Azacitidina Accord
UcrainaIndice
ISTRUZIONI PER L'USO MEDICINALE DEL MEDICINALE Azacitidina Accord (Azacitidine Accord)
Composizione:
Principio attivo: azacitidina;
1 flaconcino contiene azacitidina 150 mg;
Eccipiente: mannite (E 421).
Forma farmaceutica. Polvere per sospensione iniettabile.
Principali caratteristiche fisico-chimiche: polvere liofilizzata bianca o massa all'interno di un flaconcino di vetro trasparente.
Gruppo farmacoterapeutico. Agenti antineoplastici. Analoghi delle pirimidine.
Codice ATC L01B C07.
Proprietà farmacologiche.
Farmacodinamica.
Meccanismo d'azione
Si ritiene che l'azacitidina eserciti il suo effetto antineoplastico attraverso diversi meccanismi, inclusi l'effetto citotossico sulle cellule ematopoietiche anomale nel midollo osseo e l'ipometilazione del DNA. L'effetto citotossico dell'azacitidina può derivare da diversi meccanismi, tra cui l'inibizione della sintesi di DNA, RNA e proteine, l'incorporazione nell'RNA e nel DNA e l'attivazione delle vie di danno al DNA. Le cellule non proliferanti sono relativamente insensibili all'azacitidina. L'incorporazione dell'azacitidina nel DNA porta all'inattivazione delle DNA-metiltransferasi, causando ipometilazione del DNA. L'ipometilazione del DNA di geni anormalmente metilati coinvolti nella regolazione del normale ciclo cellulare, nelle vie di differenziazione e morte cellulare può portare alla ri-espressione genica e al ripristino delle funzioni di soppressione tumorale nelle cellule tumorali. L'importanza relativa dell'ipometilazione del DNA rispetto alla citotossicità o ad altre attività dell'azacitidina nel raggiungimento dei risultati clinici non è stata stabilita.
Efficacia e sicurezza clinica
Popolazione adulta (SMDS, LMMC e LMA [20-30% blasti midollari])
L'efficacia e la sicurezza dell'azacitidina sono state valutate in uno studio internazionale, multicentrico, controllato, aperto, randomizzato, di fase 3, a gruppi paralleli (AZA PH GL 2003 CL 001) in adulti con: SMDS di rischio intermedio-2 e alto secondo il Sistema Internazionale di Valutazione Prognostica (IPSS), anemia refrattaria con eccesso di blasti (RAEB), anemia refrattaria con eccesso di blasti in trasformazione (RAEB-T) e leucemia mielomonocitica cronica modificata (mLMMC) secondo la classificazione FAB. I pazienti con RAEB-T (21-30% blasti) sono ora considerati pazienti con LMA secondo l'attuale classificazione OMS. Lo schema azacitidina più migliore terapia di supporto (BSC) (n = 179) è stato confrontato con schemi di trattamento convenzionali (CCR). Gli schemi CCR prevedevano l'uso di sola BSC (n = 105), citarabina a bassa dose più BSC (n = 49) o chemioterapia induttiva standard più BSC (n = 25). I pazienti sono stati precedentemente assegnati dal loro medico a uno dei tre schemi CCR prima della randomizzazione. I pazienti hanno ricevuto questo schema precedentemente assegnato se non sono stati randomizzati al gruppo azacitidina. Secondo i criteri di inclusione, i pazienti dovevano avere uno stato funzionale 0-2 secondo la classificazione del gruppo cooperativo oncologico orientale (ECOG). I pazienti con SMDS secondario sono stati esclusi dallo studio. Il punto finale primario dello studio era la sopravvivenza globale. L'azacitidina è stata somministrata sottocute alla dose di 75 mg/m² ogni giorno per sette giorni, seguita da un intervallo di 21 giorni (ciclo di trattamento di 28 giorni); la mediana del numero di cicli è stata di 9 (intervallo = 1-39), e la media del numero di cicli è stata di 10,2. Nella popolazione per protocollo (ITT), l'età media era di 69 anni (intervallo da 38 a 88 anni).
Nell'analisi della popolazione ITT con 358 pazienti (179 nel gruppo azacitidina e 179 nel gruppo CCR), il trattamento con azacitidina è stato associato a una mediana di sopravvivenza di 24,46 mesi rispetto a 15,02 mesi per quelli trattati con CCR; la differenza è stata di 9,4 mesi, con un valore p log-rank stratificato = 0,0001. Il rapporto di rischio (HR) per l'effetto del trattamento è stato di 0,58 (IC 95%: 0,43; 0,77). I tassi di sopravvivenza a due anni sono stati del 50,8% nei pazienti trattati con azacitidina rispetto al 26,2% nei pazienti trattati con CCR (p < 0,0001).
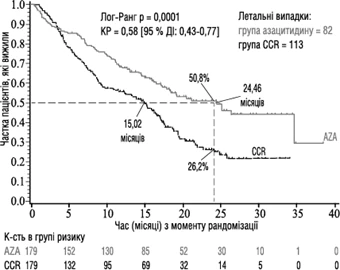
Abbreviazioni: AZA = azacitidina; CCR = schemi di trattamento convenzionali; IC = intervallo di confidenza;
HR = rapporto di rischio.
Un aumento della sopravvivenza nel gruppo azacitidina è stato osservato costantemente indipendentemente dallo schema CCR utilizzato nel gruppo di controllo (sola BSC, citarabina a bassa dose più BSC o chemioterapia induttiva standard più BSC).
Durante l'analisi delle sottogruppi citogenetici IPSS, risultati simili riguardo alla mediana della sopravvivenza globale sono stati osservati in tutti i gruppi (citogenetica buona, intermedia, scarsa, inclusa la monosomia 7).
Durante l'analisi dei sottogruppi per età, è stato osservato un aumento della sopravvivenza globale media in tutti i gruppi (< 65 anni, ≥ 65 anni e ≥ 75 anni).
Il trattamento con azacitidina è stato associato a un tempo mediano all'evento avverso fatale o trasformazione in LMA di 13,0 mesi rispetto a 7,6 mesi per quelli trattati con CCR, corrispondente a un miglioramento di 5,4 mesi con un valore p log-rank stratificato = 0,0025.
Il trattamento con azacitidina è stato anche associato a una riduzione della citopenia e dei sintomi ad essa correlati. Il trattamento con azacitidina ha portato a una riduzione della necessità di trasfusioni di eritrociti (globuli rossi) e piastrine. Tra i pazienti nel gruppo azacitidina dipendenti da trasfusioni di eritrociti all'inizio, il 45,0% è diventato indipendente da trasfusioni di eritrociti durante il periodo di trattamento rispetto all'11,4% dei pazienti nei gruppi combinati CCR (differenza statisticamente significativa (p < 0,0001) del 33,6% (IC 95%: 22,4; 44,6). Nei pazienti dipendenti da trasfusioni di eritrociti all'inizio e divenuti indipendenti, la durata media del periodo di indipendenza da trasfusioni di eritrociti è stata di 13 mesi nel gruppo azacitidina.
La risposta al trattamento è stata valutata dallo sperimentatore o da un Comitato Indipendente di Revisione (IRC). La risposta globale (remissione completa [CR] + remissione parziale [PR]), determinata dallo sperimentatore, è stata del 29% nel gruppo azacitidina e del 12% nel gruppo combinato CCR (p = 0,0001). La risposta globale (CR + PR), determinata dal comitato IRC nello studio AZA PH GL 2003 CL 001, è stata del 7% (12/179) nel gruppo azacitidina rispetto all'1% (2/179) nel gruppo combinato CCR (p=0,0113). Le differenze tra le valutazioni di risposta del comitato IRC e degli sperimentatori sono state conseguenza dei criteri del Gruppo di Lavoro Internazionale (IWG), che richiedevano un miglioramento dei parametri ematici periferici e il mantenimento di tali miglioramenti per almeno 56 giorni. Il vantaggio in termini di sopravvivenza è stato anche dimostrato nei pazienti che non hanno raggiunto una risposta completa/parziale dopo il trattamento con azacitidina. Il miglioramento ematologico (maggiore o minore), come definito dall'IRC, è stato raggiunto nel 49% dei pazienti trattati con azacitidina rispetto al 29% dei pazienti trattati con CCR combinato (p < 0,0001).
Nei pazienti con una o più anomalie citogenetiche all'inizio, la percentuale di pazienti con risposta citogenetica significativa è stata simile nei gruppi azacitidina e CCR combinato.
La risposta citogenetica minore è stata statisticamente significativa (p=0,0015) più alta nel gruppo azacitidina (34%) rispetto al gruppo CCR combinato (10%).
Popolazione adulta di età ≥ 65 anni con LMA con > 30% blasti midollari
I risultati riportati di seguito si riferiscono alla popolazione per protocollo studiata nello studio AZA-AML-001 (indicazioni approvate riportate nella sezione «Modalità di somministrazione e posologia»).
