukleotydu fosforanu (NADPH), co oznacza, że metabolizm azytsytydynu nie jest pośredniczony przez izoenzymy cytochromu P450. Dane badania in vitro azytsytydynu w hodowlach hepatocytów ludzkich wskazują, że w stężeniach od 1,0 µM do 100 µM (czyli około 30 razy wyższych niż osiągane klinicznie stężenia), azytsytydyn nie indukuje CYP 1A2, 2C19 ani 3A4 czy 3A5. W badaniach oceny hamowania szeregu izoenzymów P450 (CYP 1A2, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1 i 3A4) azytsytydyn w dawkach do 100 µM nie powodował hamowania. W związku z tym indukcja lub hamowanie enzymu CYP przez azytsytydyn w klinicznie osiągalnych stężeniach w osoczu krwi jest mało prawdopodobna.
Wydalanie
Azytsytydyn szybko wydala się z osocza krwi ze średnią połowiczny okresem eliminacji (t1/2) wynoszącym po podaniu podskórnemu 41 ± 8 minut. Po podaniu podskórnemu dawki 75 mg/m² azytsytydynu raz dziennie przez siedem dni nie obserwowano akumulacji. Wydalanie z moczem jest główną drogą eliminacji azytsytydynu i/lub jego metabolitów. Po dożylnej i podskórnej podaniu 14C-azytsytydynu 85 i 50 % podanej znaczonej substancji wykryto odpowiednio w moczu, podczas gdy < 1 % wykryto w kale.
Populacje specjalne
Wpływ niewydolności wątroby (patrz sekcja „Sposób stosowania i dawki”), płci, wieku lub pochodzenia rasowego na farmakokinetykę azytsytydynu nie był oficjalnie badany.
U dzieci
W badaniu AZA-JMML-001 analizę farmakokinetyczną przeprowadzono u 10 pacjentów z MDS i 18 dzieci z JMML w dniu 7 cyklu 1 (patrz sekcja „Właściwości farmakologiczne”). Mediana wieku (zakres) pacjentów z MDS wyniosła 13,3 (1,9-15) roku, a pacjentów z JMML – 2,1 (0,2-6,9) roku.
Po dożylnej podaniu dawki 75 mg/m² azytsytydyn szybko osiągał stężenie Cmax w ciągu 0,083 godziny zarówno w populacjach MDS, jak i JMML. Średnia geometryczna wartość Cmax wyniosła 1797,5 i 1066,3 ng/ml, a średnia geometryczna wartość AUC0-∞ wyniosła 606,9 i 240,2 ng*godz/ml dla pacjentów z MDS i JMML odpowiednio. Średnia geometryczna objętość rozkładu u pacjentów z MDS i JMML wyniosła odpowiednio 103,9 i 61,1 l. Stwierdzono, że ogólna ekspozycja na azytsytydyn w osoczu krwi była wyższa u pacjentów z MDS; jednak umiarkowana lub wysoka zmienność między pacjentami została zauważona zarówno dla parametrów AUC, jak i Cmax.
Średnia geometryczna okresu półtrwania t1/2 wyniosła 0,4 i 0,3 godziny, a średnia geometryczna klirensu – 166,4 i 148,3 l/godz dla MDS i JMML odpowiednio.
Dane farmakokinetyczne badania AZA-JMML-001 zostały połączone i porównane z danymi farmakokinetycznymi sześciu dorosłych pacjentów z MDS, którzy otrzymywali 75 mg/m² azytsytydynu dożylnie w badaniu AZA-2002-BA-002. Średnie wartości Cmax i AUC0-t azytsytydynu były podobne u dorosłych pacjentów i dzieci po dożylnej podaniu (2750 ng/ml vs 2841 ng/ml i 1025 ng*godz/ml vs 882,1 ng*godz/ml odpowiednio).
W badaniu AZA-AML-004 analizę farmakokinetyczną przeprowadzono u 6 z 7 pacjentów-dzieci, którzy mieli co najmniej jedną zmierzoną stężenie farmakokinetyczne po podaniu dawki leku (patrz sekcja „Właściwości farmakologiczne”). Mediana (zakres) wieku pacjentów z AML wyniosła 6,7 (2-12) lat.
Po podaniu wielokrotnych dawek leku 100 mg/m² średnie geometryczne wartości Cmax i AUC0-t w dniu 7 cyklu 1 wyniosły odpowiednio 1557 ng/ml i 899,6 ng*godz/ml, z dużą zmiennością międzyosobniczą (współczynnik zmienności (CV) 201,6 % i 87,8 % odpowiednio). Azytsytydyn szybko osiągał stężenie Cmax z medianą czasu 0,090 godziny po podaniu dożylnej i zmniejszał się ze średnią geometryczną wartością okresu półtrwania t1/2 wynoszącą 0,380 godziny. Średnie geometryczne wartości klirensu i objętości rozkładu wyniosły odpowiednio 127,2 l/godz i 70,2 l.
Ekspozycja farmakokinetyczna (azytsytydynu) obserwowana u dzieci z AML w nawrocie molekularnym po CR1 była porównywalna z ekspozycją uzyskaną z połączonych danych dla 10 dzieci z MDS i 18 dzieci z JMML, a także porównywalna z ekspozycją azytsytydynu u dorosłych z MDS.
Uchybienia czynności nerek
Uchybienia czynności nerek nie mają istotnego wpływu na ekspozycję farmakokinetyczną azytsytydynu po jednorazowym lub wielokrotnym podaniu podskórnym. Po podaniu podskórnemu pojedynczej dawki 75 mg/m² średnie wartości ekspozycji (AUC i Cmax) u pacjentów z łagodnym, umiarkowanym i ciężkim uchybieniem czynności nerek wzrastały odpowiednio o 11-21 %, 15-27 % i 41-66 % w porównaniu z pacjentami z normalną czynnością nerek. Jednak ekspozycja znajdowała się w tym samym ogólnym zakresie ekspozycji obserwowanym u osób z normalną czynnością nerek. Azytsytydyn można przepisywać pacjentom z uchybieniem czynności nerek bez korekty dawki początkowej, pod warunkiem, że pacjenci są monitorowani pod kątem objawów toksyczności, ponieważ azytsytydyn i/lub jego metabolity są głównie wydalane przez nerki.
Farmakogenomika
Wpływ znanych polimorfizmów cytydynadezaminazy na metabolizm azytsytydynu nie był oficjalnie badany.
Dane kliniczne.
Wskazania.
