RECOMBINATE
Włochy
Spis treści
- Ulotka: informacja dla użytkownika
- Recombinate 250 IU/5 ml proszek i rozpuszczalnik do sporządzenia roztworu do wstrzykiwań, 500 IU/5 ml proszek i rozpuszczalnik do sporządzenia roztworu do wstrzykiwań, 1000 IU/5 ml proszek i rozpuszczalnik do sporządzenia roztworu do wstrzykiwań
- 1. Co to jest Recombinate i do czego służy
- 2. Co należy wiedzieć przed zastosowaniem Recombinate
- 3. Jak stosować Recombinate
- 4. Możliwe działania niepożądane
- 5. Jak przechowywać Recombinate
- 6. Skład opakowania i inne informacje
- 1. NAZWA PRODUKTU LECZNICZEGO
- 2. SKŁAD JAKOŚCIOWY I ILOŚCIOWY
- 3. POSTAĆ LĘKU
- 4. INFORMACJE KLINICZNE
- 5. WŁAŚCIWOŚCI FARMACOLOGICZNE
- 6. DANE FARMACEUTYCZNE
- 7. WŁAŚCICIEL ZEZWOLENIA NA WPUSZCZENIE DO OBROTU
- 8. NUMER W REJESTRACJI PRODUKTU LECZNICZEGO
- 9. DATA PIERWSZEGO POZWOLENIA / ODNOWIONEGO POZWOLENIA
- 10. DATA PRZEGLĄDU TEKSTU
- Ulotka: informacje dla użytkownika
- Recombinate 250 IU/10 ml proszek i rozpuszczalnik do sporządzenia roztworu do wstrzykiwań, 500 IU/10 ml proszek i rozpuszczalnik do sporządzenia roztworu do wstrzykiwań, 1000 IU/10 ml proszek i rozpuszczalnik do sporządzenia roztworu do wstrzykiwań
- 1. Co to jest Recombinate i do czego służy
- 2. Co należy wiedzieć przed zastosowaniem Recombinate
- 3. Jak stosować Recombinate
- 4. Możliwe działania niepożądane
- 5. Jak przechowywać Recombinate
- 6. Skład opakowania i inne informacje
- 1. NAZWA PRODUKTU LECZNICZEGO
- 2. SKŁAD JAKOŚCIOWY I ILOŚCIOWY
- 3. POSTAĆ LEKU
- 4. INFORMACJE KLINICZNE
- 5. WŁAŚCIWOŚCI FARMACOLOGICZNE
- 6. DANE FARMACEUTYCZNE
- 7. WŁAŚCICIEL AUTORIZACJI DO WPUSZCZENIA NA RYNEK
- 8. NUMER W ZEZWOLENIA NA WPUSZCZENIE DO OBROTU
- 9. DATA PIERWSZEGO POZWOLENIA / ODNOWIENIA POZWOLENIA
- 10. DATA REWIZJI TEKSTU
Ulotka: informacja dla użytkownika
Recombinate 250 IU/5 ml proszek i rozpuszczalnik do sporządzenia roztworu do wstrzykiwań, 500 IU/5 ml proszek i rozpuszczalnik do sporządzenia roztworu do wstrzykiwań, 1000 IU/5 ml proszek i rozpuszczalnik do sporządzenia roztworu do wstrzykiwań
octocog alfa (rekombinowany czynnik VIII krzepnięcia krwi)
Przed zastosowaniem tego leku należy uważnie przeczytać ulotkę,
ponieważ zawiera ona ważne informacje dla Ciebie.
- Zachowaj tę ulotkę. Może Ci być potrzebna w przyszłości.
- W przypadku pytań skontaktuj się z lekarzem, farmaceutą lub pielęgniarką.
- Ten lek został przepisany wyłącznie dla Ciebie. Nie przekazuj go innym osobom, nawet jeśli objawy choroby są takie same jak u Ciebie, ponieważ może to być dla nich niebezpieczne.
- Jeśli wystąpią jakieś niepożądane działania, w tym te, których nie ma w tej ulotce, skontaktuj się z lekarzem, farmaceutą lub pielęgniarką.
Zawartość ulotki:
- Co to jest Recombinate i do czego służy
- Co należy wiedzieć przed zastosowaniem Recombinate
- Jak stosować Recombinate
- Możliwe działania niepożądane
- Jak przechowywać Recombinate
- Zawartość opakowania i inne informacje
1. Co to jest Recombinate i do czego służy
Recombinate należy do grupy farmakoterapeutycznej zwanej czynnik VIII krzepnięcia krwi.
Recombinate stosuje się u pacjentów z hemofilią A (wrodzony niedobór czynnika VIII) w celu:
- zapobiegania krwawieniom
- leczenia krwawień (np. krwawień z mięśni, jamy ustnej, krwawień w miejscu zabiegu chirurgicznego)
Recombinate nie zawiera czynnika von Willebranda i dlatego nie jest wskazany w chorobie von Willebranda (szczególnym zaburzeniu krzepnięcia krwi).
2. Co należy wiedzieć przed zastosowaniem Recombinate
Nie należy stosować Recombinate
- W przypadku nadwrażliwości na octocog alfa, białka pochodzące od myszy, bydła lub chomika albo na którykolwiek z pozostałych składników tego leku (wymienionych w punkcie 6). W razie wątpliwości należy skonsultować się z lekarzem.
Ostrzeżenia i środki ostrożności
W przypadku reakcji alergicznych:
- Istnieje rzadka możliwość wystąpienia reakcji anafilaktycznej (ciężkiej i nagłej reakcji alergicznej) na Recombinate. Należy znać wczesne objawy reakcji alergicznych, takie jak wysypka, pokrzywka, obrzęk, swędzenie całego ciała, obrzęk warg i języka, trudności z oddychaniem, duszność, ucisk w klatce piersiowej, ogólne złe samopoczucie i zawroty głowy. Te objawy mogą być wczesnym sygnałem szoku anafilaktycznego, którego objawy mogą obejmować również silne zawroty głowy, utratę przytomności oraz bardzo duże trudności z oddychaniem.
- W przypadku wystąpienia któregokolwiek z tych objawów należy natychmiast przerwać wlew. W przypadku ciężkich objawów, w tym trudności z oddychaniem i omdlenia (lub uczucia zemdlania), konieczne jest szybkie leczenie w trybie nagłym.
Kiedy konieczna jest kontrola:
- Lekarz może uznać za stosowne wykonanie badań w celu zapewnienia, że aktualna dawka jest wystarczająca do osiągnięcia i utrzymania odpowiedniego poziomu czynnika VIII. Jest to szczególnie ważne w przypadku większych zabiegów chirurgicznych. W przypadku trwających krwawień:
- Powstawanie inhibitorów (przeciwciał) to znana komplikacja, która może wystąpić podczas leczenia wszystkimi lekami zawierającymi czynnik VIII. Inhibitory, zwłaszcza o wysokim stężeniu, uniemożliwiają prawidłowe działanie leczenia, dlatego u pacjenta lub u dziecka będzie przeprowadzany dokładny monitoring w celu wykrycia rozwoju tych inhibitorów. Jeśli Recombinate nie kontroluje krwawienia u pacjenta lub u jego dziecka, należy natychmiast powiadomić lekarza.
Inne leki i Recombinate
Nie zaobserwowano niekorzystnych interakcji z innymi lekami.
Należy poinformować lekarza lub farmaceutę, jeśli pacjent przyjmuje, niedawno przyjmował lub może przyjmować inne leki.
Ciąża i karmienie piersią
Nie ma doświadczeń dotyczących stosowania Recombinate w czasie ciąży ani karmienia piersią, ponieważ hemofilia A jest rzadka u kobiet. Dlatego należy poinformować lekarza w przypadku ciąży lub karmienia piersią. Lekarz zadecyduje, czy Recombinate może być stosowany w czasie ciąży i karmienia piersią.
Kierowanie pojazdami i użytkowanie maszyn
Nie stwierdzono wpływu na zdolność prowadzenia pojazdów i użytkowania maszyn.
Recombinate zawiera sód
Ten lek zawiera 35 mg (1,5 mmol) sodu (główny składnik soli kuchennej) w każdym fiolce o zawartości 250 JI, 500 JI i 1000 JI. Odpowiada to 1,8% maksymalnej zalecanej dziennej dawki 2 g sodu dla dorosłego. Należy to wziąć pod uwagę u pacjentów przestrzegających diety ubogiej w sól.
3. Jak stosować Recombinate
Stosuj ten lek zawsze dokładnie zgodnie z instrukcjami lekarza doświadczonych w leczeniu pacjentów z hemofilią A.
Dawkowanie w profilaktyce przeciwhemoragicznej
Jeśli stosujesz Recombinate w celu zapobiegania (profilaktyki) krwawień, lekarz obliczy dawkę i poda Ci ją. Lekarz ustali dawkę zgodnie z Twoimi indywidualnymi potrzebami. Zwykle dawka wynosi 20–40 IU octocog alfa na kilogram masa ciała, podawana w odstępach co 2–3 dni. Jednak w niektórych przypadkach, szczególnie u młodszych pacjentów, mogą być wymagane krótsze odstępy lub wyższe dawki.