L'efficacia e la sicurezza dell'azacitidina sono state valutate in uno studio internazionale, multicentrico, controllato, aperto, di fase 3, a gruppi paralleli, in pazienti di età ≥ 65 anni con LMA de novo o secondaria con > 30% blasti midollari secondo la classificazione OMS, che non erano candidati al trapianto di cellule staminali ematopoietiche (HSCT). Azacitidina più BSC (n = 241) è stata confrontata con uno schema CCR. Lo schema CCR includeva solo BSC (n = 45), citarabina a bassa dose più BSC (n = 158) o chemioterapia intensiva standard con citarabina e antracicline più BSC (n = 44). I pazienti sono stati precedentemente assegnati dal loro medico a uno dei tre schemi di trattamento CCR prima della randomizzazione. I pazienti hanno ricevuto lo schema precedentemente scelto se non sono stati randomizzati al gruppo azacitidina. Come parte dei criteri di inclusione, i pazienti dovevano avere uno stato funzionale 0-2 secondo la scala ECOG e anomalie citogenetiche di rischio intermedio o basso. Il punto finale primario dello studio era la sopravvivenza globale.
L'azacitidina è stata somministrata sottocute alla dose di 75 mg/m²/giorno per 7 giorni, seguita da un intervallo di 21 giorni (ciclo di trattamento di 28 giorni), con una mediana di 6 cicli di trattamento (intervallo: da 1 a 28): pazienti che hanno ricevuto solo BSC, con mediana di 3 cicli (intervallo: da 1 a 20); pazienti che hanno ricevuto citarabina a bassa dose, con mediana di 4 cicli (intervallo da 1 a 25); e pazienti che hanno ricevuto chemioterapia intensiva standard, con mediana di 2 cicli (intervallo: da 1 a 3, ciclo di induzione più 1 o 2 cicli di consolidamento).
I parametri basali individuali erano comparabili tra i gruppi azacitidina e CCR. L'età mediana dei partecipanti era di 75,0 anni (intervallo: da 64 a 91 anni), il 75,2% erano di razza caucasica e il 59,0% erano uomini. All'inizio, il 60,7% è stato classificato come pazienti con LMA non specificata, il 32,4% come pazienti con LMA con alterazioni correlate alla displasia mieloide, il 4,1% aveva neoplasie mieloidi correlate alla terapia e il 2,9% aveva LMA con anomalie genetiche ricorrenti secondo la classificazione OMS.
Nell'analisi della popolazione ITT con 488 pazienti (241 nel gruppo azacitidina e 247 nel gruppo CCR), il trattamento con azacitidina è stato associato a una mediana di sopravvivenza di 10,4 mesi rispetto a 6,5 mesi per quelli trattati con CCR – la differenza è stata di 3,8 mesi, con un valore p log-rank stratificato = 0,1009 (bicaudale). Il rapporto di rischio per l'effetto del trattamento è stato di 0,85 (IC 95% = 0,69; 1,03). La sopravvivenza a un anno è stata del 46,5% nei pazienti trattati con azacitidina rispetto al 34,3% nei pazienti trattati con lo schema CCR.
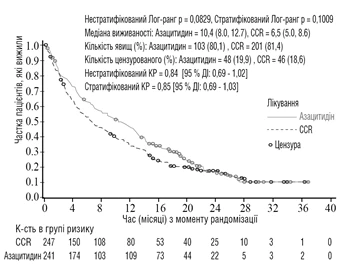
Abbreviazioni: CCR = schemi di trattamento convenzionali; IC = intervallo di confidenza; HR = rapporto di rischio.
Secondo il modello di Cox per i rischi proporzionali, aggiustato per i fattori prognostici basali predefiniti, è stato determinato un HR per azacitidina rispetto allo schema CCR di 0,80 (IC 95% = 0,66; 0,99; p = 0,0355).
Inoltre, sebbene nello studio non fosse possibile dimostrare una differenza statisticamente significativa confrontando azacitidina con i gruppi CCR precedentemente assegnati, la sopravvivenza dei pazienti trattati con azacitidina è stata più lunga rispetto a tali varianti di trattamento CCR, come sola BSC, bassa dose di citarabina più BSC, ed è stata simile a quella della chemioterapia intensiva standard più BSC.
In tutti i sottogruppi predefiniti per età [(< 75 anni e ≥ 75 anni), sesso, razza, stato funzionale ECOG (0, o 1 e 2), rischio citogenetico iniziale (medio e basso), regione geografica, classificazione OMS della LMA (inclusa LMA con alterazioni correlate alla displasia mieloide), conteggio iniziale di leucociti (≤ 5 x 10⁹/l e > 5 x 10⁹/l), percentuale iniziale di blasti midollari (≤ 50% e > 50%) e storia pregressa di malattia SMDS], è stata osservata una tendenza al vantaggio in termini di sopravvivenza globale a favore di azacitidina. In diversi sottogruppi predefiniti, l'HR per la sopravvivenza globale ha raggiunto la significatività statistica, inclusi pazienti con rischio citogenetico basso, pazienti con LMA con alterazioni correlate alla displasia mieloide, pazienti di età < 75 anni, pazienti di sesso femminile e pazienti di razza caucasica.
Le risposte ematologiche e citogenetiche sono state valutate dallo sperimentatore e da un comitato esperto istituzionale (IRC) rispetto a risultati analoghi. La frequenza globale di risposta (remissione completa [CR] + remissione completa con recupero ematologico incompleto [CRi]), come determinata dall'IRC, è stata del 27,8% nel gruppo azacitidina e del 25,1% nel gruppo terapia combinata CCR (p = 0,5384). Nei pazienti che hanno raggiunto CR o CRi, la durata media della remissione è stata di 10,4 mesi (IC 95% = 7,2; 15,2) per i pazienti trattati con azacitidina e di 12,3 mesi (IC 95% = 9,0; 17,0) per i pazienti trattati con CCR. Il miglioramento della sopravvivenza è stato anche dimostrato nei pazienti che non hanno raggiunto una risposta completa con azacitidina rispetto allo schema CCR.
Il trattamento con azacitidina ha migliorato i parametri del sangue periferico e ha portato a una riduzione della necessità di trasfusioni di eritrociti e piastrine. Un paziente è stato considerato dipendente da trasfusioni di eritrociti o piastrine all'inizio se aveva ricevuto una o più trasfusioni di eritrociti o piastrine entro 56 giorni (8 settimane) dall'inizio o prima della randomizzazione. Un paziente è stato considerato indipendente da trasfusioni di eritrociti o piastrine durante il periodo di trattamento se non ha ricevuto trasfusioni di eritrociti o piastrine in qualsiasi periodo consecutivo di 56 giorni durante il periodo di osservazione.
Tra i pazienti nel gruppo azacitidina dipendenti da trasfusioni di eritrociti all'inizio, il 38,5% (IC 95% = 31,1; 46,2) di questi pazienti è diventato indipendente da trasfusioni di eritrociti durante il periodo di trattamento rispetto al 27,6% (IC 95% = 20,9; 35,1) dei pazienti nei gruppi combinati CCR. Nei pazienti dipendenti da trasfusioni di eritrociti all'inizio e divenuti indipendenti durante il trattamento, la durata media dell'indipendenza da trasfusioni di eritrociti è stata di 13,9 mesi nel gruppo azacitidina; nel gruppo CCR tale indipendenza non è stata raggiunta.
Tra i pazienti nel gruppo azacitidina dipendenti da trasfusioni di piastrine all'inizio, il 40,6% (IC 95% = 30,9; 50,8) dei pazienti è diventato indipendente da trasfusioni di piastrine durante il periodo di trattamento rispetto al 29,3% (IC 95% = 19,7; 40,4) dei pazienti nei gruppi combinati CCR. Nei pazienti dipendenti da trasfusioni di piastrine all'inizio e divenuti indipendenti dopo il trattamento, la durata media dell'indipendenza da trasfusioni di piastrine è stata di 10,8 mesi nel gruppo azacitidina e di 19,2 mesi nel gruppo CCR.
La qualità della vita legata alla salute (HRQoL) è stata valutata mediante il Questionario Fondamentale sulla Qualità della Vita dell'Organizzazione Europea per la Ricerca e il Trattamento del Cancro (EORTC QLQ-C30). I dati HRQoL possono essere analizzati per una sottopopolazione della popolazione studiata completa. Nonostante le limitazioni nell'analisi, i dati ottenuti indicano che i pazienti non riscontrano un significativo peggioramento della qualità della vita durante il trattamento con azacitidina.