Azytsytydyn Accord wskazany jest w leczeniu dorosłych pacjentów, u których nie wskazano przeszczepu komórek macierzystych (HSCT), z następującymi chorobami:
- zespołami mielodysplastycznymi (MDS) o średnim i wysokim stopniu ryzyka wg Międzynarodowego Systemu Prognozowania (IPSS),
- przewlekłym mielomonocytarnym białaczką (CMML) z 10–29 % blastów szpiku kostnego bez zaburzeń mieloproliferyjnych,
- ostrym białaczką mieloidalnym (AML) z 20–30 % blastów i dysplazją wieloliniową zgodnie z klasyfikacją Światowej Organizacji Zdrowia (WHO),
- AML z > 30 % blastów szpiku kostnego według klasyfikacji WHO.
Przeciwwskazania.
Podwyższona wrażliwość na substancję czynną lub na którąkolwiek z substancji pomocniczych.
Zaawansowane nowotwory wątroby (patrz sekcja „Szczególne ostrzeżenia i środki ostrożności”).
Karmienie piersią (patrz sekcja „Stosowanie w czasie ciąży lub karmienia piersią”).
Szczególne ostrzeżenia i środki ostrożności.
Zalecenia dotyczące bezpiecznego obchodzenia się z lekiem
Azytsytydyn Accord jest lekiem cytotoksycznym, dlatego należy zachować ostrożność podczas pracy z preparatem i przygotowywania zawiesiny azytsytydyny, tak jak w przypadku innych potencjalnie toksycznych związków. Należy stosować odpowiednie procedury dotyczące bezpiecznego obchodzenia się z lekami przeciwnowotworowymi oraz ich utylizacji.
W przypadku kontaktu rozpuszczonego azytsytydynu z skórą należy natychmiast dokładnie przemyć miejsce wodą z mydłem. W przypadku kontaktu z błonami śluzowymi należy dokładnie przemyć wodą.
Procedura rozcieńczania
Azytsytydyn Accord należy rozcieńczyć wodą do wstrzykiwań. Okres przydatności po rozcieńczeniu można wydłużyć, stosując schłodzoną wodę do wstrzykiwań (2 °C do 8 °C). Szczegółowe informacje dotyczące przechowywania rozcieńczonego produktu znajdują się poniżej.
Należy przygotować następujące materiały:
- Fiolki azytsytydyny; fiolki wody do wstrzykiwań; niesterylne rękawiczki chirurgiczne; chłonne serwetki alkoholowe; strzykawki do wstrzykiwań o pojemności 5 ml z igłą(-ami).
- Należy nabrać do strzykawki odpowiednią objętość wody do wstrzykiwań (patrz tabela poniżej) i koniecznie usunąć powietrze, które dostało się do wnętrza strzykawki.
| Pojemność fiolki |
Objętość wody do wstrzykiwań |
Końcowa stężenie |
| 150 mg |
6 ml |
25 mg/ml |
- Igrę szpryca zawierającego wodę do wstrzykiwań należy włożyć przez gumową pokrywkę fiolki z azytsytydyną, a następnie wlać wodę do wstrzykiwań do fiolki.
- Po usunięciu szpryca i igły fiolkę należy silnie wstrząsać aż do uzyskania jednorodnej, mętnej zawiesiny. Po rozcieńczeniu każdy ml zawiesiny zawiera 25 mg azytsytydyny (100 mg/4 ml lub 150 mg/6 ml). Rozcieńczony lek stanowi jednorodną, mętną zawiesinę bez agregatów. Lek należy odrzucić, jeśli zawiera duże cząstki lub agregaty. Nie wolno filtrować zawiesiny po rozcieńczeniu, ponieważ może to prowadzić do usunięcia substancji czynnej. Należy pamiętać, że niektóre łączniki, stożki i zamknięte systemy mogą zawierać filtry; w związku z tym nie należy stosować takich systemów do podawania leku po rozcieńczeniu.
- Należy oczyścić gumową pokrywkę i włożyć nowy szpryc z igłą do fiolki. Następnie fiolkę należy odwrócić do góry nogami, upewniając się, że koniec igły znajduje się poniżej poziomu cieczy. Następnie tłok należy delikatnie pociągnąć do tyłu, aby wciągnąć ilość leku niezbędną do podania odpowiedniej dawki, upewniając się, że całe powietrze wprowadzone do szpryca zostało usunięte. Następnie szpryc z igłą należy wyjąć z fiolki, a igłę zutylizować.
- Nową igłę do podania podskórnie (zalecana 25 kalibru) należy mocno zamocować do szpryca. Nie należy przepłukiwać igły powietrzem przed wstrzyknięciem, aby zmniejszyć częstość miejscowych reakcji w miejscu wstrzyknięcia.
- Jeśli konieczne jest podanie więcej niż jednej fiolki, należy powtórzyć wszystkie powyższe czynności przygotowania zawiesiny. W przypadku dawek wymagających więcej niż jednej fiolki, dawkę należy podzielić równo, np. dawka 150 mg = 6 ml, 2 szprycy po 3 ml w każdym szprycu. Z powodu strat leku w fiolce i igle może być niemożliwe pobranie całej zawiesiny z fiolki.
- Zawartość szpryca dawkowego należy ponownie zawiesić bezpośrednio przed podaniem. Szpryc wypełniony rozcieńczoną zawiesiną należy ogrzać do temperatury około 20–25 °C w ciągu 30 minut przed podaniem. Jeśli upłynie więcej niż 30 minut, zawiesinę należy odpowiednio zutylizować i przygotować nową dawkę. Aby ponownie zawiesić zawartość, należy silnie przetaczać szpryc między dłońmi, aż do uzyskania jednorodnej, mlecznej zawiesiny. Lek należy odrzucić, jeśli zawiera duże cząstki lub agregaty.
Przechowywanie rozcieńczonego leku
Warunki przechowywania po rozcieńczeniu leku podano w sekcji „Okres przydatności do użycia”.
Obliczanie dawki indywidualnej
Dawkę całkowitą zgodnie z powierzchnią ciała (BSA) można obliczyć w następujący sposób:
Dawka całkowita (mg) = dawka (mg/m²) × BSA (m²)
Poniższa tabela podana jest wyłącznie jako przykład obliczania indywidualnych dawek azytsytydynu na podstawie średniej powierzchni ciała 1,8 m².
| Dawka, mg/m2 (% zalecanej dawki początkowej) |
Całkowita dawka, wyliczona na podstawie wartości PPM 1,8 m2 |
Potrzebna liczba fiol |
Całkowita potrzebna objętość rozcieńczonej zawiesiny |
| Fiolka 150 mg |
|||
| 75 mg/m2 (100 %) |
135 mg |
1 fiolka |
5,4 ml |
| 37,5 mg/m2 (50 %) |
67,5 mg |
1 fiolka |
2,7 ml |
| 25 mg/m2 (33 %) |
45 mg |
1 fiolka |
1,8 ml |
Sposób stosowania
Rozcieńczony Azytsytydyn Accord należy podawać podskórnie (wstrzykując igłę pod kątem 45–90°) za pomocą igły kalibru 25 w ramię, udo lub brzuch.