Jeśli uważasz, że działanie Recombinate jest niewystarczające, należy porozmawiać o tym z lekarzem.
Dawkowanie w leczeniu krwawień
Jeśli stosujesz Recombinate w celu leczenia epizodów krwawienia, lekarz obliczy dawkę dostosowaną do Twoich indywidualnych potrzeb, korzystając z poniższego wzoru:
IU potrzebne = masa ciała (kilogramy) x pożądane zwiększenie czynnika VIII (% normy) x 0,5
Poniższa tabela stanowi wskazówkę dotyczącą minimalnych poziomów czynnika VIII we krwi. W przypadku wymienionych zdarzeń krwotocznych aktywność czynnika VIII nie powinna spaść poniżej poziomu wskazanego (w % normy) w odpowiednim okresie.
W niektórych sytuacjach może być konieczne podanie większych ilości niż obliczone, szczególnie w przypadku obecności inhibitora o niskim mianie.
| Stopień krwawienia / Typ zabiegu chirurgicznego | Szczytowa aktywność czynnika AHF wymagana we krwi po infuzji (% normy lub j/dL osocza) | Częstotliwość infuzji |
| Stopień krwawienia Wczesne wewnętrrzne krwawienie do stawu lub krwawienie mięśniowe lub do jamy ustnej Rozszerzone wewnętrrzne krwawienie do stawu; krwawienie mięśniowe lub krwiak Krwawienia zagrażające życiu, takie jak krwawienie do mózgu, krwawienie do gardła lub ciężkie krwawienia brzuszne | 20 - 40 30 - 60 60 - 100 | Podawać co 12 - 24 godziny przez jeden do trzech dni, aż do ustąpienia epizodu krwawienia (na podstawie bólu) lub pełnego wyleczenia. Powtarzać infuzję co 12 - 24 godziny, zazwyczaj przez trzy dni lub dłużej, aż do ustąpienia bólu lub odzyskania funkcji. Powtarzać infuzję co 8 - 24 godziny, aż do ustąpienia zagrożenia. |
| Zabieg chirurgiczny Typ zabiegu Małe zabiegi chirurgiczne, w tym ekstrakcje zębów Duże zabiegi chirurgiczne | 30 - 60 80 - 100 (przed i po zabiegu) | Jedna infuzja, a także doustna terapia przeciwwijątkowa, podana w ciągu godziny przed zabiegiem, jest wystarczająca w około 70% przypadków. Co 24 godziny, co najmniej przez jeden dzień, aż do gojenia się rany. Powtarzać infuzję co 8 - 24 godziny, w zależności od postępu gojenia się rany. |
Stosowanie u dzieci
Recombinate może być stosowany zarówno u dorosłych, jak i u dzieci we wszystkich grupach wiekowych, w tym u noworodków. Zalecenia dotyczące dawkowania w leczeniu krwawień są takie same dla dorosłych i dzieci. W przypadku profilaktyki krwawień (zapobiegania), w niektórych przypadkach mogą być konieczne krótsze odstępy między dawkami lub większe dawki w porównaniu do standardowej dawki 20–40 j.m. czynnika VIII na kg masy ciała co 2–3 dni.
Monitorowanie przez lekarza
Lekarz przeprowadzi odpowiednie badania laboratoryjne, aby upewnić się, że osiągnięto odpowiedni poziom czynnika VIII. Jest to szczególnie ważne w przypadku większych zabiegów chirurgicznych.
Pacjenci z inhibitorami czynnika VIII
Jeśli stężenie czynnika VIII we krwi nie osiąga oczekiwanych wartości lub jeśli krwawienie nie jest odpowiednio kontrolowane pomimo zwiększenia dawki, należy podejrzewać obecność inhibitorów czynnika VIII. Obecność inhibitorów czynnika VIII zostanie sprawdzona przez lekarza.
W przypadku rozwoju inhibitorów czynnika VIII może być konieczne podanie większych ilości Recombinate w celu kontrolowania krwawienia. Jeśli dawka ta nie pozwala skutecznie kontrolować krwawienia, lekarz może rozważyć zastosowanie innego produktu. Nie zwiększaj całkowitej dawki Recombinate w celu kontrolowania krwawienia bez wcześniejszej konsultacji z lekarzem.
Sposób i droga podania
Recombinate podaje się dożylnie (w żyłę) po przygotowaniu roztworu za pomocą rozpuszczalnika zawartego w opakowaniu:
- jako wstrzyknięcie wykonywane przez lekarza lub pielęgniarkę
- jako wlewanie (infuzja) wykonywane przez lekarza lub pielęgniarkę
Szybkość podania należy dostosować do samopoczucia pacjenta. Preparat może być podawany z maksymalną szybkością do 10 ml na minutę.
Częstotliwość podawania
Lekarz ustali i poinformuje Cię o częstotliwości podawania Recombinate, zależnie od skuteczności leczenia w Twoim przypadku.
Czas trwania leczenia
Ogólnie terapia zastępcza Recombinate jest leczeniem na całe życie.
Jeśli podasz zbyt dużą dawkę Recombinate
- Nie odnotowano objawów przedawkowania rekombinowanym czynnikiem VIII krzepnięcia krwi. W przypadku wątpliwości skonsultuj się z lekarzem.
Jeśli zapomnisz podać dawkę Recombinate
- Nie podawaj podwójnej dawki, aby uzupełnić pominiętą dawkę.
- Natychmiast przejdź do kolejnej, regularnej dawki i kontynuuj podawanie w regularnych odstępach zgodnie z zaleceniem lekarza.
Jeśli przerwiesz leczenie Recombinate
Nie przerywaj stosowania Recombinate bez konsultacji z lekarzem ze względu na możliwość wystąpienia krwawień stanowiących zagrożenie życia.
Jeśli masz jakiekolwiek wątpliwości dotyczące stosowania tego leku, skorzystaj z porady lekarza, farmaceuty lub pielęgniarki.
4. Możliwe działania niepożądane
Tak jak wszystkie leki, ten lek może powodować działania niepożądane, choć nie u wszystkich osób
one występują.
Podczas stosowania tego produktu obserwowano następujące działania niepożądane: nudności, wymioty,
ból brzucha, napady ciepła, lekkie zmęczenie, zawroty głowy, uczucie ogólnego niedoboru samopoczucia, bóle głowy,
przemijające wysypki skórne (zaczerwienienie skóry), krwiaki, reakcje w miejscu wstrzyknięcia, potliwość,
dreszcze, drżenie, gorączkę, ból nóg, zimne ręce i stopy, uczucie mrowienia w rękach lub stopach,
ból gardła, infekcje uszu, niepowodzenie testów słuchowych, epistaksję i bladość skóry.
Rzadko odnotowano zdarzenia niepożądane spowodowane nadwrażliwością, w tym: uogólnone pokrzywki i świąd
(wysypkę skórną z silnym świądem i powstawaniem grudek), wysypkę, trudności w oddychaniu, kaszel,
ból lub uczucie ucisku w klatce piersiowej, świsty w klatce piersiowej, niskie ciśnienie krwi (hipotensję),
utratę przytomności, przyspieszone bicie serca, poważne reakcje nadwrażliwości, które mogą prowadzić
do trudności z połykaniem i/lub oddychaniem, obrzęk i zaczerwienienie twarzy i/lub rąk (anafilaksję).
W przypadku wystąpienia reakcji alergicznych lub anafilaktycznych należy natychmiast przerwać
wstrzykiwanie/infuzję i skontaktować się z lekarzem.
U dzieci, które wcześniej nie były leczone lekami zawierającymi czynnik VIII, tworzenie się przeciwciał
hamujących (patrz punkt 2) może być bardzo częste (u więcej niż 1 na 10 pacjentów); natomiast u pacjentów,
którzy wcześniej otrzymywali leczenie czynnikiem VIII (ponad 150 dni leczenia) ryzyko to jest rzadkie
(u mniej niż 1 na 100 pacjentów). Jeśli do tego dojdzie, lek u Ciebie lub u Twojego dziecka może przestać
właściwie działać i może wystąpić trwające krwawienie. W takim przypadku należy niezwłocznie skontaktować się
z lekarzem.
Zgłaszanie działań niepożądanych
Jeśli wystąpi u Ciebie jakiekolwiek działanie niepożądane, w tym również takie, których nie ma w tej ulotce,
skontaktuj się z lekarzem, farmaceutą lub pielęgniarką.
Możesz również zgłaszać działania niepożądane bezpośrednio za pośrednictwem krajowego systemu zgłaszania działań niepożądanych.
Zgłaszając działania niepożądane, możesz pomóc w dostarczeniu dodatkowych informacji na temat bezpieczeństwa
tego leku.
5. Jak przechowywać Recombinate
- Przechowuj ten lek w miejscu niedostępnym dla dzieci.
- Przechowuj w lodówce (2 °C – 8 °C).
- Nie zamrażaj.
- Przechowuj w opakowaniu zewnętrznym, aby chronić lek przed światłem.