Popolazione pediatrica
Lo studio AZA-JMML-001 è stato uno studio internazionale, multicentrico, aperto, di fase 2, per valutare la farmacocinetica, la farmacodinamica, la sicurezza e l'attività dell'azacitidina prima del trapianto di cellule staminali ematopoietiche (HSCT) in pazienti pediatrici con SMDS o LMMC giovanile recentemente diagnosticati e in evoluzione. L'obiettivo primario dello studio clinico era valutare l'effetto dell'azacitidina sulla risposta al trattamento al ciclo 3, giorno 28.
I pazienti (con SMDS: n = 10; con LMMC giovanile: n = 18, da 3 mesi a 15 anni; 71% maschi) hanno ricevuto trattamento endovenoso con azacitidina alla dose di 75 mg/m² ogni giorno dal giorno 1 al giorno 7 di un ciclo di 28 giorni per un minimo di tre cicli e un massimo di sei cicli.
L'inclusione di pazienti con SMDS nello studio è stata interrotta dopo l'arruolamento di 10 pazienti con SMDS a causa dell'insufficiente efficacia del trattamento: in questi 10 pazienti non è stata registrata alcuna risposta confermata.
Nel gruppo di studio LMMC giovanile sono stati inclusi 18 pazienti (13 con mutazioni somatiche di PTPN11, 3 con mutazioni di NRAS, 1 con mutazione di KRAS e 1 con diagnosi clinica di neurofibromatosi di tipo 1 [NF-1]). Sedici pazienti hanno completato tre cicli di terapia e 5 di loro hanno completato 6 cicli. Complessivamente, 11 pazienti con LMMC giovanile hanno mostrato una risposta clinica al ciclo 3, giorno 28; di questi 11 partecipanti allo studio, 9 (50%) hanno avuto una risposta clinica confermata (3 partecipanti con cCR e 6 partecipanti con cPR). Nella coorte di pazienti con LMMC giovanile trattati con azacitidina, 7 (43,8%) pazienti hanno avuto una risposta sostenuta a livello di piastrine (conteggio ≥ 100 x 10⁹/l), e 7 (43,8%) pazienti hanno richiesto trasfusioni di sangue prima dell'HSCT. 17 su 18 pazienti sono passati alla procedura di HSCT.
A causa della progettazione dello studio (piccolo numero di pazienti e diversi fattori che ostacolano l'analisi), non è possibile trarre conclusioni da questo studio clinico sul fatto che l'uso di azacitidina prima della procedura di HSCT migliori i risultati di sopravvivenza nei pazienti con LMMC giovanile.
Lo studio AZA-AML-004 è stato uno studio multicentrico, aperto, di fase 2, per valutare la sicurezza, la farmacodinamica e l'efficacia dell'azacitidina rispetto all'assenza di trattamento antineoplastico in bambini e giovani adulti con LMA in recidiva molecolare dopo CR1.
Sette pazienti (mediana di età 6,7 anni [intervallo da 2 a 12 anni]; 71,4% maschi) hanno ricevuto somministrazione endovenosa di azacitidina alla dose di 100 mg/m² ogni giorno dal giorno 1 al giorno 7 di ogni ciclo di 28 giorni per un massimo di 3 cicli.
Cinque pazienti secondo la valutazione avevano segni di malattia residua minima (MRD) al giorno 84, con 4 pazienti che hanno raggiunto o stabilità molecolare (n = 3) o miglioramento molecolare (n = 1), e un paziente con recidiva clinica. Sei su sette pazienti (90% [IC 95% = 0,4; 1,0]), che hanno ricevuto azacitidina, hanno superato l'HSCT.
A causa della piccola dimensione del campione, non è possibile stabilire l'efficacia dell'azacitidina in bambini con LMA. Le informazioni sulla sicurezza sono riportate nella sezione «Effetti indesiderati».
Farmacocinetica.
Assorbimento
Dopo somministrazione sottocutanea di una dose singola di 75 mg/m², l'azacitidina è stata rapidamente assorbita con una concentrazione plasmatica di picco di 750 ± 403 ng/ml, osservata a 0,5 ore dopo la somministrazione della dose (primo punto di campionamento).
La biodisponibilità assoluta dell'azacitidina dopo somministrazione sottocutanea rispetto a quella endovenosa (dose singola di 75 mg/m²) è stata di circa l'89%, in base all'area sotto la curva farmacocinetica (AUC).
L'area sotto la curva e la concentrazione massima in plasma (Cmax) dopo somministrazione sottocutanea di azacitidina sono state approssimativamente proporzionali nell'intervallo di dosi da 25 a 100 mg/m².
Distribuzione
Dopo somministrazione endovenosa, il volume medio di distribuzione è stato di 76 ± 26 l e la clearance sistemica di 147 ± 47 l/ora.
Biotrasformazione
Secondo dati ottenuti in vitro, il metabolismo dell'azacitidina non è mediato dagli isoenzimi del citocromo P450 (CYP), dalle UDP-glucuronosiltransferasi (UGT), dalle solfotransferasi (SULT) e dalle glutatione transferasi (GST).
L'azacitidina subisce idrolisi spontanea e deaminazione mediata dalla citidina deaminasi. Nelle frazioni S9 di fegato umano, la formazione di metaboliti non dipendeva dal nicotinammide adenina dinucleotide fosfato ridotto (NADPH), indicando che il metabolismo dell'azacitidina non è mediato dagli isoenzimi del citocromo P450. I dati dello studio in vitro con azacitidina in epatociti umani coltivati indicano che, a concentrazioni da 1,0 µM a 100 µM (cioè circa 30 volte superiore alle concentrazioni clinicamente raggiungibili), l'azacitidina non induce CYP 1A2, 2C19 o 3A4 o 3A5. Negli studi di valutazione dell'inibizione di diversi isoenzimi P450 (CYP 1A2, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1 e 3A4), l'azacitidina a dosi fino a 100 µM non ha causato inibizione. Pertanto, l'induzione o l'inibizione degli enzimi CYP da parte dell'azacitidina a concentrazioni clinicamente raggiungibili nel plasma è improbabile.
Eliminazione
L'azacitidina è rapidamente eliminata dal plasma con un'emivita media (t1/2) di 41 ± 8 minuti dopo somministrazione sottocutanea. Dopo somministrazione sottocutanea di 75 mg/m² di azacitidina una volta al giorno per sette giorni, non si verifica accumulo. L'escrezione urinaria è la principale via di eliminazione dell'azacitidina e/o dei suoi metaboliti. Dopo somministrazione endovenosa e sottocutanea di azacitidina marcata con 14C, l'85% e il 50% della sostanza marcata somministrata è stato recuperato nell'urina rispettivamente, mentre meno dell'1% è stato recuperato nelle feci.
Popolazioni speciali
L'effetto dell'insufficienza epatica (vedi sezione «Modalità di somministrazione e posologia»), sesso, età o appartenenza razziale sulla farmacocinetica dell'azacitidina non è stato studiato ufficialmente.
Popolazione pediatrica
Nello studio AZA-JMML-001, l'analisi farmacocinetica è stata condotta in 10 pazienti con SMDS e 18 bambini con LMMC al giorno 7 del ciclo 1 (vedi sezione «Proprietà farmacologiche»). La mediana di età (intervallo) dei pazienti con SMDS era di 13,3 (1,9-15) anni e dei pazienti con LMMC era di 2,1 (0,2-6,9) anni.
Dopo somministrazione endovenosa di una dose di 75 mg/m², l'azacitidina ha raggiunto rapidamente la concentrazione Cmax entro 0,083 ore sia nelle popolazioni SMDS che LMMC. Il valore geometrico medio di Cmax è stato di 1797,5 e 1066,3 ng/ml e il valore geometrico medio di AUC0-∞ è stato di 606,9 e 240,2 ng*h/ml per i pazienti con SMDS e LMMC rispettivamente. Il volume medio di distribuzione geometrico nei pazienti con SMDS e LMMC è stato di 103,9 e 61,1 l rispettivamente. È risultato che l'esposizione totale all'azacitidina nel plasma era più alta nei pazienti con SMDS; tuttavia, una variabilità moderata o alta tra pazienti è stata osservata sia per i parametri AUC che per Cmax.
Il valore medio geometrico dell'emivita t1/2 è stato di 0,4 e 0,3 ore e il valore medio geometrico della clearance è stato di 166,4 e 148,3 l/ora per SMDS e LMMC rispettivamente.
I dati farmacocinetici dello studio AZA-JMML-001 sono stati combinati e confrontati con i dati farmacocinetici di sei pazienti adulti con SMDS che hanno ricevuto 75 mg/m² di azacitidina endovena nello studio AZA-2002-BA-002. I valori medi di Cmax e AUC0-t di azacitidina sono stati simili in pazienti adulti e bambini dopo somministrazione endovenosa (2750 ng/ml vs 2841 ng/ml e 1025 ng*h/ml vs 882,1 ng*h/ml rispettivamente).