Dawki przekraczające 4 ml należy wstrzykiwać w dwóch oddzielnych miejscach.
Miejsca wstrzyknięć należy zmieniać. Nowe wstrzyknięcia należy wykonywać w odległości co najmniej 2,5 cm od poprzedniego miejsca i nigdy w bolące obszary, obszary z sinicami, zaczerwienione lub z twardymi zmianami.
Utylizacja
Nie wykorzystany lek lub odpady należy usunąć zgodnie z lokalnymi przepisami.
Interakcje z innymi lekami i inne rodzaje interakcji.
Zgodnie z danymi uzyskanymi in vitro, metabolizm azytsytydynu nie jest pośrednictwem izoenzymów cytochromu P450 (CYP), UDP-glukuronozylotransferaz (UGT), sulfotransferaz (SULT) i glutationotransferaz (GST), dlatego interakcje związane z tymi enzymami metabolizującymi in vivo uznaje się za mało prawdopodobne.
Klinicznie istotne hamujące lub indukcyjne działanie azytsytydynu na enzymy cytochromu P450 jest mało prawdopodobne (patrz sekcja „Właściwości farmakologiczne”).
Nie przeprowadzono oficjalnych badań klinicznych dotyczących interakcji z azytsytydynem.
Szczególne środki ostrożności dotyczące stosowania.
Toxyczność hematologiczna
Leczenie azytsytydyną wiąże się z występowaniem anemii, neutropenii oraz trombocytopenii, szczególnie w pierwszych dwóch cyklach leczenia (patrz sekcja „Powikłania uboczne”). Przed każdym cyklem leczenia, a także w razie potrzeby, należy wykonywać rozwinięte badanie krwi w celu monitorowania odpowiedzi na leczenie i toksyczności. Po podaniu zalecanej dawki w pierwszym cyklu, dawkę w kolejnych cyklach należy zmniejszyć lub odłożyć podanie leku, w zależności od najniższych wartości liczby komórek krwiotwórczych i hematologicznej odpowiedzi na leczenie (patrz sekcja „Sposób i dawka stosowania”). Pacjentom należy zalecić natychmiastowe zgłaszanie wystąpienia gorączki. Pacjenci i lekarze powinni również zachować ostrożność wobec objawów i oznak krwawienia.
Upośledzenie funkcji wątroby
Nie przeprowadzono oficjalnych badań z udziałem pacjentów z upośledzeniem funkcji wątroby. Podczas leczenia azytsytydyną obserwowano postępującą śpiączkę wątrobą i zgon u pacjentów z dużym obciążeniem nowotworowym spowodowanym chorobą przerzutową, szczególnie u pacjentów z początkowym stężeniem albumin w surowicy < 30 g/l. Azytsytydyn jest przeciwwskazany u pacjentów z zaawansowanymi złośliwymi nowotworami wątroby (patrz sekcja „Przeciwskazania”).
Upośledzenie funkcji nerek
U pacjentów otrzymujących azytsytydynę dożylnie w połączeniu z innymi lekami chemioterapeutycznymi obserwowano zaburzenia funkcji nerek, od podwyższenia stężenia kreatyniny w surowicy po niewydolność nerek i śmierć. Ponadto u pięciu pacjentów z przewlekłą białaczką szpikową (CML), którzy otrzymywali leczenie azytsytydyną i etopozydem, rozwinął się kwasicytowy kanalik nerkowy, charakteryzujący się obniżeniem stężenia bikarbonianu w surowicy do < 20 mmol/l, w połączeniu z podwyższoną zasadowością moczu i hipokaliemią (stężenie potasu w surowicy < 3 mmol/l). W przypadku nieuzasadnionego obniżenia stężenia bikarbonianu w surowicy (< 20 mmol/l) lub wzrostu stężenia kreatyniny lub AKP w surowicy należy rozważyć zmniejszenie dawki lub odłożenie podania leku (patrz sekcja „Sposób i dawka stosowania”).
Pacjentom należy zalecić natychmiastowe zgłaszanie oligurii i anurii.
Chociaż nie stwierdzono klinicznie istotnych różnic w częstości występowania działań niepożądanych między osobami z prawidłową funkcją nerek a pacjentami z niewydolnością nerek, pacjentów z niewydolnością nerek należy starannie monitorować pod kątem objawów toksyczności, ponieważ azytsytydyn i/lub jego metabolity są wydalane głównie z moczem (patrz sekcja „Sposób i dawka stosowania”).
Badania laboratoryjne
Przed rozpoczęciem terapii oraz przed każdym cyklem leczenia należy wykonać badania funkcji wątroby, stężenia kreatyniny i bikarbonianu w surowicy. Badanie rozwinięte krwi należy wykonywać przed rozpoczęciem terapii oraz w razie potrzeby do monitorowania odpowiedzi na leczenie i toksyczności, ale co najmniej przed każdym cyklem leczenia (patrz również sekcja „Powikłania uboczne”).
Choroby serca i płuc
Pacjenci z ciężką przewlekłą niewydolnością serca w wywiadzie, klinicznie niestabilną chorobą serca lub chorobą płuc byli wykluczeni z podstawowych badań rejestracyjnych (AZA PH GL 2003 CL 001 oraz AZA-AML-001), dlatego bezpieczeństwo i skuteczność azytsytydyny u tych pacjentów nie są ustalone. Najnowsze dane z badań klinicznych u pacjentów z chorobami sercowo-naczyniowymi lub płucnymi w wywiadzie wykazały istotne zwiększenie częstości powikłań sercowych podczas stosowania azytsytydyny (patrz sekcja „Powikłania uboczne”). Dlatego zaleca się ostrożne przepisywanie azytsytydyny tym pacjentom. Przed rozpoczęciem i w trakcie leczenia należy ocenić funkcje serca i płuc.
Faszcyt necrotyczny
U pacjentów otrzymujących azytsytydynę zgłaszano przypadki faszcytu necrotycznego, w tym zakończone śmiercią. U pacjentów, u których rozwinął się faszcyt necrotyczny, należy przerwać leczenie azytsytydyną i natychmiast rozpocząć odpowiednie leczenie.