- Nie stosuj tego leku po dacie wygaśnięcia podanej na etykiecie i na pudełku. Data wygaśnięcia znajduje się na pudełku po skrócie „Waz.”. Data wygaśnięcia odnosi się do ostatniego dnia danego miesiąca. W okresie ważności produkt może być przechowywany w temperaturze 15°C–25°C przed użyciem przez maksymalnie sześć miesięcy. Nie umieszczaj ponownie w lodówce po przechowywaniu w temperaturze 15°C–25°C. Po odtworzeniu, Recombinate należy podawać w temperaturze pokojowej w ciągu trzech godzin.
Przechowywanie po odtworzeniu
- Ten produkt przeznaczony jest do jednorazowego użycia. Użyj produktu w ciągu trzech godzin od momentu odtworzenia.
- Nie chłodź roztworu po odtworzeniu. Nie stosuj Recombinate, jeśli roztwór zawiera osad lub jest mętny. Nie wyrzucaj leków do ścieków ani do zwykłych śmieci. Zapytaj farmaceuty, jak pozbyć się leków, których już nie używasz. Pomoże to chronić środowisko.
6. Skład opakowania i inne informacje
Co zawiera Recombinate
- Substancją czynną jest octocog alfa, rekombinowany czynnik VIII krzepnięcia 50 IU/ml, 100 IU/ml lub 200 IU/ml. Produkt występuje w trzech dawkach: 250 IU, 500 IU lub 1000 IU (jednostek międzynarodowych) na fiolkę substancji czynnej.
- Pozostałe składniki to:
- dla proszku: albumina ludzka, chlorek sodu, histydyna, makrogol 3350, chlorek wapnia dwuwodny, kwas solny (do regulacji pH) i wodorotlenek sodu (do regulacji pH).
- dla rozpuszczalnika: woda do sporządzania roztworów do wstrzykiwań.
Wygląd zewnętrzny Recombinate i zawartość opakowania
Recombinate jest dostępne w postaci proszku i rozpuszczalnika do sporządzenia roztworu do wstrzykiwań, jako sypki proszek o barwie białej lub niemal białej. Po odtworzeniu roztwór jest klarowny, bezbarwny i pozbawiony zanieczyszczeń. Rozpuszczalnik (sterylna woda do sporządzania roztworów do wstrzykiwań) jest klarownym, bezbarwnym płynem.
Opakowanie zawiera fiolkę z 250 IU lub 500 IU lub 1000 IU proszku, fiolkę z 5 ml rozpuszczalnika, urządzenie do odtworzenia (BAXJECT II), strzykawkę jednorazową ze sterylnej plastiki, minizestaw do infuzji jednorazowy, 2 waty nasączone alkoholem oraz 2 plastry.
Zamiast Baxject II może zostać dostarczone urządzenie do odtworzenia z igłą, zawierające sterylną dwukierunkową igłę (do przeniesienia rozpuszczalnika do fiolki z Recombinate) oraz sterylną igłę-filtr (do przeniesienia odtworzonego roztworu do strzykawki).
Opakowanie jednostkowe.
Właściciel pozwolenia na dopuszczenie do obrotu
Baxalta Innovations GmbH
Industriestrasse 67, A-1221 Wiedeń
Pełnomocnik w Włoszech:
Takeda Italia S.p.A.
Tel. +39 06 502601
Producent
Baxalta Belgium Manufacturing SA
Bd. René Branquart 80, B-7860 Lessines,
Belgia
Niniejszy lek jest dopuszczony do obrotu w krajach członkowskich Europejskiego Obszaru Gospodarczego pod następującymi nazwami handlowymi:
Belgia: Recombinate 250 (500, 1000) IU/5 ml
Recombinate 250 (500, 1000) IU/10 ml
Bułgaria: Recombinate 250 (500, 1000) IU/5 ml
Cypr: Recombinate 250 (500, 1000) IU
Niemcy: Recombinate Antihämophilie Faktor (rekombinant) 1000
Grecja: Recombinate 250 (500, 1000) IU
Litwa: Recombinate 250 (500, 1000) IU/5 ml
Malta: Recombinate 250 (500, 1000) IU
Holandia: Recombinate 250 (500, 1000) IE/5 ml
Recombinate 250 (500, 1000) IE/10 ml
Estonia: Recombinate 250 (500, 1000) IU/5 ml
Irlandia: Recombinate 250 (500, 1000) IU
Włochy: Recombinate 250 (500, 1000) IU/5 ml
Recombinate 250 (500, 1000) IU/10 ml
Łotwa: Recombinate 250 (500, 1000) IU/5 ml
Następujące informacje są przeznaczone wyłącznie dla personelu medycznego:
CHARAKTERYSTYKA PRODUKTU LECZNICZEGO
1. NAZWA PRODUKTU LECZNICZEGO
Recombinate 250 IU/5 ml proszek i rozpuszczalnik do sporządzenia roztworu do wstrzykiwań
Recombinate 500 IU/5 ml proszek i rozpuszczalnik do sporządzenia roztworu do wstrzykiwań
Recombinate 1000 IU/5 ml proszek i rozpuszczalnik do sporządzenia roztworu do wstrzykiwań
2. SKŁAD JAKOŚCIOWY I ILOŚCIOWY
Octocog alfa 50 IU na ml odtworzonego roztworu
Po odtworzeniu: Fiolka o pojemności 5 ml zawiera 250 IU octocog alfa
Recombinate 250 IU/5 ml zawiera nominalnie 250 IU octocog alfa, rekombinowanego czynnika VIII
krzepnięcia, w każdej fiolce.
Produkt zawiera około 50 IU/ml octocog alfa, rekombinowanego czynnika VIII krzepnięcia, po odtworzeniu za pomocą 5 ml wody do wstrzykiwań.
Octocog alfa 100 IU na ml odtworzonego roztworu
Po odtworzeniu: Fiolka o pojemności 5 ml zawiera 500 IU octocog alfa
Recombinate 500 IU/5 ml zawiera nominalnie 500 IU octocog alfa, rekombinowanego czynnika VIII
krzepnięcia, w każdej fiolce.
Produkt zawiera około 100 IU/ml octocog alfa, rekombinowanego czynnika VIII krzepnięcia, po odtworzeniu za pomocą 5 ml wody do wstrzykiwań.
Octocog alfa 200 IU na ml odtworzonego roztworu
Po odtworzeniu: Fiolka o pojemności 5 ml zawiera 1000 IU octocog alfa
Recombinate 1000 IU/5 ml zawiera nominalnie 1000 IU octocog alfa, rekombinowanego czynnika VIII
krzepnięcia, w każdej fiolce.
Produkt zawiera około 200 IU/ml octocog alfa, rekombinowanego czynnika VIII krzepnięcia, po odtworzeniu za pomocą 5 ml wody do wstrzykiwań.
Aktywność oznacza się za pomocą testu chromogenicznego zgodnie z Europejską Farmakopeą, przy użyciu kalibracji Mega FDA według standardu WHO. Aktywność specyficzna Recombinate wynosi około 4000–8000 IU/mg białka.
Recombinate zawiera rekombinowany czynnik VIII krzepnięcia (INN: octocog alfa). Octocog alfa (rekombinowany czynnik VIII krzepnięcia) to białko oczyszczone, składające się z 2332 aminokwasów. Ma sekwencję aminokwasów porównywalną do czynnika VIII oraz modyfikacje potranslacyjne podobne do cząsteczki pochodzącej z osocza. Rekombinowany czynnik VIII krzepnięcia to glikoproteina wytwarzana w komórkach ssaków, otrzymanych metodami inżynierii biologicznej z linii komórkowej jajnika chomika chińskiego.
Substancje pomocnicze o znanym działaniu:
Każda fiolka zawiera 35 mg (1,5 mmol) sodu.
Pełna lista substancji pomocniczych znajduje się w punkcie 6.1.
3. POSTAĆ LĘKU
Proszek i rozpuszczalnik do sporządzenia roztworu do wstrzykiwań.
Proszek kruchy, od białego do lekko zabarwionego. Rozpuszczalnik (woda sterylizowana do sporządzenia roztworów do wstrzykiwań) to klarowny, bezbarwny płyn.
4. INFORMACJE KLINICZNE
4.1 Wskazania terapeutyczne
Leczenie i profilaktyka krwawień u pacjentów z hemofilią A (wrodzony niedobór czynnika VIII).
Ten produkt nie zawiera czynnika von Willebranda i w związku z tym nie jest wskazany w chorobie von Willebranda.
Recombinate jest wskazany dla wszystkich grup wiekowych, od noworodków po dorosłych.
4.2 Dawkowanie i sposób podania
Leczenie powinno być nadzorowane przez lekarza z doświadczeniem w leczeniu hemofilii.