Nello studio AZA-AML-004, l'analisi farmacocinetica è stata condotta in 6 su 7 pazienti pediatrici, che avevano almeno una concentrazione farmacocinetica misurata dopo somministrazione della dose del farmaco (vedi sezione «Proprietà farmacologiche»). La mediana (intervallo) di età dei pazienti con LMA era di 6,7 (2-12) anni.
Dopo somministrazione di dosi ripetute del farmaco a 100 mg/m², le medie geometriche per i parametri Cmax e AUC0-t al giorno 7 del primo ciclo sono state rispettivamente di 1557 ng/ml e 899,6 ng*h/ml, con alta variabilità intersoggetto (coefficiente di variazione (CV) 201,6% e 87,8% rispettivamente). L'azacitidina ha raggiunto rapidamente il livello di Cmax con una mediana del tempo di 0,090 ore dopo somministrazione endovenosa e si è ridotta con un valore medio geometrico dell'emivita t1/2 di 0,380 ore. Le medie geometriche per la clearance e il volume di distribuzione sono state rispettivamente di 127,2 l/ora e 70,2 l.
L'esposizione farmacocinetica (azacitidina), osservata in bambini con LMA in recidiva molecolare dopo CR1, è stata paragonabile all'esposizione ottenuta dai dati combinati per 10 bambini con SMDS e 18 bambini con LMMC, e paragonabile all'esposizione di azacitidina in adulti con SMDS.
Compromissione renale
La compromissione della funzione renale non ha un impatto sostanziale sull'esposizione farmacocinetica dell'azacitidina dopo somministrazione sottocutanea singola o ripetuta. Dopo somministrazione sottocutanea di una dose singola di 75 mg/m², i valori medi di esposizione (AUC e Cmax) nei pazienti con compromissione renale lieve, moderata e grave sono aumentati rispettivamente dell'11-21%, 15-27% e 41-66% rispetto ai pazienti con funzione renale normale. Tuttavia, l'esposizione rientrava nello stesso intervallo generale di esposizione osservato in soggetti con funzione renale normale. L'azacitidina può essere somministrata a pazienti con compromissione renale senza aggiustamento della dose iniziale, a condizione che questi pazienti siano monitorati per segni di tossicità, poiché l'azacitidina e/o i suoi metaboliti sono principalmente eliminati dai reni.
Farmacogenomica
L'effetto dei noti polimorfismi della citidina deaminasi sul metabolismo dell'azacitidina non è stato studiato ufficialmente.
Caratteristiche cliniche.
Indicazioni.
Azacitidina Accord è indicata nel trattamento di adulti non candidabili al trapianto di cellule staminali emopoietiche (SCT) con le seguenti patologie:
- sindromi mielodisplastiche (MDS) di grado intermedio-2 e alto rischio secondo il sistema prognostico internazionale (IPSS),
- leucemia mielomonocitica cronica (CMML) con 10-29% di blasti nel midollo osseo senza disordine mieloproliferativo,
- leucemia mieloide acuta (AML) con 20-30% di blasti e displasia multilinage, secondo la classificazione dell'Organizzazione Mondiale della Sanità (OMS),
- AML con > 30% di blasti nel midollo osseo secondo la classificazione OMS.
Controindicazioni.
Ipersensibilità al principio attivo o a uno qualsiasi degli eccipienti.
Tumori maligni epatici in fase avanzata (vedere la sezione «Informazioni particolari sull'uso»).
Allattamento al seno (vedere la sezione «Uso in gravidanza o allattamento»).
Misure precauzionali particolari.
Raccomandazioni per una manipolazione sicura
Azacitidina Accord è un agente citotossico e, come per altri composti potenzialmente tossici, è necessario adottare precauzioni durante la manipolazione del medicinale e la preparazione delle sospensioni di azacitidina. Devono essere seguite procedure adeguate per la manipolazione e lo smaltimento dei farmaci antineoplastici.
Se la soluzione di azacitidina entra in contatto con la cute, lavare immediatamente e accuratamente con acqua e sapone. Se entra in contatto con le membrane mucose, risciacquare accuratamente con acqua.
Procedura di diluizione
Azacitidina Accord deve essere diluito con acqua per preparazioni iniettabili. La durata di conservazione della soluzione diluita può essere prolungata utilizzando acqua per preparazioni iniettabili refrigerata (da 2 °C a 8 °C). Ulteriori informazioni sulla conservazione del prodotto diluito sono riportate di seguito.
Preparare i seguenti materiali:
- Flaconcino(i) di azacitidina; flaconcino(i) di acqua per preparazioni iniettabili; guanti chirurgici non sterili; salviette alcoliche; siringa(e) da 5 ml con ago(i).
- Prelevare con la siringa il volume appropriato di acqua per preparazioni iniettabili (vedere la tabella qui sotto) ed eliminare obbligatoriamente l'aria presente nella siringa.
| Capacità della fiala |
Volume di acqua per preparazioni iniettabili |
Concentrazione finale |
| 150 mg |
6 ml |
25 mg/ml |
- L'ago della siringa contenente acqua per preparazioni iniettabili deve essere inserito attraverso il tappo in gomma del flaconcino di azacitidina e quindi l'acqua per preparazioni iniettabili deve essere iniettata nel flaconcino.
- Dopo aver rimosso la siringa e l'ago, il flaconcino deve essere agitato energeticamente fino a ottenere una sospensione omogenea torbida. Dopo la ricostituzione, ogni ml di sospensione conterrà 25 mg di azacitidina (100 mg/4 ml o 150 mg/6 ml). Il medicinale ricostituito è una sospensione torbida omogenea, priva di agglomerati. Il medicinale deve essere scartato se contiene particelle di grandi dimensioni o agglomerati. Non filtrare la sospensione dopo la ricostituzione poiché ciò potrebbe portare alla rimozione del principio attivo. Si deve tenere presente che alcuni dispositivi di connessione, coni e sistemi chiusi possono contenere filtri; pertanto, tali sistemi non devono essere utilizzati per la somministrazione del medicinale dopo la ricostituzione.
- Dopo aver disinfettato il tappo in gomma, inserire una nuova siringa con ago nel flaconcino. Successivamente, capovolgere il flaconcino, assicurandosi che la punta dell'ago sia al di sotto del livello del liquido. Tirare quindi indietro lo stantuffo per aspirare la quantità di medicinale necessaria per somministrare la dose corretta, assicurandosi di eliminare l'aria entrata nella siringa. Rimuovere quindi la siringa con l'ago dal flaconcino e smaltire l'ago.
- Un nuovo ago per somministrazione sottocutanea (si raccomanda calibro 25) deve essere avvitato saldamente sulla siringa. Non si deve espellere l'aria dall'ago prima dell'iniezione, al fine di ridurre la frequenza di reazioni locali nel sito di iniezione.
- Se è necessario somministrare più di un flaconcino, tutte le operazioni sopra descritte per la preparazione della sospensione devono essere ripetute. Per le dosi che richiedono più di un flaconcino, la dose deve essere suddivisa in parti uguali, ad esempio: dose di 150 mg = 6 ml, 2 siringhe da 3 ml ciascuna. A causa del residuo di medicinale presente nel flaconcino e nell'ago, potrebbe non essere possibile aspirare completamente tutta la sospensione dal flaconcino.
- Il contenuto della siringa preriempita deve essere risospeso immediatamente prima della somministrazione. La siringa riempita con la sospensione ricostituita deve essere portata a una temperatura di circa 20–25 °C entro 30 minuti dalla somministrazione. Se il tempo trascorso supera i 30 minuti, la sospensione deve essere smaltita adeguatamente e deve essere preparata una nuova dose. Per risospendere il contenuto, si raccomanda di agitare energicamente la siringa tra i palmi delle mani fino a ottenere una sospensione torbida omogenea. Il medicinale deve essere scartato se contiene particelle di grandi dimensioni o agglomerati.
Conservazione del medicinale ricostituito
Le condizioni di conservazione dopo la ricostituzione del medicinale sono riportate nella sezione «Periodo di validità».
Calcolo della dose individuale
La dose totale, in base alla superficie corporea (BSA), può essere calcolata nel seguente modo:
Dose totale (mg) = dose (mg/m²) × BSA (m²)
La tabella riportata di seguito è fornita unicamente come esempio per il calcolo delle dosi individuali di azacitidina, basandosi su un valore medio di superficie corporea di 1,8 m².
| Dosaggio, mg/m2 (% della dose iniziale raccomandata) |
Dosaggio totale, basato su un valore di AUC di 1,8 m2 |
Numero di fiale necessarie |
Volume totale necessario di sospensione ricostituita |
| Fiala 150 mg |
|||
| 75 mg/m2 (100 %) |
135 mg |
1 fiala |
5,4 ml |
| 37,5 mg/m2 (50 %) |
67,5 mg |
1 fiala |
2,7 ml |
| 25 mg/m2 (33 %) |
45 mg |
1 fiala |
1,8 ml |
Modo di somministrazione
L'azacitidina Accord diluita deve essere somministrata per via sottocutanea (inserire l'ago con un angolo di 45-90°) mediante un ago calibro 25 nella spalla, coscia o addome.