Zespół lizy guza
Pacjenci z ryzykiem wystąpienia zespołu lizy guza to osoby z dużym obciążeniem nowotworowym przed leczeniem. Takich pacjentów należy starannie monitorować i podejmować odpowiednie środki zapobiegawcze.
Zespół różnicowania
U pacjentów stosujących azytsytydynę odnotowano przypadki zespołu różnicowania (znany również jako zespół kwasu retinowego). Zespół różnicowania może być śmiertelny, a jego objawy i oznaki kliniczne obejmują niewydolność oddechową, infiltraty płucne, gorączkę, wysypkę, obrzęk płuc, obrzęk obwodowy, szybki przyrost masy ciała, wylew do opłucnej, wylew do osierdzia, hipotensję oraz zaburzenia funkcji nerek (patrz sekcja „Powikłania uboczne”). W przypadku pierwszych objawów lub oznak wskazujących na zespół różnicowania należy rozważyć leczenie wysokimi dawkami kortykosteroidów dożylnych i monitorowanie hemodynamiki. Należy rozważyć możliwość tymczasowego przerwania stosowania azytsytydyny do ustąpienia objawów, a w przypadku wznowienia leczenia należy zachować ostrożność.
Stosowanie w czasie ciąży lub karmienia piersią.
Kobiety w wieku rozrodczym / Antykoncepcja u mężczyzn i kobiet
Kobietom w wieku rozrodczym należy zalecić stosowanie skutecznych środków antykoncepcyjnych w trakcie leczenia i co najmniej przez 6 miesięcy po jego zakończeniu. Mężczyźni powinni stosować skuteczną antykoncepcję w trakcie leczenia i przez 3 miesiące po jego zakończeniu.
Ciąża
Nie ma wystarczających danych dotyczących stosowania azytsytydyny u ciężarnych kobiet. Badania na myszach wykazały toksyczność reprodukcyjną leku. Ryzyko dla człowieka nie jest znane. Na podstawie badań na zwierzętach i mechanizmu działania azytsytydyny nie należy stosować tego leku w czasie ciąży, szczególnie w I trymestrze, chyba że istnieje pilna potrzeba jego zastosowania. W każdym przypadku należy dokładnie ocenić korzyści z leczenia i potencjalne ryzyko dla płodu.
Karmienie piersią
Nie wiadomo, czy azytsytydyna/metabolity przenikają do mleka matki. Z uwagi na możliwość wystąpienia poważnych działań niepożądanych u niemowlęcia karmionego piersią, karmienie piersią jest przeciwwskazane w czasie leczenia azytsytydyną.
Fekundacja
Brak danych dotyczących wpływu azytsytydyny na płodność człowieka. U zwierząt stwierdzono działania niepożądane na płodność samców po zastosowaniu azytsytydyny. Przed rozpoczęciem leczenia mężczyźni powinni otrzymać poradę dotyczącą zabezpieczenia nasienia.
Wpływ na zdolność prowadzenia pojazdów i obsługi urządzeń.
Azytsytydyn ma nieznaczny lub umiarkowany wpływ na zdolność prowadzenia pojazdów i obsługi urządzeń. Podczas stosowania azytsytydyny zgłaszano zmęczenie. Dlatego zaleca się zachowanie ostrożności podczas prowadzenia pojazdów lub pracy z innymi urządzeniami.
Sposób stosowania i dawki
Leczenie azytsytydyną Accord należy rozpoczynać i kontrolować pod nadzorem lekarza posiadającego doświadczenie w stosowaniu leków przeciwnowotworowych. Pacjentom należy podać leki przeciwwymiotne w celu zapobiegania nudności i wymiotom.
Dawki
Zalecana dawka początkowa w pierwszym cyklu leczenia dla wszystkich pacjentów, niezależnie od początkowych hematologicznych wartości laboratoryjnych, wynosi 75 mg/m² powierzchni ciała, podawana podskórnie codziennie przez 7 dni, po czym następuje przerwa trwająca 21 dni (28-dniowy cykl leczenia).
Zaleca się, aby pacjenci otrzymywali leczenie przez co najmniej sześć cykli. Leczenie należy kontynuować tak długo, jak pacjent czerpie korzyści z terapii lub do czasu postępu choroby.
Należy obserwować pacjentów pod kątem oznak hematologicznej reakcji/toxyczności oraz toksyczności dla nerek (patrz sekcja „Szczególne ostrzeżenia i środki ostrożności podczas stosowania”); może być konieczne odłożenie rozpoczęcia kolejnego cyklu lub zmniejszenie dawki, zgodnie z opisem poniżej.
Preparatu Azytsytydyna Accord nie należy stosować jako wzajemnie zastępczego w stosunku do doustnej azytsytydyny. Ze względu na różnicę w ekspozycji zalecenia dotyczące dawki i schematu podawania doustnej azytsytydyny różnią się od zaleceń dotyczących azytsytydyny do wstrzykiwań. Osobom pracującym w ochronie zdrowia zaleca się dokładne sprawdzanie nazwy leku, dawki i sposobu stosowania.
Badania laboratoryjne
Przed rozpoczęciem terapii oraz przed każdym cyklem leczenia należy wykonać badania czynności wątroby, oznaczyć stężenie kreatyniny w surowicy i stężenie bikarbonianu w surowicy. Rozszerzony morfologiczny przegląd krwi należy wykonywać przed rozpoczęciem terapii oraz w razie potrzeby monitorować odpowiedź na leczenie i toksyczność, ale co najmniej przed każdym cyklem leczenia.
Korekta dawki z powodu toksyczności hematologicznej
Toksykność hematologiczna definiowana jest jako najniższa osiągnięta liczba (nadir) w danym cyklu, jeśli liczba płytek krwi ≤ 50,0 x 10⁹/l i/lub bezwzględna liczba neutrofili (ALN) ≤ 1 x 10⁹/l.
Odzyskanie stanu definiowane jest jako wzrost linii komórkowej (linii komórkowych), w której wystąpiła toksyczność hematologiczna, co najmniej o połowę bezwzględnej różnicy między najniższą a początkową liczbą plus najniższa liczba (tzn. liczba komórek krwi w momencie odzyskania ≥ najniższa liczba + (0,5 × [liczba początkowa – najniższa liczba])).