Monitorowanie leczenia
W trakcie leczenia zaleca się odpowiednie oznaczanie poziomów czynnika VIII w celu ustalenia dawki do podania oraz częstotliwości powtarzania infuzji. Odpowiedź poszczególnych pacjentów na czynnik VIII może się różnić, co skutkuje różnymi okresami półtrwania i odzyskiem aktywności. Dawkowanie oparte na masie ciała może wymagać dostosowania u pacjentów z niedowagą lub otyłością. Szczególnie w przypadku większych zabiegów chirurgicznych konieczne jest dokładne monitorowanie terapii zastępczej za pomocą analiz krzepnięcia (aktywność czynnika VIII w osoczu).
Dawkowanie
Dawka i długość trwania terapii zastępczej zależą od nasilenia niedoboru funkcji hemostatycznej, lokalizacji i stopnia krwawienia oraz stanu klinicznego pacjenta.
Leczenie powinno być prowadzone we współpracy z lekarzem specjalizującym się w zaburzeniach krzepnięcia oraz z laboratorium potrafiącym oznaczać stężenie AHF (antygenu czynnika VIII) we krwi.
Podawaną liczbę jednostek czynnika VIII wyraża się w Jednostkach Międzynarodowych (J.M.), odnosząc się do obecnego standardu WHO dla produktów zawierających czynnik VIII. Aktywność czynnika VIII w osoczu wyraża się zarówno w procentach (w odniesieniu do osocza ludzkiego normalnego), jak i w Jednostkach Międzynarodowych (odnosząc się do międzynarodowego standardu dla czynnika VIII osocza).
Jedna Jednostka Międzynarodowa (J.M.) aktywności czynnika VIII odpowiada ilości czynnika VIII zawartej w 1 ml normalnego osocza ludzkiego.
Terapia na żądanie
Oczekiwany wzrost in vivo poziomu szczytowego Recombinate wyrażony w J.M./dL osocza lub w % (procentach) wartości normalnej można obliczyć, mnożąc dawkę podaną na kg masy ciała (J.M./kg) przez 2.
Metodę obliczeń przedstawiono w poniższych przykładach:
Oczekiwany wzrost % FVIII = liczba podanych jednostek × 2% / J.M. / kg
masa ciała (kg)
Przykład dla dorosłego o masie ciała 70 kg: 1750 J.M. × 2% / J.M. / kg = ok. 50%
70 kg
lub
Wymagana dawka (J.M.) = masa ciała (kg) × pożądany wzrost % FVIII
2% / J.M. / kg
Przykład dla dziecka o masie ciała 40 kg: 40 kg × 70% = 1400 J.M.
2% / J.M. / kg
Chociaż dawkę można oszacować za pomocą powyższego wzoru, zaleca się zdecydowanie przeprowadzenie, jeśli to możliwe, odpowiednich badań laboratoryjnych, w tym oznaczania stężenia AHF we krwi pacjenta w odpowiednich odstępach czasu, aby upewnić się, że osiągnięto i utrzymuje się odpowiedni poziom AHF. Jeśli u pacjenta nie osiąga się pożądanego poziomu plazmatycznego AHF lub jeśli krwawienie nie ustępuje po podaniu odpowiedniej dawki, należy podejrzewać obecność inhibitora. Odpowiednimi badaniami laboratoryjnymi można wykazać i zmierzyć obecność inhibitora, wyrażając ją w Jednostkach Międzynarodowych AHF neutralizowanych przez 1 ml osocza (jednostki Bethesda) lub w całkowitej oszacowanej objętości osocza. Jeśli inhibitor występuje w ilości mniejszej niż 10 jednostek Bethesda na 1 ml, podanie dodatkowego AHF może zneutralizować inhibitor. W takim przypadku podanie dodatkowych Jednostek Międzynarodowych AHF powinno prowadzić do oczekiwanego efektu. W tych przypadkach konieczne jest monitorowanie poziomu AHF za pomocą badań laboratoryjnych. Poziomy inhibitora powyżej 10 jednostek Bethesda na 1 ml mogą uniemożliwić lub uczynić niemożliwym kontrolowanie hemostazy za pomocą AHF z powodu zbyt wysokich dawek, które byłyby wymagane.
Poniższy schemat dawkowania podany w Tabeli I może być stosowany jako wskazówka dla dorosłych i dzieci. Podawaną ilość i częstotliwość infuzji należy zawsze dostosować do skuteczności klinicznej w poszczególnych przypadkach.
W zależności od przypadku i uznania lekarza Recombinate może być stosowany również w profilaktyce (krótko- lub długoterminowej) krwawień.
W przypadku poniższych zdarzeń krwotocznych aktywność czynnika VIII nie powinna spaść poniżej podanego poziomu aktywności plazmatycznej (w <% wartości normalnej>) w odpowiednim okresie. Poniższa tabela może być wykorzystana jako wskazówka do ustalenia dawki w przypadku krwawień oraz zabiegów chirurgicznych:
Tabela I: Schemat dawkowania
| Krwawienie | ||
| Stopień krwawienia | Poziom aktywności czynnika AHF wymagany we krwi po wstrzyknięciu (% normy lub UI/dL osocza) | Częstotliwość wstrzykiwań |
| Początkowe krwawienie do stawu, krwawienie mięśniowe lub krwawienie z jamy ustnej | 20 - 40 | Rozpocząć wstrzykiwanie co 12 - 24 godziny przez jeden do trzech dni, aż do ustąpienia epizodu krwawienia (na podstawie odczuwanego bólu) lub pełnego wyleczenia |
| Obfusze krwawienie do stawu, krwawienie mięśniowe lub siniak | 30 - 60 | Powtarzać wstrzykiwanie co 12 - 24 godziny, zazwyczaj przez trzy dni lub dłużej, aż do ustąpienia bólu i odzyskania funkcji |
| Krwawienia zagrożone dla życia, takie jak krwawienie wewnątrzczaszkowe, krwawienie z gardła lub ciężkie krwawienie jamy brzusznej | 60 - 100 | Powtarzać wstrzykiwanie co 8 - 24 godziny, aż do ustąpienia zagrożenia |
| Chirurgia | ||
| Typ zabiegu | ||
| Małe operacje, w tym ekstrakcje zębów | 30 - 60 | Jednorazowe wstrzyknięcie, a także doustna terapia przeciwwijąca się rozpuszczaniu skrzepu, podana w ciągu godziny przed zabiegiem, jest wystarczająca w około 70% przypadków. Następnie co 24 godziny przez co najmniej 1 dzień, aż do wyleczenia rany. |
| Duże operacje | 80 - 100 (przed i po zabiegu) | Powtarzać wstrzykiwanie co 8 - 24 godziny, w zależności od postępu gojenia rany. |
Dane przedstawione reprezentują szczytową aktywność AHF u pacjentów z oczekiwanym średniym okresem półtrwania Faktora VIII. Jeśli uznane zostanie za konieczne, szczytową aktywność należy zmierzyć w ciągu pół godziny od podania. U pacjentów z stosunkowo krótkim okresem półtrwania Faktora VIII może być konieczne zwiększenie dawki i/lub częstotliwości podawania.
Każdy fiolka Recombinate zawiera na etykiecie aktywność rekombinowanego Faktora Antyhemofilowego (Recombinate) wyrażoną w IU na fiolkę.
Oznaczenie aktywności odnosi się do Międzynarodowego Standardu WHO dla stężeń Faktora VIII:C. Przeprowadzone badania wykazały, że w celu dokładnego oznaczenia tych poziomów aktywności test aktywności należy przeprowadzać przy użyciu probówek i pipet z tworzywa sztucznego oraz podłoża zawierającego normalne poziomy Faktora von Willebranda.
Profilaktyka
W celu długoterminowej profilaktyki krwawień u pacjentów z ciężką hemofilią A typowe dawki wynoszą od 20 do 40 IU Faktora VIII na kg masy ciała co 2–3 dni.
Pacjentów należy monitorować pod kątem rozwoju inhibitorów Faktora VIII. W przypadku nieosiągnięcia przewidywanych poziomów aktywności Faktora VIII we krwi lub niemożliwości kontrolowania epizodu krwawienia odpowiednią dawką, należy przeprowadzić test w celu wykrycia ewentualnej obecności inhibitora Faktora VIII. U pacjentów z wysokimi poziomami inhibitora terapia oparta na Faktorze VIII może nie być skuteczna, należy więc rozważyć inne opcje terapeutyczne. Leczenie takich pacjentów powinno być powierzone lekarzom doświadczonym w leczeniu pacjentów z hemofilią.
Zobacz również punkt 4.4.
Populacja pediatryczna
Recombinate może być stosowany u dzieci w każdym wieku, w tym u noworodków (badania dotyczące bezpieczeństwa i skuteczności przeprowadzono zarówno u dzieci wcześniej leczonych, jak i u dzieci nieleczonych wcześniej: zobacz punkt 5.1). W leczeniu na żądanie dawkowanie u pacjentów pediatrycznych nie różni się od dawkowania u dorosłych. W długoterminowej profilaktyce krwawień u pacjentów z ciężką hemofilią A w niektórych przypadkach mogą być konieczne krótsze odstępy między dawkami lub większe dawki niż typowa dawka 20–40 IU Faktora VIII na kg masy ciała co 2–3 dni.