Le dosi superiori a 4 ml devono essere somministrate in due siti separati.
I siti di iniezione devono essere ruotati. Le nuove iniezioni devono essere effettuate ad almeno 2,5 cm di distanza dal sito precedente e mai in aree dolenti, livide, arrossate o indurite.
Smaltimento
Qualsiasi farmaco non utilizzato o rifiuto deve essere smaltito in conformità con i requisiti locali.
Interazioni con altri medicinali ed altre forme di interazione.
Sulla base dei dati ottenuti in vitro, il metabolismo dell'azacitidina non è mediato dagli isoenzimi del citocromo P450 (CYP), dalle UDP-glucuronosiltransferasi (UGT), dalle sulfotransferasi (SULT) e dalle glutatione transferasi (GST); pertanto, interazioni legate a questi enzimi metabolizzanti in vivo sono considerate improbabili.
È improbabile un effetto inibitorio o induttivo clinicamente significativo dell'azacitidina sui citocromi P450 (vedere la sezione «Proprietà farmacologiche»).
Non sono stati condotti studi clinici formali sulle interazioni con l'azacitidina.
Caratteristiche di impiego.
Tossicità ematologica
Il trattamento con azacitidina è associato ad anemia, neutropenia e trombocitopenia, in particolare durante i primi due cicli (vedere sezione «Effetti indesiderati»). Un emocromo completo per il monitoraggio della risposta al trattamento e della tossicità deve essere effettuato secondo necessità, ma almeno prima di ogni ciclo di trattamento. Dopo la somministrazione della dose raccomandata per il primo ciclo, la dose per i cicli successivi deve essere ridotta o la somministrazione deve essere rinviata in base ai valori più bassi di conteggio delle cellule ematiche e alla risposta ematologica al trattamento (vedere sezione «Modalità di somministrazione e posologia»). Ai pazienti deve essere raccomandato di segnalare immediatamente eventuali episodi di febbre. Pazienti e medici devono inoltre prestare attenzione ai segni e sintomi di emorragia.
Alterazioni della funzione epatica
Non sono stati condotti studi formali su pazienti con compromissione della funzione epatica. Sono stati riportati casi di coma epatico progressivo con esito fatale durante il trattamento con azacitidina in pazienti con un elevato carico tumorale dovuto a malattia metastatica, in particolare in pazienti con livello sierico iniziale di albumina < 30 g/l. L’azacitidina è controindicata nei pazienti con tumori maligni epatici in stadio avanzato (vedere sezione «Controindicazioni»).
Alterazioni della funzione renale
In pazienti che hanno ricevuto azacitidina per via endovenosa in combinazione con altri agenti chemioterapici, sono state riportate alterazioni della funzione renale, che vanno dall’aumento della creatinina sierica fino all’insufficienza renale e all’esito fatale. Inoltre, in cinque pazienti con leucemia mieloide cronica (LMC) trattati con azacitidina ed etoposide, si è sviluppato un acidosi tubulare renale, definita come riduzione del livello di bicarbonato sierico a < 20 mmol/l in associazione con alcalinità urinaria aumentata e ipokaliemia (livello di potassio sierico < 3 mmol/l). In caso di riduzione inspiegabile del bicarbonato sierico (< 20 mmol/l) o aumento della creatinina o delle AST sieriche, la dose di azacitidina deve essere ridotta o la somministrazione deve essere sospesa (vedere sezione «Modalità di somministrazione e posologia»).
Ai pazienti deve essere consigliato di segnalare immediatamente oliguria e anuria.
Sebbene non siano state osservate differenze clinicamente significative nella frequenza degli effetti indesiderati tra pazienti con funzione renale normale e pazienti con insufficienza renale, i pazienti con insufficienza renale devono essere attentamente monitorati per segni di tossicità, poiché l’azacitidina e/o i suoi metaboliti sono principalmente eliminati attraverso i reni (vedere sezione «Modalità di somministrazione e posologia»).
Analisi di laboratorio
Prima dell’inizio della terapia e prima di ogni ciclo di trattamento devono essere effettuati test della funzionalità epatica, analisi del livello di creatinina sierica e bicarbonato sierico. Un emocromo completo deve essere effettuato prima dell’inizio della terapia e secondo necessità per monitorare la risposta al trattamento e la tossicità, ma almeno prima di ogni ciclo di trattamento (vedere anche sezione «Effetti indesiderati»).
Malattie cardiache e polmonari
I pazienti con insufficienza cardiaca congestizia grave anamnestica, malattia cardiaca clinicamente instabile o malattia polmonare sono stati esclusi dagli studi registrativi fondamentali (AZA PH GL 2003 CL 001 e AZA-AML-001); pertanto, la sicurezza ed efficacia dell’azacitidina in questi pazienti non sono state stabilite. Dati recenti di studi clinici su pazienti con anamnesi di malattie cardiovascolari o polmonari hanno mostrato un significativo aumento della frequenza di complicanze cardiache con l’uso di azacitidina (vedere sezione «Effetti indesiderati»). Pertanto, si raccomanda di somministrare azacitidina con cautela a tali pazienti. Prima dell’inizio e durante il trattamento deve essere effettuata una valutazione della funzionalità cardiaca e polmonare.
Fascite necrotizzante
In pazienti trattati con azacitidina sono stati riportati casi di fascite necrotizzante, inclusi esiti fatali. Nei pazienti che sviluppano fascite necrotizzante, il trattamento con azacitidina deve essere interrotto e deve essere avviato immediatamente un trattamento appropriato.
Sindrome da lisi tumorale
I pazienti a rischio di sviluppare la sindrome da lisi tumorale sono quelli con un elevato carico tumorale prima del trattamento. Tali pazienti devono essere attentamente monitorati e devono essere adottate misure preventive adeguate.
Sindrome da differenziazione
Sono stati riportati casi di sindrome da differenziazione (nota anche come sindrome da acido retinoico) tra i pazienti in trattamento con azacitidina. Tale sindrome può essere fatale e i sintomi e segni clinici comprendono distress respiratorio, infiltrati polmonari, febbre, eruzione cutanea, edema polmonare, edema periferico, rapido aumento di peso, versamento pleurico, versamento pericardico, ipotensione e compromissione della funzione renale (vedere sezione «Effetti indesiderati»). In caso di comparsa dei primi sintomi o segni indicativi di sindrome da differenziazione, si deve prendere in considerazione il trattamento con alte dosi di corticosteroidi per via endovenosa e il monitoraggio emodinamico. Si deve valutare la possibilità di interrompere temporaneamente il trattamento con azacitidina iniettabile fino alla scomparsa dei sintomi; in caso di ripresa del trattamento, si deve procedere con cautela.
Uso durante la gravidanza o l’allattamento.
Donne in età fertile/Contraccezione negli uomini e nelle donne
Le donne in età fertile devono utilizzare metodi contraccettivi efficaci durante il trattamento e per almeno 6 mesi dopo la fine del trattamento. Si raccomanda agli uomini di utilizzare un metodo contraccettivo efficace durante il trattamento e per almeno 3 mesi dopo la sua conclusione.
Gravidanza
Non esistono dati adeguati sull’uso di azacitidina in donne in gravidanza. Studi su topi hanno mostrato tossicità riproduttiva del farmaco. Il rischio potenziale di questo medicinale per l’essere umano è sconosciuto. Sulla base dei risultati degli studi sugli animali e del meccanismo d’azione, l’azacitidina
non deve essere utilizzata durante la gravidanza, in particolare durante il I trimestre, a meno che non vi sia un’assoluta necessità. In ogni singolo caso, devono essere attentamente valutati i benefici del trattamento e i possibili rischi per il feto.
Allattamento
Non è noto se azacitidina/metaboliti passino nel latte materno. A causa della possibilità di reazioni avverse gravi nel neonato allattato, l’allattamento al seno è controindicato durante la terapia con azacitidina.
Fertilità
Non esistono dati sull’effetto dell’azacitidina sulla fertilità umana. Negli animali sono stati documentati effetti avversi sulla fertilità maschile con l’uso di azacitidina. Prima dell’inizio del trattamento, ai pazienti maschi deve essere consigliato di consultare uno specialista per la conservazione dello sperma.
Capacità di guidare veicoli o utilizzare macchinari.
L’azacitidina ha un’influenza lieve o moderata sulla capacità di guidare veicoli o di utilizzare macchinari. È stato riportato affaticamento con l’uso di azacitidina. Pertanto, si raccomanda di prestare cautela durante la guida di automezzi o l’uso di macchinari.