Pacjenci bez zmniejszonych początkowych parametrów morfologii krwi (tzn. liczba leukocytów (WBC) ≥ 3,0 x 10⁹/l i ALN ≥ 1,5 x 10⁹/l oraz liczba płytek krwi ≥ 75,0 x 10⁹/l) przed pierwszym cyklem leczenia
Jeśli po leczeniu azytsytydyną Accord wystąpi toksyczność hematologiczna, kolejny cykl terapii należy odłożyć do odzyskania liczby płytek krwi i ALN. Jeśli odzyskanie nastąpi w ciągu 14 dni, korekta dawki nie jest wymagana. Jednak jeśli odzyskanie nie nastąpi w ciągu 14 dni, dawkę należy zmniejszyć zgodnie z poniższą tabelą. Po zmianie dawki długość cyklu powinna wrócić do 28 dni.
| Nadir cyklu |
Dawka w następnym cyklu, jeśli odzyskanie* nie osiągnięte w ciągu 14 dni (%) |
|
| AKN (x 109/l) |
Trzomocyty (x 109/l) |
|
| ≤ 1,0 |
≤ 50,0 |
50 % |
| > 1,0 |
> 50,0 |
100 % |
*Odzyskanie = ilość ≥ najniższa ilość + (0,5 x [początkowa ilość – najniższa ilość])
Pacjenci z obniżoną początkową liczbą komórek krwi (tj. leukocyty < 3,0 x 10⁹/l lub ANC < 1,5 x 10⁹/l lub trombocyty < 75,0 x 10⁹/l) przed pierwszym leczeniem
Jeśli po leczeniu Azytsytydynem Accord spadek liczby leukocytów, ANC lub trombocytów w porównaniu z okresem przed leczeniem wynosi ≤ 50 % lub przekracza 50 %, ale występuje poprawa różnicowania dowolnej linii komórkowej, kolejny cykl nie powinien być odłożony i nie ma potrzeby dostosowywania dawki.
Jeśli spadek liczby leukocytów, ANC lub trombocytów przekracza 50 % w porównaniu z wartościami przed leczeniem i nie ma poprawy w różnicowaniu linii komórkowej, następny cykl leczenia Azytsytydynem Accord powinien być odłożony, aż do odzyskania liczby trombocytów i ANC. Jeśli odzyskanie zostanie osiągnięte w ciągu 14 dni, nie jest wymagane dostosowanie dawki. Jeśli jednak odzyskanie nie zostanie osiągnięte w ciągu 14 dni, należy określić nasycenie komórkowe szpiku kostnego. Jeśli nasycenie komórkowe szpiku kostnego wynosi > 50 %, nie jest wymagane dostosowanie dawki. Jeśli nasycenie komórkowe szpiku kostnego wynosi ≤ 50 %, leczenie powinno być odłożone i dawkę należy zmniejszyć zgodnie z poniższą tabelą:
| Zasilenie komórkami szpiku kostnego |
Dawka w następnym cyklu, jeśli odnowa nie została osiągnięta w ciągu 14 dni (%) |
|
| Odnowa* ≤ 21 dni |
Odnowa* > 21 dni |
|
| 15–50 % |
100 % |
50 % |
| < 15 % |
100 % |
33 % |
*Odzyskanie = ilość ≥ najniższa ilość + (0,5 x [początkowa ilość – najniższa ilość]).
Po modyfikacji dawki długość następnego cyklu powinna powrócić do 28 dni.
Osobne grupy pacjentów
Pacjenci w podeszłym wieku
Nie zaleca się specjalnej korekty dawki dla osób w podeszłym wieku. Ponieważ u pacjentów w podeszłym wieku częściej występuje obniżona funkcja nerek, może być wskazane monitorowanie czynności nerek.
Pacjenci z zaburzeniami czynności nerek
Azytsytydyn Accord można podawać pacjentom z zaburzeniami czynności nerek bez korekty dawki początkowej (patrz sekcja „Właściwości farmakologiczne”). W przypadku niejasnego spadku stężenia bikarbonianu w surowicy poniżej 20 mmol/l dawkę należy zmniejszyć o 50% w następnym cyklu. W przypadku niejasnego wzrostu stężenia kreatyniny w surowicy lub azotu mocznika we krwi (BUN) ≥ dwukrotnego wzrostu wartości wyjściowych i powyżej górnej granicy normy (GGN), następny cykl należy odłożyć, aż wartości powrócą do normy lub wartości wyjściowej, a następnie dawkę należy zmniejszyć o 50% w następnym cyklu leczenia (patrz sekcja „Szczególne ostrzeżenia i środki ostrożności”).
Pacjenci z zaburzeniami czynności wątroby
Nie przeprowadzono formalnych badań z udziałem pacjentów z zaburzeniami czynności wątroby (patrz sekcja „Szczególne ostrzeżenia i środki ostrożności”). Pacjentów z ciężką niewydolnością wątroby należy dokładnie monitorować pod kątem działań niepożądanych. U pacjentów z zaburzeniami czynności wątroby nie zaleca się zmiany dawki początkowej; dalsze zmiany dawki powinny opierać się na badaniach hematologicznych. Azytsytydyn Accord jest przeciwwskazany u pacjentów z zaawansowanymi nowotworami wątroby (patrz sekcje „Przeciwwskazania” i „Szczególne ostrzeżenia i środki ostrożności”).
Dzieci
Nie ustalono bezpieczeństwa i skuteczności azytsytydynu u dzieci w wieku od 0 do 17 lat. Dostępne dane są opisane w sekcjach „Właściwości farmakologiczne” i „Działania niepożądane”, jednak nie można podać zaleceń dotyczących dawkowania.
Sposób podania
Po rozcieńczeniu każdy ml zawiesiny zawiera 25 mg azytsytydynu. Rozcieńczony Azytsytydyn Accord należy podawać podskórnie w okolice barku, uda lub brzucha. Miejsca wstrzyknięć należy zmieniać. Kolejne wstrzyknięcia należy wykonywać w odległości co najmniej 2,5 cm od poprzedniego miejsca i nigdy w obszary bolesne, posiniaczone, zaczerwienione lub z twardymi zmianami.
Nie należy filtrować zawiesiny po rozcieńczeniu. Instrukcje dotyczące przygotowania roztworu przed podaniem znajdują się w sekcji „Szczególne środki ostrożności”.
Dzieci.
Nie należy stosować u dzieci (do 18. roku życia), ponieważ bezpieczeństwo i skuteczność leku u tej grupy pacjentów nie zostały ustalone.
Przedawkowanie.
Podczas badań klinicznych zgłoszono jeden przypadek przedawkowania azytsytydynu. U pacjenta wystąpiła biegunka, nudności i wymioty po jednorazowym dożylnej podaniu dawki leku około 290 mg/m², co stanowiło niemal czterokrotność zalecanej dawki początkowej.