Sposób podania
Preparat należy podawać dożylnie po odtworzeniu za pomocą rozpuszczalnika dostarczonego w zestawie (zobacz punkt 6.6). Odtworzonego produktu nie należy chłodzić. Zaleca się podawanie Recombinate w temperaturze pokojowej nie później niż w ciągu 3 godzin od odtworzenia. Prędkość podania powinna zapewniać komfort pacjenta, maksymalnie do 10 ml/min. Przed i podczas podania Recombinate należy monitorować puls pacjenta. W przypadku wystąpienia istotnego wzrostu tętna zmniejszenie szybkości wlewu lub tymczasowe przerwanie wstrzykiwania zwykle szybko powoduje ustąpienie objawów (zobacz punkty 4.4 i 4.8).
Instrukcje dotyczące odtworzenia leku przed podaniem znajdują się w punkcie 6.6.
4.3 Przeciwwskazania
Nadwrażliwość na substancję czynną lub którykolwiek z substancji pomocniczych wymienionych w punkcie 6.1. Znana reakcja alergiczna na białka wołowe, mysie lub chomikowe.
4.4 Specjalne ostrzeżenia i środki ostrożności stosowania
Śledzenie
W celu poprawy śledzenia leków biologicznych nazwa i numer serii podanego leku powinny być jasno zarejestrowane.
Nadwrażliwość
Zgłaszano przypadki ciężkich reakcji alergicznych na Recombinate. Pacjentów z znaną nadwrażliwością na białka mysie, wołowe lub chomikowe należy leczyć ostrożnie. Pacjentów należy poinformować o wczesnych objawach reakcji nadwrażliwościowych, takich jak pokrzywka, uogólniona pokrzywka, uczucie ściskania w klatce piersiowej, świsty w klatce piersiowej, hipotensja i anafilaksja. W przypadku wystąpienia reakcji alergicznej lub anafilaktycznej należy natychmiast przerwać wstrzykiwanie lub wlew. W przypadku wstrząsu należy zastosować standardowe leczenie wstrząsu.
Inhibitory
Tworzenie się przeciwciał neutralizujących (inhibitorów) przeciwko Faktorowi VIII stanowi znaną komplikację w leczeniu pacjentów z hemofilią A. Takie inhibitory są zazwyczaj immunoglobulinami IgG skierowanymi przeciwko działaniu krzepnącemu Faktora VIII i są wyrażane w jednostkach Bethesdy (UB) na ml osocza metodą zmodyfikowaną. Ryzyko rozwoju inhibitorów jest powiązane z ciężkością choroby i czasem ekspozycji na Faktor VIII, jest większe w pierwszych 50 dniach ekspozycji, ale utrzymuje się przez całe życie, mimo że jest to niepowszechne ryzyko.
Znaczenie kliniczne rozwoju inhibitorów zależy od ich miana: inhibitory o niskim mianie występujące tymczasowo lub stale utrzymujące się na niskim poziomie mają mniejszy wpływ na ryzyko niewystarczającej odpowiedzi klinicznej niż inhibitory o wysokim mianie.
Ogólnie rzecz biorąc, wszyscy pacjenci leczeni produktami zawierającymi Faktor VIII krzepnięcia powinni być dokładnie monitorowani pod kątem rozwoju inhibitorów za pomocą odpowiednich obserwacji klinicznych i badań laboratoryjnych. Jeśli nie uzyskuje się oczekiwanych poziomów aktywności Faktora VIII we krwi lub jeśli krwawienie nie jest kontrolowane odpowiednią dawką, należy przeprowadzić badanie w celu ustalenia obecności inhibitorów Faktora VIII. U pacjentów z wysokimi poziomami inhibitora terapia Faktorem VIII może nie być skuteczna i należy rozważyć inne opcje terapeutyczne. Opieka nad tymi pacjentami powinna być powierzona lekarzom doświadczonym w leczeniu hemofilii i inhibitorów Faktora VIII.
Zdarzenia kardiovaskularne
U pacjentów z istniejącymi czynnikami ryzyka kardiovaskularnego terapia zastępcza FVIII może zwiększać ryzyko kardiovaskularne.
Powikłania związane z cewnikiem
Jeśli konieczne jest użycie centralnego urządzenia dostępu do żył (CVAD), należy wziąć pod uwagę ryzyko powikłań związanych z CVAD, w tym infekcji lokalnych, bakteriemii i zakrzepicy w miejscu założenia cewnika.
Populacja pediatryczna
Ostrzeżenia i środki ostrożności stosowania u pacjentów pediatrycznych nie różnią się od tych u pacjentów dorosłych.
Ten lek zawiera 35 mg (1,5 mmol) sodu w każdej fiolce o pojemności 250 IU, 500 IU i 1000 IU, co odpowiada 1,8% maksymalnej zalecanej dziennej dawki sodu według WHO, wynoszącej 2 g sodu dla dorosłego. Należy to wziąć pod uwagę u pacjentów poddawanych diecie ubogosodowej.
4.5 Interakcje z innymi lekami i inne formy interakcji
Nie przeprowadzono badań interakcji.
4.6 Niepłodność, ciąża i laktacja
Nie przeprowadzono badań rozrodu na zwierzętach z Faktorem VIII. Biorąc pod uwagę, że hemofilia A u kobiet jest zjawiskiem rzadkim, nie są dostępne dane eksperymentalne dotyczące stosowania Faktora VIII w czasie ciąży lub karmienia piersią. Faktor VIII należy zatem podawać w czasie ciąży i karmienia piersią tylko w przypadku wyraźnych wskazań.
4.7 Wpływ na zdolność prowadzenia pojazdów i obsługi urządzeń
Nie zaobserwowano wpływu na zdolność prowadzenia pojazdów i obsługi urządzeń.
4.8 Działania niepożądane
Tabela podsumowująca działania niepożądane
Poniższa tabela zawiera działania niepożądane zgłoszone w oparciu o doniesienia spontaniczne i badania kliniczne.
Poniższa tabela została sporządzona na podstawie klasyfikacji MedDRA według układów i narządów (SOC i Preferred Term).
Częstotliwość została oszacowana według następujących kryteriów: bardzo często (≥ 1/10), często (≥ 1/100, < 1/10), niezbyt często (≥ 1/1000, < 1/100), rzadko (≥ 1/10 000, < 1/1000), bardzo rzadko (< 1/10 000), nieznana (częstotliwość nie może być określona na podstawie dostępnych danych).
| Klasyfikacja wg systemów i narządów według MedDRA | Częstość | Preferowany termin MedDRA |
| Infekcje i inwazje | niepowszechna 1 | Infekcja ucha 1 |
| Choroby układu krwiotwórczego i chłonnego | niepowszechna (PTPs)1bardzo powszechna (PUPs)1 | Inhibicja czynnika VIII 1 |
| Zaburzenia układu odpornościowego | nieznana | Reakcja anafilaktyczna Nadwrażliwość2 |
| Choroby układu nerwowego | niepowszechna | Zawroty głowy Tremor |
| nieznana | Utrata przytomności Omdlenie Bóle głowy Parestezja | |
| Choroby serca | nieznana | Cyanosis Tachykardia |
| Choroby układu naczyniowego | niepowszechna | Krwawienie z nosa Gorączka Ematoma Hipotensja Bladość Odczucie zimna w kończynach |
| Choroby układu oddechowego, klatki piersiowej i śródpiersia | niepowszechna | Ból gardła i krtani |
| nieznana | Oddychanie ciężkie Kaszel Szumy oddechowe | |
| Choroby układu pokarmowego | niepowszechna | Nudności |
| nieznana | Wymioty Ból brzucha | |
| Choroby skóry i tkanki podskórnej | niepowszechna | Podrażnienie skóry Swędzenie Wysypka Wysypka makulopapularna |
| nieznana | Angioobrzęk Pokrzywka Łuszczenie się skóry Rumień | |
| Choroby układu mięśniowo-szkieletowego i tkanki łącznej | niepowszechna | Ból kończyn |
| Choroby układu ogólnego i stanów związanych z miejscem podania | powszechna | Dreszcze |
| niepowszechna | Odczucie zmęczenia Niedomaga gorączkowa | |
| nieznana | Niepokój Reakcje w miejscu wstrzyknięcia Ból w klatce piersiowej Odczucie ucisku w klatce piersiowej | |
| Badania diagnostyczne 1 | niepowszechna | Wynik testu stymulacji dźwiękowej nieprawidłowy |
Częstotliwość oparta jest na badaniach przeprowadzonych z wykorzystaniem wszystkich produktów zawierających czynnik VIII, które obejmowały pacjentów z ciężką hemofilią A. PTPs = pacjenci wcześniej leczeni, PUPs = pacjenci wcześniej nieleczoni.
Wczesne objawy reakcji nadwrażliwości to np. pokrzywka, duszność, kaszel, uczucie ucisku w klatce piersiowej, świsty w płucach, anafilaksja, wysypka, hipotensja, świąd, dreszcze, rumień, gorączka, sinica, tachykardia, wymioty, omdlenia, ból głowy. Zaleca się ostrożność u pacjentów znanym z alergicznych reakcji na składniki preparatu (patrz punkty 4.3. i 4.4.).