Modalità e posologia.
Il trattamento con Azacitidina Accord deve essere iniziato e monitorato sotto la supervisione di un medico esperto nell'uso di agenti chemioterapici. Ai pazienti deve essere somministrata una premedicazione con farmaci antiemetici per prevenire nausea e vomito.
Dosi
La dose raccomandata iniziale per il primo ciclo di trattamento per tutti i pazienti, indipendentemente dai valori ematologici iniziali, è di 75 mg/m² di superficie corporea, somministrata per via sottocutanea una volta al giorno per 7 giorni consecutivi, seguita da un intervallo di 21 giorni (ciclo di trattamento di 28 giorni).
Si raccomanda che i pazienti ricevano almeno sei cicli di trattamento. Il trattamento deve essere proseguito finché il paziente continua a trarne beneficio o fino alla progressione della malattia.
I pazienti devono essere monitorati attentamente per rilevare segni di reazione/tossicità ematologica e tossicità renale (vedere il paragrafo «Speciali avvertenze e precauzioni per l’uso»); potrebbe essere necessario ritardare l’inizio del ciclo successivo o ridurre la dose, come descritto di seguito.
Il medicinale Azacitidina Accord non deve essere considerato intercambiabile con l’azacitidina orale. A causa delle differenze nell’esposizione sistemica, le raccomandazioni relative alla dose e allo schema di somministrazione dell’azacitidina orale differiscono da quelle dell’azacitidina per iniezione. Si raccomanda ai professionisti sanitari di verificare attentamente il nome del medicinale, la dose e la via di somministrazione.
Analisi di laboratorio
Prima dell’inizio della terapia e prima di ogni ciclo di trattamento devono essere effettuati esami della funzionalità epatica, nonché la misurazione del livello ematico di creatinina e bicarbonato. Un emocromo completo deve essere eseguito prima dell’inizio della terapia e, se necessario, durante il monitoraggio della risposta al trattamento e della tossicità, ma almeno prima di ogni ciclo di trattamento.
Adattamento della dose per tossicità ematologica
La tossicità ematologica è definita come il valore minimo raggiunto (nadir) in un determinato ciclo, quando il livello delle piastrine è ≤ 50,0 x 10⁹/l e/o il conteggio assoluto dei neutrofili (ANC) ≤ 1 x 10⁹/l.
Il recupero è definito come un aumento della linea cellulare (o delle linee cellulari) in cui si è verificata tossicità ematologica, pari almeno alla metà della differenza assoluta tra il valore iniziale e il valore minimo, più il valore minimo stesso (ovvero: il valore cellulare al recupero ≥ valore minimo + (0,5 x [valore iniziale – valore minimo])).
Pazienti senza alterazioni ematologiche iniziali (cioè conteggio dei leucociti (WBC) ≥ 3,0 x 10⁹/l e ANC ≥ 1,5 x 10⁹/l, e conteggio delle piastrine ≥ 75,0 x 10⁹/l) prima del primo ciclo di trattamento
Se si verifica tossicità ematologica dopo il trattamento con Azacitidina Accord, il ciclo successivo di terapia deve essere ritardato fino al recupero del conteggio delle piastrine e dell’ANC. Se il recupero avviene entro 14 giorni, non è necessario alcun aggiustamento della dose. Tuttavia, se il recupero non si verifica entro 14 giorni, la dose deve essere ridotta secondo la tabella riportata di seguito. Dopo ogni modifica della dose, la durata del ciclo deve tornare a essere di 28 giorni.
| Nadir del ciclo |
Dosaggio nel ciclo successivo se il recupero* non è stato raggiunto entro 14 giorni (%) |
|
| ANC (x 109/l) |
Plaquettes (x 109/l) |
|
| ≤ 1,0 |
≤ 50,0 |
50 % |
| > 1,0 |
> 50,0 |
100 % |
*Recupero = quantità ≥ quantità minima + (0,5 x [quantità iniziale – quantità minima])
Pazienti con basse quantità iniziali di cellule ematiche (cioè leucociti < 3,0 x 109/l o ANC < 1,5 x 109/l o piastrine < 75,0 x 109/l) prima del primo trattamento
Se dopo il trattamento con Azacitidina Accord la riduzione dei leucociti, dell'ANC o delle piastrine rispetto ai livelli precedenti al trattamento è ≤ 50 % oppure supera il 50 % ma si osserva un miglioramento della differenziazione di una qualsiasi linea cellulare, il ciclo successivo non deve essere ritardato e non è necessaria alcuna modifica della dose.
Se la riduzione dei leucociti, dell'ANC o delle piastrine supera il 50 % rispetto ai livelli precedenti al trattamento e non si osserva alcun miglioramento della differenziazione della linea cellulare, il ciclo successivo di trattamento con Azacitidina Accord deve essere ritardato fino al recupero di piastrine e ANC. Se il recupero avviene entro 14 giorni, non è necessaria alcuna modifica della dose. Tuttavia, se il recupero non avviene entro 14 giorni, è necessario valutare la cellularità del midollo osseo. Se la cellularità del midollo osseo è > 50 %, non è necessaria alcuna modifica della dose. Se la cellularità del midollo osseo è ≤ 50 %, il trattamento deve essere ritardato e la dose ridotta secondo la tabella riportata di seguito:
| Cellularità del midollo osseo |
Dose nel ciclo successivo se non si raggiunge il recupero entro 14 giorni (%) |
|
| Recupero* ≤ 21 giorni |
Recupero* > 21 giorni |
|
| 15-50 % |
100 % |
50 % |
| < 15 % |
100 % |
33 % |
*Ripristino = quantità ≥ quantità più bassa + (0,5 x [quantità iniziale – quantità più bassa]).
Dopo la modifica della dose, la durata del ciclo successivo deve tornare a 28 giorni.
Popolazioni speciali
Pazienti anziani
Non è raccomandata una particolare correzione della dose per le persone anziane. Poiché nei pazienti anziani è più frequente un ridotto funzionamento renale, può essere utile monitorare la funzionalità renale.
Pazienti con compromissione della funzionalità renale
Azacitidina può essere somministrata a pazienti con compromissione della funzionalità renale senza correzione della dose iniziale (vedere il paragrafo «Proprietà farmacologiche»). Se si verifica una riduzione inspiegabile del livello di bicarbonato nel siero al di sotto di 20 mmol/l, la dose deve essere ridotta del 50% durante il ciclo successivo. In caso di aumento inspiegabile della creatinina nel siero o dell'azoto ureico nel sangue (BUN) ≥ doppio rispetto ai valori iniziali e superiore al limite superiore della norma (LSN), il ciclo successivo deve essere rinviato finché i valori non tornino alla norma o al livello iniziale, e la dose deve essere ridotta del 50% durante il successivo ciclo di trattamento (vedere il paragrafo «Informazioni importanti sull’uso del medicinale»).
Pazienti con compromissione della funzionalità epatica
Non sono stati condotti studi formali su pazienti con compromissione della funzionalità epatica (vedere il paragrafo «Informazioni importanti sull’uso del medicinale»). I pazienti con grave insufficienza epatica devono essere attentamente monitorati per effetti indesiderati. Per i pazienti con compromissione della funzionalità epatica non è raccomandata una modifica della dose iniziale; ulteriori aggiustamenti della dose devono basarsi sui parametri ematologici di laboratorio. Azacitidina Accord è controindicata nei pazienti con tumori epatici maligni in stadio avanzato (vedere i paragrafi «Controindicazioni» e «Informazioni importanti sull’uso del medicinale»).
Popolazione pediatrica
La sicurezza e l'efficacia di azacitidina nei bambini di età compresa tra 0 e 17 anni non sono ancora state stabilite. I dati attualmente disponibili sono descritti nei paragrafi «Proprietà farmacologiche» e «Effetti indesiderati», ma non è possibile fornire raccomandazioni sulla posologia.
Modalità di somministrazione
Dopo la ricostituzione, ogni ml di sospensione conterrà 25 mg di azacitidina. Azacitidina Accord ricostituita deve essere somministrata per via sottocutanea nella spalla, coscia o addome. I siti di iniezione devono essere alternati. Le nuove iniezioni devono essere effettuate a una distanza di almeno 2,5 cm dal sito precedente e in nessun caso in aree dolenti, con ematomi, arrossate o indurite.
Dopo la ricostituzione, la sospensione non deve essere filtrata. Le istruzioni per la ricostituzione del medicinale prima dell’uso sono riportate nel paragrafo «Precauzioni particolari per la conservazione e l’uso».
Popolazione pediatrica.
Non deve essere utilizzato nei bambini (di età inferiore a 18 anni) poiché la sicurezza ed efficacia del medicinale in questa categoria di pazienti non sono state stabilite.