W przypadku przedawkowania należy monitorować stan pacjenta za pomocą badań krwi i, w razie potrzeby, podjąć leczenie wspierające. Nie istnieje znany specyficzny antydotum na przedawkowanie azytsytydynu.
Efekty uboczne
Podsumowanie profilu bezpieczeństwa
Obydwie populacje dorosłych z zespołem mielodysplastycznym (MDS), przewlekłym mielomonocyticznym białaczką (CMML) oraz ostrym białaczką mieloidalną (AML) (20–30 % komórek blastycznych szpiku kostnego)
Efekty uboczne, które mogą być związane z leczeniem azytsytydyną, zaobserwowano u 97 % pacjentów.
Najczęstsze poważne efekty uboczne zgłaszane w badaniu podstawowym (AZA PH GL 2003 CL 001) obejmowały febrilną neutropenię (8,0 %) oraz anemię (2,3 %), o których donoszono również w badaniach pomocniczych (CALGB 9221 i CALGB 8921). Inne poważne niepożądane reakcje w tych trzech badaniach obejmowały infekcje, takie jak sepsa neutropenijna (0,8 %) i zapalenie płuc (2,5 %) (niektóre z letalnym skutkiem), trombocytopenię (3,5 %), reakcje nadwrażliwości (0,25 %) oraz krwawienia (np. krwotok do mózgu [0,5 %], krwawienia przewodu pokarmowego [0,8 %] i krwotoki śródczaszkowe [0,5 %]).
Najczęstszymi efektami ubocznymi podczas leczenia azytsytydyną były reakcje hematologiczne (71,4 %), w tym trombocytopenia, neutropenia i leukopenia (zazwyczaj stopnia 3–4), zaburzenia przewodu pokarmowego (60,6 %), w tym nudności, wymioty (zazwyczaj stopnia 1–2) oraz reakcje w miejscu wstrzyknięcia (77,1 %; zazwyczaj stopnia 1–2).
Populacja dorosłych w wieku ≥ 65 lat z AML z > 30 % komórek blastycznych szpiku kostnego
Najczęstsze poważne efekty uboczne (≥ 10 %) obserwowane w badaniu AZA-AML-001 w grupie leczonej azytsytydyną to febrilna neutropenia (25,0 %), zapalenie płuc (20,3 %) oraz gorączka (10,6 %). Inne, mniej częste poważne efekty uboczne w grupie leczonej azytsytydyną obejmowały sepsę (5,1 %), anemię (4,2 %), sepsę neutropenijną (3,0 %), infekcję dróg moczowych (3,0 %), trombocytopenię (2,5 %), neutropenię (2,1 %), cellulitis (2,1 %), zawroty głowy (2,1 %) oraz duszność (2,1 %).
Najczęściej występujące efekty uboczne (≥ 30 %) podczas leczenia azytsytydyną to zaburzenia przewodu pokarmowego, w tym zaparcia (41,9 %), nudności (39,8 %) oraz biegunka (36,9 %; zazwyczaj stopnia 1–2); ogólne zaburzenia oraz reakcje w miejscu wstrzyknięcia, w tym gorączka (37,7 %; zazwyczaj stopnia 1–2) oraz zjawiska hematologiczne, w tym febrilna neutropenia (32,2 %) i neutropenia (30,1 %; zazwyczaj stopnia 3–4).
Tabelaryczne zestawienie efektów ubocznych
Poniższa tabela 1 zawiera efekty uboczne związane z leczeniem azytsytydyną, zgłoszone podczas głównych badań klinicznych u pacjentów z MDS i AML oraz w okresie po rejestracji.
Częstotliwość efektów ubocznych określono następująco: bardzo częste (≥ 1/10), częste (≥ 1/100 do < 1/10); rzadkie (≥ 1/1000 do < 1/100); pojedyncze (≥ 1/10 000 do < 1/1000); rzadkie ( < 1/10 000); częstotliwość nieznana (nie można oszacować na podstawie dostępnych danych). W każdej grupie częstotliwości niepożądane efekty są wymienione w kolejności zmniejszającej się częstości występowania. Efekty uboczne przedstawione w poniższej tabeli odpowiadają najwyższej zaobserwowanej częstości w dowolnym z głównych badań klinicznych.
Efekty uboczne zgłaszane u pacjentów z MDS lub AML leczonych azytsytydyną
(badania kliniczne oraz okres po rejestracji)
Tabela 1
| Klasa układów narządów |
Bardzo często |
Często |
Nieczęsto |
Pojedyncze |
Częstotliwość nieznana |
| Infekcje i inwazje |
zapalenie płuc* (w tym bakteryjne, wirusowe i grzybicze), zapalenie nosowo-gardła |
sepsa* (w tym bakteryjna, wirusowa i grzybicza), sepsa neutropenijna*, infekcja dróg oddechowych (w tym infekcje górnych dróg oddechowych i zapalenie oskrzeli), infekcja dróg moczowych, cellulitis, zapalenie zatok, zapalenie gardła, katar, opryszcz zwykły, infekcja skóry |
fascjoliza nekrotyczna* |
||
| Łagodne, złośliwe i niejednoznaczne nowotwory (w tym torbiele i polipy) |
zespół różnicowania*, a |
||||
| Ze strony krwi i układu limfatycznego |
gorączka neutropenijna*, neutropenia, leukopenia, trombocytopenia, anemia |
pancytopenia*, niedostateczność szpiku kostnego |
|||
| Ze strony układu odpornościowego |
reakcje nadwrażliwości |
||||
| Ze strony przemiany materii i odżywiania |
anoreksja, zmniejszony apetyt, hipokaliemia |
odwodnienie |
zespół lizy guza |
||
| Zaburzenia psychiczne |
bezsenność |
dezorientacja, niepokój |
|||
| Ze strony układu nerwowego |
zawroty głowy, ból głowy |
krwawienie śródczaszkowe*, omdlenie, senność, osłabienie |
|||
| Ze strony narządu wzroku |
krwawienie do oka, krwawienie spojówek |
||||
| Zaburzenia serca |
wysięk do worka osierdziowego |
zapalenie osierdzia |
|||
| Zaburzenia naczyniowe |
hipotensja*, nadciśnienie, hipotensja ortostatyczna, siniak |
||||
| Zaburzenia oddechowe, choroby jamy piersiowej i śródpiersia |
udrudnione oddychanie, krwawienie z nosa |
wysięk do jamy opłucnowej, duszność przy wysiłku, ból gardła i krtani |
choroba płuc międzywątrobowa |
||
| Ze strony przewodu pokarmowego |
biegunka, wymioty, zaparcia, nudności, ból brzucha (w tym dyskomfort w górnej części brzucha i brzuchu) |
krwawienie przewodu pokarmowego* (w tym krwawienie z jamy ustnej), krwawienia z hemoroidów, zapalenie błony śluzowej jamy ustnej, krwawienie dziąseł, wzdęcia |
|||
| Ze strony wątroby i dróg żółciowych |
niewydolność wątroby*, postępująca śpiączka wątrobową |
||||
| Ze strony skóry i tkanki podskórnej |
plamki posocznicze, świąd (w tym uogólniony), wysyp, siniaki |
purpura, łysienie, pokrzywka, zaczerwienienie, wysyp makularny |
ostra gorączkowa neutrofilowa dermatopatia, gangrenozna piodermia |
zapalenie naczyń skóry |
|
| Ze strony układu mięśniowo-szkieletowego i tkanki łącznej |
artrodynia, ból układu ruchowego (w tym ból pleców, kości i kończyn) |
skurcze mięśni, miodynia |
|||
| Zaburzenia ze strony nerek i dróg moczowych |
niewydolność nerek*, krwiomocz, podwyższenie stężenia kreatyniny w surowicy |
kwasica kanalikowa nerek |
|||
| Zaburzenia ogólne i w miejscu podania |
gorączka*, zmęczenie, osłabienie, ból w klatce piersiowej, zaczerwienienie w miejscu wstrzyknięcia, ból w miejscu wstrzyknięcia, reakcja w miejscu wstrzyknięcia (nieokreślona) |
siniaki, siniaki, zgrubienia, wysyp, świąd, zapalenie, zmiana koloru, guzki i krwawienia (w miejscu wstrzyknięcia), niedomaganie, dreszcze, krwawienie w miejscu wprowadzenia cewnika |
nekroza miejsca podania (w miejscu podania) |
||
| Badania laboratoryjne |
spadek masy ciała |
* – w rzadkich przypadkach zgłaszano skutki śmiertelne.