Opis wybranych niepożądanych odczynów
Rozwój przeciwciał neutralizujących (inhibitorów) może wystąpić u pacjentów z hemofilią A leczonych czynnikiem VIII, w tym Recombinate. Obecność inhibitorów może objawiać się niewystarczającą odpowiedzią kliniczną. W takich przypadkach zaleca się kontakt z wyspecjalizowanym ośrodkiem hemofilii.
Populacja pediatryczna
Podczas badań klinicznych nie stwierdzono różnic zależnych od wieku w zakresie niepożądanych odczynów, z wyjątkiem rozwoju inhibitorów u wcześniejszych nieleczonych dzieci (PUPs).
Zgłaszanie podejrzewanych niepożądanych odczynów
Zgłaszanie podejrzewanych niepożądanych odczynów, które występują po dopuszczeniu leku do obrotu, jest ważne, ponieważ pozwala na ciągłe monitorowanie stosunku korzyści do ryzyka. Operatorzy medyczni powinni zgłaszać wszelkie podejrzewane niepożądane odczyny za pośrednictwem strony https://www.aifa.gov.it/content/segnalazioni-reazioni-avverse . .
4.9 Przedawkowanie
Nie znane są objawy przedawkowania.
5. WŁAŚCIWOŚCI FARMACOLOGICZNE
5.1 Właściwości farmakodynamiczne
Kategoria farmakoterapeutyczna: leki przeciwwysiękowe: czynnik VIII krzepnięcia krwi. Kod ATC: B02BD02.
Skład czynnik VIII / czynnik von Willebranda składa się z dwóch cząsteczek (czynnik VIII i czynnik von Willebranda) o różnych funkcjach fizjologicznych.
Po wstrzyknięciu do pacjenta z hemofilią czynnik VIII wiąże się w krwiobiegu z czynnikiem von Willebranda.
Aktywowany czynnik VIII działa jako kofaktor aktywowanego czynnika IX, przyspieszając przekształcanie czynnika X w aktywowany czynnik X; ten z kolei przekształca protrombinę w trombinę, która z kolei przekształca fibrynogen w fibrynę, prowadząc do powstania skrzepu. Hemofilia A to dziedziczny zaburzony krzepnięcia krwi sprzężony z płcią, spowodowany obniżonym poziomem czynnika VIII:C i prowadzący do rozległych krwawień do stawów, mięśni lub narządów wewnętrznych, zarówno samoistnych, jak i przypadkowych lub spowodowanych urazami chirurgicznymi. Terapia zastępcza pozwala na zwiększenie stężenia czynnika VIII we krwi, umożliwiając tymczasową korektę niedoboru czynnika oraz korektę skłonności do krwawień.
Należy zaznaczyć, że roczna liczba epizodów krwawień (ABR) nie jest porównywalna między różnymi preparatami czynnika VIII ani między różnymi badaniami klinicznymi.
Populacja pediatryczna
Recombinate został zbadany u 71 dzieci nigdy wcześniej nie leczonych (PUPs), których średnia wieku w momencie pierwszej infuzji Recombinate wynosiła 10 miesięcy (zakres: 2 dni – 50 miesięcy). Produkt okazał się dobrze tolerowany i nie był związany z istotnymi niepożądanymi skutkami krótkoterminowymi. Jego skuteczność kliniczna była porównywalna do innych cząsteczek FVIII o pełnej długości łańcucha, zarówno w leczeniu krwawień ostrych, jak i w profilaktyce chirurgicznej (10 pacjentów poddano zabiegom chirurgicznym). Długoterminowe obserwacje u pacjentów wykazały częstość zdarzeń niepożądanych związanych z produktem na poziomie 0,86/1000 infuzji, z których żadne nie było poważne ani zagrażające życiu.
5.2 Właściwości farmakokinetyczne
Badania farmakokinetyczne przeprowadzone u 69 pacjentów wcześniej leczonych wykazały, że średni okres półtrwania Recombinate w krwiobiegu wynosi 14,6 ± 4,9 godziny (n = 67), wartość ta nie różni się istotnie statystycznie od okresu półtrwania HemofilM, czynnika antyhemofilowego (ludzkiego) uzyskanego z osocza (pdAHF), którego średni okres półtrwania wynosi 14,7 ± 5,1 godziny (n = 61). Rzeczywiste odzyskanie po wstrzyknięciu dawki 50 IU/kg po podaniu Recombinate wyniosło 123,9 ± 47,7 IU/dL (n = 23), co jest istotnie wyższe niż rzeczywiste odzyskanie po podaniu HemofilM, wynoszące 101,7 ± 31,6 IU/dL (n = 61). Jednakże obliczony stosunek odzyskania rzeczywistego do oczekiwanego (czyli wzrost aktywności czynnika VIII o 2% na 1 IU rAHF/kg masy ciała) dla Recombinate (121,2 ± 48,9%) jest podobny do wartości dla HemofilM (123,4 ± 16,4%).
Łącznie przeprowadzono 494 badania odzyskania u 68 pacjentów nigdy wcześniej nie leczonych.
Dwieście dwanaście badań odzyskania przeprowadzono w czasie leczenia epizodów krwawień; średnie rzeczywiste odzyskanie ± SD wyniosło 70,0 ± 37,9 IU/dL (N = 208, cztery odzyskania pominięto w analizie, ponieważ wartości znajdowały się poza zakresem referencyjnym). Wysoka zmienność wynika z szerokiego zakresu rzeczywiście podanych dawek, od 13,8 do 103,2 IU/kg (średnio ± SD: 36,0 ± 16,2, mediana: 30,2 IU/kg). Uwzględniając zmienne dawki, obliczono stosunek odzyskania rzeczywistego do przewidywanego, który wyniósł średnio 1,0 ± 0,3.
Łącznie przeprowadzono 68 badań odzyskania u pacjentów otrzymujących kolejne infuzje w ramach ciągłego leczenia istniejących epizodów krwawienia. Poziom rzeczywistego odzyskania FVIII został skorygowany o poziom FVIII przed infuzją. Średnie rzeczywiste odzyskanie ± SD wyniosło 88,6 ± 38,2 IU/dL (N = 66, dwa odzyskania pominięto w analizie, ponieważ wartości znajdowały się poza zakresem referencyjnym). Ponownie szeroki zakres rzeczywiście podanych dawek, od 18,5 do 85,7 IU/kg (średnio ± SD: 38,6 ± 15,9, mediana: 32,1 IU/kg), spowodował znaczną zmienność obserwowanych poziomów odzyskania. Średni ± SD stosunek odzyskania rzeczywistego do przewidywanego wyniósł 1,0 ± 0,3, mediana: 1,0.
Łącznie przeprowadzono 214 badań odzyskania u pacjentów w stanie ustabilizowanym; średnie rzeczywiste odzyskanie wyniosło 71,6 ± 29,7 IU/dL (N = 209, pięć odzyskań pominięto w analizie, ponieważ wartości znajdowały się poza zakresem referencyjnym). Dawkowane dawki wahały się od 10,4 do 68,1 IU/kg (średnio ± SD: 38,0 ± 12,7, mediana: 36,1 IU/kg). Średni ± SD stosunek odzyskania rzeczywistego do przewidywanego wyniósł 1,0 ± 0,3.
5.3 Dane przedkliniczne dotyczące bezpieczeństwa
Recombinate działa jak endogenny czynnik VIII. Dawkowanie wielokrotnie wyższe niż zalecane u ludzi w przeliczeniu na kg masy ciała nie wykazało toksycznych skutków w badaniach na zwierzętach laboratoryjnych. Recombinate został poddany badaniom mutagenności zarówno in vitro przy dawkach znacznie przekraczających stężenie AHF we krwi, jak i in vivo przy dawkach do 10-krotnie wyższych niż maksymalna dawka sugerowana w klinice, bez wywoływania mutacji odwracalnych, aberracji chromosomowych ani wzrostu liczby mikrojąder w policromatycznych erytrocytach szpiku kostnego.
Ponieważ doświadczenie kliniczne nie dostarcza żadnych dowodów na działanie rakotwórcze i mutagenne, nie uznano za konieczne przeprowadzanie długoterminowych badań oceniających potencjalną rakotwórczość u zwierząt.
6. DANE FARMACEUTYCZNE
6.1 Wykaz substancji pomocniczych
Proszek:
Albumina ludzka
Chlorek sodu
Histydyna
Macrogol 3350
Chlorek wapnia dwuwodny
Kwas solny (do regulacji pH)
Wodorotlenek sodu (do regulacji pH)
Roztwórnik:
Woda do sporządzania zastrzyków
6.2 Niekompatybilności
W przypadku braku badań zgodności, tego leku nie wolno mieszać z innymi lekami.
Należy wyłącznie używać zestawu do infuzji zawartego w opakowaniu, ponieważ leczenie może się nie udać z powodu adsorpcji czynnika VIII krzepnięcia krwi do powierzchni wewnętrznej niektórych urządzeń do infuzji.