Sovradosaggio.
Durante gli studi clinici è stato riportato un caso di sovradosaggio con azacitidina. Un paziente ha manifestato diarrea, nausea e vomito dopo una somministrazione endovenosa singola di circa 290 mg/m², dose che supera di quasi quattro volte la dose iniziale raccomandata.
In caso di sovradosaggio, lo stato del paziente deve essere monitorato tramite esami ematici e, se necessario, deve essere istituito un trattamento di supporto. Non esiste un antidoto specifico noto per il sovradosaggio di azacitidina.
Effetti indesiderati
Riepilogo del profilo di sicurezza
Popolazione adulta con sindrome mielodisplastica (MDS), leucemia mielomonocitica cronica (CMML) e leucemia mieloide acuta (AML) (20-30% di blasti nel midollo osseo)
Gli effetti indesiderati probabilmente o verosimilmente correlati all'uso di azacitidina sono stati osservati nel 97% dei pazienti.
Le reazioni avverse gravi più comuni riportate nello studio principale (AZA PH GL 2003 CL 001) comprendevano neutropenia febbrile (8,0%) e anemia (2,3%), entrambe riportate anche negli studi di supporto (CALGB 9221 e CALGB 8921). Altri eventi avversi gravi osservati in questi tre studi comprendevano infezioni, come sepsi neutropenica (0,8%) e polmonite (2,5%) (alcune con esito fatale), trombocitopenia (3,5%), reazioni di ipersensibilità (0,25%) e manifestazioni emorragiche (ad esempio emorragia cerebrale [0,5%], emorragie gastrointestinali [0,8%] ed emorragie intracraniche [0,5%]).
Gli effetti indesiderati più comuni durante il trattamento con azacitidina erano reazioni ematologiche (71,4%), comprese trombocitopenia, neutropenia e leucopenia (generalmente di grado 3-4), disturbi gastrointestinali (60,6%), compresi nausea, vomito (generalmente di grado 1-2) e reazioni nel sito di somministrazione (77,1%; generalmente di grado 1-2).
Popolazione adulta di età ≥ 65 anni con AML con > 30% di blasti nel midollo osseo
Le reazioni avverse gravi più comuni (≥ 10%) osservate nello studio AZA-AML-001 nel gruppo trattato con azacitidina sono state neutropenia febbrile (25,0%), polmonite (20,3%) e febbre (10,6%). Altri eventi avversi gravi meno frequenti nel gruppo trattato con azacitidina comprendevano setticemia (5,1%), anemia (4,2%), sepsi neutropenica (3,0%), infezione del tratto urinario (3,0%), trombocitopenia (2,5%), neutropenia (2,1%), cellulite (2,1%), capogiri (2,1%) e dispnea (2,1%).
Gli effetti indesiderati più frequentemente osservati (≥ 30%) con il trattamento con azacitidina sono stati disturbi gastrointestinali, compresi costipazione (41,9%), nausea (39,8%) e diarrea (36,9%; generalmente di grado 1-2); disturbi generali e reazioni nel sito di somministrazione, inclusa febbre (37,7%; generalmente di grado 1-2); e manifestazioni ematologiche, comprese neutropenia febbrile (32,2%) e neutropenia (30,1%; generalmente di grado 3-4).
Elenco tabulato degli effetti indesiderati
La Tabella 1 riportata di seguito contiene gli effetti indesiderati associati al trattamento con azacitidina, osservati negli studi clinici principali su pazienti con MDS e AML e durante il periodo post-marketing.
La frequenza degli effetti indesiderati è definita come segue: molto frequente (≥ 1/10), frequente (da ≥ 1/100 a < 1/10), non frequente (da ≥ 1/1.000 a < 1/100), raro (da ≥ 1/10.000 a < 1/1.000), molto raro (< 1/10.000), frequenza non nota (non può essere stimata sulla base dei dati disponibili). All'interno di ogni gruppo di frequenza, gli effetti indesiderati sono elencati in ordine decrescente di gravità. Gli effetti indesiderati sono riportati nella tabella seguente in base alla frequenza più alta osservata in uno qualsiasi degli studi clinici principali.
Effetti indesiderati riportati in pazienti con MDS o AML trattati con azacitidina
(studi clinici e periodo post-marketing)
Tabella 1
| Classe di sistema d'organo |
Molto frequenti |
Frequenti |
Non comuni |
Rari |
Frequenza non nota |
| Infestazioni e infezioni |
polmonite* (inclusa batterica, virale e micotica), nasofaringite |
sepsi* (inclusa batterica, virale e micotica), sepsi neutropenica*, infezione delle vie respiratorie (inclusa infezione delle vie respiratorie superiori e bronchite), infezione delle vie urinarie, cellulite, diverticolite, infezione micotica del cavo orale, sinusite, faringite, rinite, herpes semplice, infezione cutanea |
fascite necrotizzante* |
||
| Neoplasie benigne, maligne e di localizzazione imprecisata (inclusi cisti e polipi) |
sindrome da differenziazione*, a |
||||
| Patologie del sistema emolinfopoietico |
neutropenia febbrile*, neutropenia, leucopenia, trombocitopenia, anemia |
pancitopenia*, insufficienza del midollo osseo |
|||
| Patologie del sistema immunitario |
reazioni di ipersensibilità |
||||
| Disturbi del metabolismo e della nutrizione |
anoressia, riduzione dell'appetito, ipokaliemia |
disidratazione |
sindrome da lisi tumorale |
||
| Disturbi psichiatrici |
insonnia |
confusione mentale, ansia |
|||
| Patologie del sistema nervoso |
capogiri, mal di testa |
emorragia intracranica*, sincope, sonnolenza, letargia |
|||
| Patologie dell'occhio |
emorragia oculare, emorragia congiuntivale |
||||
| Patologie cardiache |
versamento pericardico |
pericardite |
|||
| Patologie vascolari |
ipotensione*, ipertensione, ipotensione ortostatica, ematoma |
||||
| Patologie respiratorie, toraciche e del mediastino |
dispnea, epistassi |
versamento pleurico, dispnea da sforzo, dolore faringo-laringeo |
malattia interstiziale del polmone |
||
| Patologie gastrointestinali |
diarrea, vomito, costipazione, nausea, dolore addominale (inclusi fastidio nell'area superiore dell'addome e nell'addome) |
emorragia gastrointestinale* (inclusa emorragia orale), emorragia emorroidaria, stomatite, sanguinamento gengivale, dispepsia |
|||
| Patologie epatiche e della colecisti |
insufficienza epatica*, coma epatico progressivo |
||||
| Patologie della cute e del tessuto sottocutaneo |
petecchie, prurito (incluso generalizzato), eruzione cutanea, ecchimosi |
purpura, alopecia, orticaria, eritema, eruzione maculosa |
eritema multiforme acuto febbrile, piodermite gangrenosa |
vasculite cutanea |
|
| Patologie del sistema muscoloscheletrico e del tessuto connettivo |
artralgia, dolore del sistema muscoloscheletrico (incluso dolore alla schiena, alle ossa e alle estremità) |
crampi muscolari, mialgia |
|||
| Patologie renali e urinarie |
insufficienza renale*, ematuria, aumento della creatinina sierica |
acidosi tubulare renale |
|||
| Alterazioni generali e condizioni in sede di somministrazione |
febbre*, affaticamento, astenia, dolore al petto, eritema in sede di iniezione, dolore in sede di iniezione, reazione in sede di iniezione (non specificata) |
ecchimosi, ematoma, indurimento, eruzione cutanea, prurito, infiammazione, alterazione del colore, noduli ed emorragia (in sede di iniezione), malessere, brividi, emorragia in sede di inserzione del catetere |
necrosi del sito di somministrazione (in sede di iniezione) |
||
| Esami di laboratorio |
diminuzione del peso corporeo |
* – in rari casi sono stati riportati esiti letali.
a – vedere sezione «Informazioni particolari di utilizzo».
Descrizione delle singole reazioni avverse
Reazioni avverse ematologiche
Le reazioni avverse ematologiche più comunemente riportate (≥ 10 %) associate al trattamento con azacitidina comprendono anemia, trombocitopenia, neutropenia, neutropenia febbrile e leucopenia, generalmente di grado 3 o 4. Il rischio di questi eventi è maggiore durante i primi due cicli di trattamento, dopo i quali si verificano con minore frequenza nei pazienti con recupero della funzionalità ematologica. La maggior parte delle reazioni avverse ematologiche può essere gestita mediante monitoraggio regolare dell’emocromo completo, posticipando l’amministrazione di azacitidina nel ciclo successivo, prescrivendo profilatticamente antibiotici e/o fattori di crescita (ad esempio G-CSF (fattore stimolante le colonie di granulociti)) per il trattamento della neutropenia, e trasfusioni di sangue per controllare anemia o trombocitopenia, se necessario.