a – patrz sekcja „Szczególne ostrzeżenia i środki ostrożności dotyczące stosowania”.
Opis wybranych niepożądanych reakcji
Zaburzenia hematologiczne
Zaburzenia hematologiczne, o których najczęściej zgłaszano (≥ 10 %), związane z leczeniem azytsytydyną, obejmowały anemię, trombocytopenię, neutropenię, febrilną neutropenię i leukopenię, które zazwyczaj miały charakter ciężki (stopień 3 lub 4). Ryzyko wystąpienia tych zjawisk jest większe w trakcie pierwszych dwóch cykli leczenia, po czym stają się one rzadsze u pacjentów z odzyskaną funkcją hematologiczną. Większość hematologicznych niepożądanych reakcji kontrolowano poprzez regularne monitorowanie rozwiniętego morfologii krwi, opóźnienie podania azytsytydyny w kolejnym cyklu, profilaktyczne stosowanie antybiotyków i/lub wspomaganie czynnikami wzrostu (np. G-CSF – granulocytarny czynnik stymulujący wzrost kolonii) w celu leczenia neutropenii oraz przetaczanie krwi w celu kontrolowania anemii lub trombocytopenii, jeśli było to konieczne.
Infekcje
Mielosupresja może prowadzić do neutropenii i zwiększonego ryzyka infekcji. Obserwowano poważne niepożądane reakcje, takie jak sepsa, w tym sepsa neutropeniczną, oraz zapalenie płuc u pacjentów leczonych azytsytydyną, niektóre z końcem śmiertelnym. Infekcje można leczyć za pomocą leków przeciwinfekcyjnych oraz wspomagania czynnikami wzrostu (np. G-CSF) w celu leczenia neutropenii.
Krwawienia
U pacjentów leczonych azytsytydyną może wystąpić krwawienie. Zgłaszano poważne niepożądane reakcje, takie jak krwawienia przewodu pokarmowego i krwawienia wewnątrzczaszkowe. Pacjenci powinni być monitorowani pod kątem objawów i oznak krwawienia, szczególnie pacjenci z istniejącą lub leczeniem wywołaną trombocytopenią.
Podwyższona wrażliwość
Zgłaszano poważne reakcje nadwrażliwości u pacjentów leczonych azytsytydyną. W przypadku reakcji anafilaktycznej leczenie azytsytydyną należy natychmiast przerwać i rozpocząć odpowiednie leczenie objawowe.
Zaburzenia skórne i podskórne
Większość reakcji skórnych i podskórnych była związana z miejscem wstrzyknięcia. Żadna z tych niepożądanych reakcji nie doprowadziła do przerwania stosowania azytsytydyny ani do zmniejszenia dawki azytsytydyny w badaniach podstawowych. Większość niepożądanych reakcji pojawiała się w trakcie pierwszych dwóch cykli leczenia i miała tendencję do osłabienia w kolejnych cyklach. Reakcje podskórne, takie jak wysypka/reakcja zapalna/świerzbienie w miejscu wstrzyknięcia, wysypka, zaczerwienienie i zmiany skórne, mogą wymagać leczenia lekami wspomagającymi, takimi jak leki przeciwhistaminowe, kortykosteroidy i niesteroidowe leki przeciwzapalne (NLPZ). Należy odróżniać te reakcje skórne od infekcji tkanek miękkich, które czasem pojawiają się w miejscu wstrzyknięcia. Zgłaszano infekcje tkanek miękkich, w tym cellulitis i faszcytę nekrotyczną, które w rzadkich przypadkach prowadziły do skutków śmiertelnych podczas stosowania azytsytydyny w okresie pozarejestracyjnym. Strategia postępowania w przypadku infekcyjnych niepożądanych reakcji opisana jest w sekcji „Niepożądane reakcje” – „Infekcje”.
Zaburzenia przewodu pokarmowego
Najczęstsze niepożądane reakcje ze strony przewodu pokarmowego związane z leczeniem azytsytydyną obejmowały zaparcia, biegunkę, nudności i wymioty. Te niepożądane reakcje leczono objawowo za pomocą leków przeciwwymiotnych w celu kontrolowania nudności i wymiotów; leków przeciwbiegunkowych w przypadku biegunki oraz środków przeciwbólowych i/lub środków łagodzących stolec w przypadku zaparć.
Zaburzenia nerek
U pacjentów leczonych azytsytydyną zgłaszano zaburzenia funkcji nerek, począwszy od podwyższenia stężenia kreatyniny w surowicy i hematurii, po kwasicę kanalikową, niewydolność nerek i skutki śmiertelne (patrz sekcja „Szczególne ostrzeżenia i środki ostrożności dotyczące stosowania”).