6.3 Okres ważności
3 lata. Po odtworzeniu, Recombinate nie powinien być przechowywany w lodówce i należy podać w ciągu trzech godzin.
6.4 Szczególne środki ostrożności dotyczące przechowywania
Przechowywać w lodówce (2°C – 8°C).
Nie zamarzać.
Przechowywać w opakowaniu zewnętrznym, aby chronić lek przed światłem.
W okresie ważności lek może być przechowywany do sześciu miesięcy w temperaturze 15°C – 25°C przed użyciem.
Nie wolno ponownie chłodzić po przechowywaniu w temperaturze 15–25°C.
Warunki przechowywania po odtworzeniu leku podano w punkcie 6.3.
6.5 Rodzaj i zawartość opakowania
Jedno opakowanie zawiera fiolkę z proszkiem, fiolkę z 5 ml rozpuszczalnika (obie ze szkła typu I z korkami gumowymi) oraz urządzenie do odtworzenia (BAXJECT II) + jednorazową strzykawkę plastikową + minizestaw do infuzji + 2 waty alkoholowe + 2 plastry.
Alternatywnie, zamiast BAXJECT II, opakowanie może zawierać urządzenie z igłą do odtworzenia, składające się z dwustronnej sterylnej igły (do przetransferowania rozpuszczalnika do fiolki z Recombinate) oraz sterylnej igły filtracyjnej (do przetransferowania odtworzonego roztworu do strzykawki).
Opakowanie jednostkowe.
6.6 Szczególne środki ostrożności dotyczące usuwania i manipulowania
Preparat należy podawać dożylnie po odtworzeniu za pomocą sterylnej wody do sporządzania zastrzyków zawartej w opakowaniu. Należy użyć jednorazowej plastikowej strzykawki zawartej w opakowaniu.
- Użyć w ciągu 3 godzin od odtworzenia.
- Nie chłodzić po odtworzeniu.
- Nieużywany lek oraz odpady pochodzące z tego leku należy usuwać zgodnie z obowiązującymi przepisami lokalnymi.
- Roztwór powinien być klarowny lub lekko mleczny. Nie należy stosować roztworów mętnych lub z osadami. Przed podaniem odtworzone produkty należy sprawdzić wizualnie pod kątem obecności cząstek lub nietypowego zabarwienia.
- Nie należy stosować, jeśli produkt, jego sterylny system barierowy lub opakowanie są uszkodzone lub wykazują jakiekolwiek oznaki degradacji.
| Rekonstytucja: Używać techniki bezpyłowej | |
| Rekonstytucja za pomocą BAXJECT II | Rekonstytucja za pomocą igieł |
1. Przygotować Recombinate (w postaci proszku) i Wodę do wstrzykiwań (rozpuszczalnik) do temperatury 15°C–25°C. 2. Usunąć pokrywki z fiolki zawierającej liofilizat i fiolki z rozpuszczalnikiem. 3. Dezynfekować septa za pomocą waty nasączonej alkoholem. Postawić fiolki na płaskiej powierzchni. 4. Otworzyć opakowanie urządzenia BAXJECT II, usuwając papierową osłonę, nie dotykając wnętrza (Ryc. a). Nie wyciągać urządzenia z opakowania. 5. Odwrócić pudełko, wsunąć przezroczysty końcówkę plastikową przez septum fiolki z rozpuszczalnikiem. Chwycić brzeg pudełka i wyciągnąć je, zwalniając urządzenie BAXJECT II (Ryc. b). Nie usuwać niebieskiej osłonki z urządzenia BAXJECT II. 6. Trzymając BAXJECT II podłączony do fiolki z rozpuszczalnikiem, odwrócić układ tak, aby fiolka z rozpuszczalnikiem znalazła się w górnej części urządzenia. Wprowadzić białą plastikową końcówkę przez septum Recombinate. Rozpuszczalnik zostanie wessany do fiolki pod zmniejszonym ciśnieniem zawierającej Recombinate (Ryc. c). 7. Delikatnie wymieszać, aż do całkowitego rozpuszczenia się substancji. Upewnić się, że Recombinate jest całkowicie rozpuszczony, w przeciwnym razie substancja czynna nie przejdzie przez filtr urządzenia. Produkt rozpuszcza się szybko (zwykle w mniej niż minutę). Ryc. a Ryc. b Ryc. c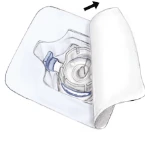    | 1. Przygotować Recombinate (w postaci proszku) i Wodę do wstrzykiwań (rozpuszczalnik) do temperatury 15°C–25°C. 2. Usunąć pokrywki z fiolki zawierającej liofilizat i fiolki z rozpuszczalnikiem. 3. Dezynfekować septa za pomocą waty nasączonej alkoholem. Postawić fiolki na płaskiej powierzchni. 4. Usunąć osłonę z jednego końca dwustronnej igły i włożyć odsłonięty koniec igły przez septum fiolki z rozpuszczalnikiem. 5. Usunąć osłonę z drugiego końca dwustronnej igły. Odwrócić fiolkę z rozpuszczalnikiem nad fiolką z Recombinate trzymaną w pionie i szybko wsunąć wolny koniec igły w środek septum fiolki z Recombinate. Rozpuszczalnik przepłynie do fiolki z liofilizatem dzięki podciśnieniu. 6. Rozłączyć obie fiolki, usuwając igłę z septum fiolki z rozpuszczalnikiem, a następnie usunąć igłę z fiolki z Recombinate. Delikatnie wymieszać, aż do całkowitego rozpuszczenia się substancji. Upewnić się, że Recombinate jest całkowicie rozpuszczony, w przeciwnym razie substancja czynna pozostanie w filtrze igły. |
| Podanie: Używać techniki bezpyłowej | |
Zaleca się rozpoczęcie podania nie później niż w ciągu trzech godzin od momentu rekonstytucji. Odtworzony produkt nie powinien być chłodzony. Przed podaniem leki do stosowania parenteralnego należy poddać kontroli wizualnej pod kątem obecności cząstek lub nieprawidłowego zabarwienia, o ile pozwala na to roztwór i opakowanie. Dla Recombinate dopuszczalne jest zabarwienie od bezbarwnego do lekko żółtawego. 1. Usunąć niebieską osłonkę z BAXJECT II. NIE WSYSIAC POWIETRZA DO SZYPRYNGI. Podłączyć szprygę do BAXJECT II (Ryc. d). 2. Odwrócić układ (tak aby fiolka z koncentratem znalazła się w górnej części urządzenia). Wessanie koncentratu do szpryngi poprzez powolne wyciąganie tłoczka (Ryc. e). 3. Odłączyć szprygę. 4. Podłączyć zestaw do podania do szpryngi. Wprowadzić dożylnie. Preparat można podawać z prędkością do 10 ml na minutę. Przed i podczas podawania Recombinate należy kontrolować puls pacjenta. W przypadku wystąpienia istotnego wzrostu częstości tętna, zmniejszenie szybkości infuzji lub tymczasowa przerwa w podawaniu leku zwykle szybko powodują ustąpienie objawów (patrz punkty 4.4 i 4.8). Ryc. d Ryc. e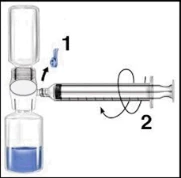 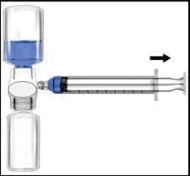 | Zaleca się rozpoczęcie podania nie później niż w ciągu trzech godzin od momentu rekonstytucji. Odtworzony produkt nie powinien być chłodzony. Przed podaniem leki do stosowania parenteralnego należy poddać kontroli wizualnej pod kątem obecności cząstek lub nieprawidłowego zabarwienia, o ile pozwala na to roztwór i opakowanie. Dla Recombinate dopuszczalne jest zabarwienie od bezbarwnego do lekko żółtawego. 1. Podłączyć igłę filtracyjną do jednorazowej szpryngi i wyciągnąć tłoczek, aby wprowadzić powietrze do szpryngi. 2. Wprowadzić igłę filtracyjną do odtworzonego Recombinate. 3. Wprowadzić powietrze do fiolki, a następnie wessanie odtworzonego materiału do szpryngi. 4. Usunąć i wyrzucić igłę filtracyjną. Podłączyć zestaw do podania do szpryngi. Wprowadzić dożylnie. Preparat można podawać z prędkością do 10 ml na minutę. Przed i podczas podawania Recombinate należy kontrolować puls pacjenta. W przypadku wystąpienia istotnego wzrostu częstości tętna, zmniejszenie szybkości infuzji lub tymczasowa przerwa w podawaniu leku zwykle szybko powodują ustąpienie objawów (patrz punkty 4.4 i 4.8). 5. Aby wessanie zawartości każdej fiolki odtworzonego Recombinate, należy użyć nowej, nieużywanej igły filtracyjnej. |
7. WŁAŚCICIEL ZEZWOLENIA NA WPUSZCZENIE DO OBROTU
Baxalta Innovations GmbH
Industriestrasse 67, A-1221 Wiedeń