Infezioni
La mielosoppressione può portare a neutropenia e aumentato rischio di infezione. Sono state riportate gravi reazioni avverse, come sepsi, inclusa sepsi neutropenica, e polmonite, in pazienti trattati con azacitidina, alcune con esito letale. Le infezioni possono essere trattate con agenti anti-infettivi e supporto con fattori di crescita (ad esempio G-CSF) per il trattamento della neutropenia.
Sanguinamento
Nei pazienti in trattamento con azacitidina può verificarsi sanguinamento. Sono state riportate gravi reazioni avverse, come emorragie gastrointestinali ed emorragie intracraniche. I pazienti devono essere monitorati per segni e sintomi di sanguinamento, in particolare quelli con trombocitopenia preesistente o correlata al trattamento.
Ipersensibilità
Sono state riportate gravi reazioni di ipersensibilità in pazienti trattati con azacitidina. In caso di reazione anafilattica, il trattamento con azacitidina deve essere immediatamente interrotto e deve essere iniziato un appropriato trattamento sintomatico.
Reazioni avverse cutanee e del tessuto sottocutaneo
La maggior parte delle reazioni cutanee e del tessuto sottocutaneo era correlata al sito di iniezione. Nessuna di queste reazioni avverse ha portato all’interruzione o alla riduzione della dose di azacitidina negli studi principali. La maggior parte delle reazioni avverse si verificava durante i primi due cicli di trattamento e tendeva a diminuire nei cicli successivi. Le reazioni avverse del tessuto sottocutaneo, come eruzione cutanea/infiammazione/prurito al sito di iniezione, eruzione cutanea, eritema e lesioni cutanee, possono richiedere trattamento con farmaci concomitanti come antistaminici, corticosteroidi e farmaci antiinfiammatori non steroidei (FANS). Queste reazioni cutanee devono essere distinte dalle infezioni dei tessuti molli, che talvolta si verificano al sito di iniezione. Sono state riportate infezioni dei tessuti molli, inclusi cellulite e fascite necrotizzante, che in rari casi hanno portato a esiti letali durante l’uso post-commercializzazione di azacitidina. La gestione clinica delle reazioni avverse infettive è descritta nella sezione «Reazioni avverse» «Infezioni».
Reazioni avverse gastrointestinali
Le reazioni avverse gastrointestinali più comuni associate al trattamento con azacitidina comprendono costipazione, diarrea, nausea e vomito. Queste reazioni avverse sono state trattate sintomaticamente con agenti antiemetici per controllare nausea e vomito; agenti antidiarroici in caso di diarrea; e lassativi e/o agenti per ammorbidire le feci in caso di costipazione.
Reazioni avverse renali
Nei pazienti trattati con azacitidina sono stati riportati disturbi della funzionalità renale, che vanno dall’aumento della creatinina sierica e ematuria fino all’acidosi tubulare renale, insufficienza renale ed esiti letali (vedere sezione «Informazioni particolari di utilizzo»).
Reazioni avverse epatiche
Nei pazienti con un elevato carico tumorale dovuto a malattia metastatica sono stati riportati insufficienza epatica, coma epatico progressivo ed esito letale durante il trattamento con azacitidina (vedere sezione «Informazioni particolari di utilizzo»).
Disturbi cardiaci
I dati di uno studio clinico che includeva pazienti con anamnesi di malattie cardiovascolari o polmonari hanno mostrato un aumento dei disturbi cardiaci nei pazienti con diagnosi recente di AML in trattamento con azacitidina (vedere sezione «Informazioni particolari di utilizzo»).
Pazienti anziani
Informazioni limitate sulla sicurezza di azacitidina sono disponibili per pazienti di età ≥ 85 anni (14 pazienti [5,9 %] di età ≥ 85 anni sono stati inclusi nello studio AZA-AML-001).
Pediatria
Nello studio AZA-JMML-001, 28 bambini (di età compresa tra 1 mese e < 18 anni) sono stati trattati con azacitidina per MDS (n = 10) o leucemia mielomonocitica giovanile (JMML) (n = 18) (vedere sezione «Proprietà farmacologiche»).
Tutti e 28 i pazienti hanno manifestato almeno un evento avverso, e 17 (60,7 %) hanno manifestato almeno un evento correlato al trattamento. Le reazioni avverse più comuni nell’intera popolazione pediatrica sono state ipertermia, manifestazioni ematologiche, inclusi anemia, trombocitopenia e neutropenia febbrile, e disturbi gastrointestinali, tra cui costipazione e vomito.
In tre partecipanti sono stati osservati eventi correlati al trattamento che hanno portato all’interruzione del farmaco (ipertermia, progressione della malattia e dolore addominale).
Nello studio AZA-AML-004, 7 bambini (di età compresa tra 2 e 12 anni) sono stati trattati con azacitidina per AML in ricaduta molecolare dopo la prima remissione completa [CR1] (vedere sezione «Proprietà farmacologiche»).
Tutti e sette i pazienti hanno manifestato almeno un evento avverso correlato al trattamento. Le reazioni avverse più comunemente riportate sono state neutropenia, nausea, leucopenia, trombocitopenia, diarrea e aumento dei livelli di alanina aminotransferasi (ALT). In due pazienti si è verificato un evento correlato al trattamento che ha portato all’interruzione del farmaco (neutropenia febbrile, neutropenia).
Durante lo studio clinico condotto su un numero limitato di bambini trattati con azacitidina, non sono emersi nuovi segnali di allarme. Il profilo generale di sicurezza era coerente con quello della popolazione adulta.
Segnalazione delle reazioni avverse sospette
La segnalazione delle reazioni avverse dopo l’immissione in commercio del medicinale è di fondamentale importanza. Permette un monitoraggio continuo del rapporto beneficio/rischio del medicinale. Il personale medico e farmaceutico, così come i pazienti o i loro rappresentanti legali, devono segnalare tutti i casi sospetti di reazioni avverse e di mancata efficacia del medicinale attraverso il Sistema Informativo Automatizzato di Farmacovigilanza al seguente indirizzo: https://aisf.dec.gov.ua.
Durata della conservazione.
3 anni.
Dopo ricostituzione
Quando il medicinale Azacitidina Accord viene ricostituito con acqua per preparazioni iniettabili non refrigerata, la stabilità chimica e fisica della soluzione ricostituita durante l’uso è stata dimostrata a 25 °C per 60 minuti e a una temperatura compresa tra 2 °C e 8 °C per 8 ore.
La durata della conservazione della soluzione ricostituita può essere prolungata ricostituendo il prodotto con acqua per preparazioni iniettabili refrigerata (da 2 °C a 8 °C). Quando il medicinale Azacitidina Accord viene ricostituito con acqua per preparazioni iniettabili refrigerata (da 2 °C a 8 °C), la stabilità chimica e fisica della soluzione ricostituita durante l’uso è stata dimostrata a una temperatura compresa tra 2 °C e 8 °C per 22 ore.
Dal punto di vista microbiologico, il prodotto ricostituito deve essere utilizzato immediatamente. Se non utilizzato immediatamente, l’utente è responsabile della durata e delle condizioni di conservazione prima dell’uso, che non devono superare 8 ore a una temperatura compresa tra 2 °C e 8 °C nel caso di ricostituzione con acqua non refrigerata, e 22 ore nel caso di ricostituzione con acqua refrigerata (da 2 °C a 8 °C).
Condizioni di conservazione.
Il medicinale non richiede particolari condizioni di conservazione.
Conservare in luogo inaccessibile ai bambini.
Incompatibilità.
Non mescolare con altri medicinali, eccetto quelli indicati nella sezione «Modalità e posologia».
Confezionamento. 150 mg in un flaconcino, 1 flaconcino per confezione.
Categoria di dispensazione. Sotto prescrizione medica.
Produttore.
Accord Healthcare Polska Sp. z o.o. Sklad Importera/Accord Healthcare Polska Sp. z o.o. Magazyn Importera.
Indirizzo del produttore e sede operativa.
ul. Lutomierska 50, Pabianice, 95-200, Polonia/ul. Lutomierska 50, Pabianice, 95-200, Poland.
Titolo della richiesta. Accord Healthcare S.L.U.
Le richieste relative a problemi di qualità del medicinale, sicurezza d’uso, uso improprio o reclami sono accettate 24 ore su 24 (24/7) al numero: +380993100335 oppure via e-mail all’indirizzo: [email protected].
Indirizzo del titolare dell’autorizzazione all’immissione in commercio. World Trade Center, Moll de Barcelona, s/n, Edifici Est 6a planta, 08039 Barcelona, Spagna/World Trade Center, Moll de Barcelona, s/n, Edifici Est 6a planta, 08039 Barcelona, Spain.