Zaburzenia wątroby
U pacjentów z dużym obciążeniem nowotworowym z powodu choroby przerzutowej zgłaszano niewydolność wątroby, postępującą śpiączkę wątrobową i skutki śmiertelne podczas leczenia azytsytydyną (patrz sekcja „Szczególne ostrzeżenia i środki ostrożności dotyczące stosowania”).
Zaburzenia serca
Dane z badania klinicznego obejmującego pacjentów z wywiadem chorób układu sercowo-naczyniowego lub płucnych wykazały zwiększenie liczby zaburzeń serca u pacjentów z nowo zdiagnozowaną AML leczonych azytsytydyną (patrz sekcja „Szczególne ostrzeżenia i środki ostrożności dotyczące stosowania”).
Pacjenci starsi
Dostępne są ograniczone informacje dotyczące bezpieczeństwa azytsytydyny u pacjentów w wieku ≥ 85 lat (w badaniu AZA-AML-001 udział wzięło 14 [5,9 %] pacjentów w wieku ≥ 85 lat).
Dzieci
W badaniu AZA-JMML-001 28 dzieci (w wieku od 1 miesiąca do < 18 lat) otrzymywało leczenie azytsytydyną z powodu MDS (n = 10) lub młodzieńczego mielomonocytarnego leukemia (JMML) (n = 18) (patrz sekcja „Właściwości farmakologiczne”).
U wszystkich 28 pacjentów zaobserwowano co najmniej jedno niepożądane zjawisko, a u 17 (60,7 %) – co najmniej jedno zjawisko związane z leczeniem. Najczęstsze niepożądane reakcje w ogólnej populacji dziecięcej to hipertermia, zaburzenia hematologiczne, w tym anemia, trombocytopenia i febrilna neutropenia, oraz zaburzenia ze strony przewodu pokarmowego, w tym zaparcia i wymioty.
U trzech uczestników zaobserwowano zjawiska związane z leczeniem, które doprowadziły do przerwania przyjmowania leku (hipertermia, postęp choroby i ból brzucha).
W badaniu AZA-AML-004 7 dzieci (w wieku od 2 do 12 lat) otrzymywało leczenie azytsytydyną z powodu AML w trakcie molekularnego nawrotu po pierwszej remisji całkowitej [CR1] (patrz sekcja „Właściwości farmakologiczne”).
U wszystkich siedmiu pacjentów odnotowano co najmniej jedną niepożądaną reakcję związaną z leczeniem. Najczęściej zgłaszanymi niepożądanymi reakcjami były neutropenia, nudności, leukopenia, trombocytopenia, biegunka i podwyższenie poziomu alaninotransferazy (ALT). U dwóch pacjentów wystąpiła niepożądana reakcja związana z leczeniem, która doprowadziła do przerwania przyjmowania leku (febrilna neutropenia, neutropenia).
Podczas badania klinicznego u ograniczonej liczby dzieci leczonych azytsytydyną nie wykryto nowych sygnałów ryzyka. Ogólny profil bezpieczeństwa odpowiadał profilowi populacji dorosłej.
Zgłaszanie podejrzewanych niepożądanych reakcji
Zgłaszanie niepożądanych reakcji po rejestracji leku ma istotne znaczenie. Umożliwia to monitorowanie stosunku korzyści do ryzyka związanego z zastosowaniem tego leku. Osoby pracujące w zawodach medycznych i farmaceutycznych, a także pacjenci lub ich opiekunowie prawni powinni zgłaszać wszystkie przypadki podejrzewanych niepożądanych reakcji oraz brak skuteczności leku poprzez Automatyczny System Informacyjny Nadzoru Farmakologicznego pod adresem: https://aisf.dec.gov.ua.
Okres ważności.
3 lata.
Po rozcieńczeniu
W przypadku rozcieńczenia leku Azytsytydyna Accord wodą do wstrzykiwań niechłodzoną, chemiczna i fizyczna stabilność rozcieńczonego leku podczas stosowania została potwierdzona w temperaturze 25 °C przez 60 minut oraz w temperaturze od 2 °C do 8 °C przez 8 godzin.
Okres ważności rozcieńczonego leku może być wydłużony przez jego rozcieńczenie wodą do wstrzykiwań schłodzoną (od 2 °C do 8 °C). W przypadku rozcieńczenia leku Azytsytydyna Accord wodą do wstrzykiwań schłodzoną (od 2 °C do 8 °C) chemiczna i fizyczna stabilność rozcieńczonego leku podczas stosowania została potwierdzona w temperaturze od 2 °C do 8 °C przez 22 godziny.
Z mikrobiologicznego punktu widzenia rozcieńczony produkt należy użyć natychmiast. Jeżeli nie zostanie użyty natychmiast, użytkownik ponosi odpowiedzialność za czas i warunki przechowywania w okresie jego stosowania, który nie powinien przekraczać 8 godzin w temperaturze od 2 °C do 8 °C w przypadku rozcieńczenia wodą do wstrzykiwań niechłodzoną, oraz 22 godzin w przypadku rozcieńczenia wodą do wstrzykiwań schłodzoną (od 2 °C do 8 °C).
Warunki przechowywania.
Lek nie wymaga specjalnych warunków przechowywania.
Przechowywać w miejscu niedostępnym dla dzieci.
Niezgodność.
Nie mieszać z innymi lekami, z wyjątkiem tych, które są wymienione w sekcji „Sposób podania i dawki”.
Opakowanie. 150 mg w fiolce, 1 fiolka w opakowaniu.
Kategoria wydania. Na receptę.
Producent.
Accord Healthcare Polska Sp. z o.o. Magazyn Importera / Accord Healthcare Polska Sp. z o.o. Magazyn Importera.
Adres producenta i miejsce prowadzenia działalności.
ul. Lutomierska 50, Pabianice, 95-200, Polska / ul. Lutomierska 50, Pabianice, 95-200, Poland.
Wnioskodawca. Accord Healthcare S.L.U. / Accord Healthcare S.L.U.
Zgłoszenia dotyczące niezadowalającej jakości leku; pytań dotyczących bezpieczeństwa stosowania i nieprawidłowego stosowania leku lub reklamacji są przyjmowane całodobowo (24/7) pod numerem telefonu: +380993100335 lub drogą e-mailową na adres: [email protected].
Adres wnioskodawcy. World Trade Center, Moll de Barcelona, s/n, Edifici Est 6a planta, 08039 Barcelona, Hiszpania / World Trade Center, Moll de Barcelona, s/n, Edifici Est 6a planta, 08039 Barcelona, Spain.