8. NUMER W REJESTRACJI PRODUKTU LECZNICZEGO
WRA nr 028687046: „250 J.I./5 ml proszek i rozpuszczalnik do sporządzenia roztworu do wstrzykiwań” 1 fiolka z proszkiem + 1 fiolka z rozpuszczalnikiem z urządzeniem do rekonstytucji bez igły
WRA nr 028687073: „250 J.I./5 ml proszek i rozpuszczalnik do sporządzenia roztworu do wstrzykiwań” 1 fiolka z proszkiem + 1 fiolka z rozpuszczalnikiem z urządzeniem do rekonstytucji z dwugłowicową igłą + igła filtracyjna
WRA nr 028687059: „500 J.I./5 ml proszek i rozpuszczalnik do sporządzenia roztworu do wstrzykiwań” 1 fiolka z proszkiem + 1 fiolka z rozpuszczalnikiem z urządzeniem do rekonstytucji bez igły
WRA nr 028687085: „500 J.I./5 ml proszek i rozpuszczalnik do sporządzenia roztworu do wstrzykiwań” 1 fiolka z proszkiem + 1 fiolka z rozpuszczalnikiem z urządzeniem do rekonstytucji z dwugłowicową igłą + igła filtracyjna
WRA nr 028687061: „1000 J.I./5 ml proszek i rozpuszczalnik do sporządzenia roztworu do wstrzykiwań” 1 fiolka z proszkiem + 1 fiolka z rozpuszczalnikiem z urządzeniem do rekonstytucji bez igły
WRA nr 028687097: „1000 J.I./5 ml proszek i rozpuszczalnik do sporządzenia roztworu do wstrzykiwań” 1 fiolka z proszkiem + 1 fiolka z rozpuszczalnikiem z urządzeniem do rekonstytucji z dwugłowicową igłą + igła filtracyjna
9. DATA PIERWSZEGO POZWOLENIA / ODNOWIONEGO POZWOLENIA
Data pierwszego pozwolenia: 23 października 2013
Data odnowienia pozwolenia: 9 kwietnia 2018
10. DATA PRZEGLĄDU TEKSTU
Ulotka: informacje dla użytkownika
Recombinate 250 IU/10 ml proszek i rozpuszczalnik do sporządzenia roztworu do wstrzykiwań, 500 IU/10 ml proszek i rozpuszczalnik do sporządzenia roztworu do wstrzykiwań, 1000 IU/10 ml proszek i rozpuszczalnik do sporządzenia roztworu do wstrzykiwań
Octocog alfa (rekombinowany czynnik VIII krzepnięcia krwi)
Przed zastosowaniem tego leku dokładnie przeczytaj cały ten ulotnik, ponieważ zawiera on ważne informacje dla Ciebie.
- Zachowaj ten ulotnik. Może się okazać konieczne, aby przeczytać go ponownie.
- Jeżeli masz jakiekolwiek wątpliwości, skontaktuj się z lekarzem, farmaceutą lub pielęgniarką.
- Ten lek został przepisany wyłącznie dla Ciebie. Nie przekazuj go innym osobom, nawet jeśli objawy ich choroby są takie same jak Twoje, ponieważ może to być dla nich niebezpieczne.
- Jeżeli wystąpią u Ciebie jakiekolwiek działania niepożądane, w tym te, których nie ma w niniejszym ulotniku, skontaktuj się z lekarzem, farmaceutą lub pielęgniarką.
Zawartość tego ulotnika:
- Co to jest Recombinate i do czego służy
- Co należy wiedzieć przed zastosowaniem Recombinate
- Jak stosować Recombinate
- Możliwe działania niepożądane
- Jak przechowywać Recombinate
- Zawartość opakowania i inne informacje
1. Co to jest Recombinate i do czego służy
Recombinate należy do grupy farmakoterapeutycznej zwanej czynnikiem VIII krzepnięcia krwi.
Recombinate stosuje się u pacjentów z hemofilią A (wrodzony niedobór czynnika VIII) w celu:
- zapobiegania krwawieniom
- leczenia krwawień (np. krwawień mięśniowych, jamy ustnej, krwawień w miejscu operacyjnym)
Recombinate nie zawiera czynnika von Willebranda i dlatego nie jest wskazany w chorobie von Willebranda (szczególne zaburzenie krzepnięcia krwi).
2. Co należy wiedzieć przed zastosowaniem Recombinate
Nie stosować Recombinate
- Jeśli jest Pan(i) uczulony(a) na octocog alfa, białka pochodzące od myszy, bydła lub chomika lub na którykolwiek z pozostałych składników tego leku (wymienionych w punkcie 6). W przypadku wątpliwości należy skonsultować się z lekarzem.
Ostrzeżenia i środki ostrożności
W przypadku reakcji alergicznych:
- Istnieje rzadka możliwość wystąpienia reakcji anafilaktycznej (ciężkiej i nagłej reakcji alergicznej) na Recombinate. Należy znać wczesne objawy reakcji alergicznych, takie jak wysypka, pokrzywka, grudki, świąd całego ciała, obrzęk warg i języka, trudności z oddychaniem, duszność, ucisk w klatce piersiowej, ogólne uczucie niedoboru i zawroty głowy. Te objawy mogą być wczesnym sygnałem szoku anafilaktycznego, którego przebieg może również obejmować silne zawroty głowy, utratę przytomności oraz skrajne trudności z oddychaniem.
- W przypadku wystąpienia któregokolwiek z tych objawów należy natychmiast przerwać infuzję. W przypadku ciężkich objawów, w tym trudności z oddychaniem i omdleniem (lub uczuciem omdlenia), wymagana jest natychmiastowa pomoc w trybie nagłym.
Kiedy konieczne jest monitorowanie:
- Lekarz może uznać za stosowne przeprowadzenie badań w celu upewnienia się, że obecna dawka jest wystarczająca do osiągnięcia i utrzymania odpowiedniego poziomu czynnika VIII. Jest to szczególnie ważne w przypadku większych zabiegów chirurgicznych. W przypadku trwających krwawień:
- Powstawanie inhibitorów (przeciwciał) to znana komplikacja, która może wystąpić podczas leczenia wszystkimi lekami zawierającymi czynnik VIII. Inhibitory, zwłaszcza na wysokim poziomie, uniemożliwiają prawidłowe działanie leczenia, dlatego Pana(i) lub dziecko będą poddawane dokładnemu monitorowaniu pod kątem rozwoju tych inhibitorów. Jeśli Recombinate nie kontroluje krwawienia u Pana(i) lub u dziecka, należy natychmiast powiadomić lekarza.
Inne leki i Recombinate
Nie zaobserwowano niekorzystnych interakcji z innymi lekami.
Należy poinformować lekarza lub farmaceutę, jeśli Pana(i) stosuje, niedawno stosował(a) lub może zacząć stosować jakikolwiek inny lek.
Ciąża i karmienie piersią
Nie ma doświadczeń dotyczących stosowania Recombinate w czasie ciąży lub karmienia piersią, ponieważ hemofilia A jest rzadka u kobiet. Dlatego należy poinformować lekarza w przypadku ciąży lub karmienia piersią. Lekarz zadecyduje, czy Recombinate może być stosowany w czasie ciąży i karmienia piersią.
Kierowanie pojazdami i obsługa maszyn
Nie zaobserwowano wpływu na zdolność kierowania pojazdami lub korzystania z maszyn.
Recombinate zawiera sód
Ten lek zawiera 35 mg (1,5 mmol) sodu (główny składnik soli kuchennej) w każdym fiolce o mocy 250 J, 500 J i 1000 J. Odpowiada to 1,8% maksymalnej zalecanej dziennej dawki sodu wynoszącej 2 g dla dorosłego. Należy to uwzględnić u pacjentów przestrzegających diety ubogiej w sól.
3. Jak stosować Recombinate
Zawsze stosuj Recombinate zgodnie z dokładnymi wskazówkami lekarza doświadczonego w leczeniu pacjentów z hemofilią A.
Dawkowanie w profilaktyce przeciwhemoragicznej
Jeśli stosujesz Recombinate w celu zapobiegania (profilaktyce) krwawieniom, lekarz obliczy dawkę odpowiednią dla Ciebie i poinformuje Cię o niej. Lekarz ustali dawkę na podstawie Twoich indywidualnych potrzeb.
Zwykle dawka wynosi 20–40 IU octocog alfa na kilogram wagi ciała, podawana w odstępach co 2–3 dni. Jednak w niektórych przypadkach, szczególnie u młodszych pacjentów, mogą być wymagane krótsze odstępy lub wyższe dawki.
Jeśli podejrzewasz, że działanie Recombinate jest niewystarczające, należy porozmawiać o tym z lekarzem.
Dawkowanie w leczeniu krwawień
Jeśli stosujesz Recombinate w celu leczenia epizodów krwawień, lekarz obliczy dawkę dostosowaną do Twoich indywidualnych potrzeb, stosując poniższy wzór:
IU potrzebne = waga ciała (kilogramy) x pożądane zwiększenie czynnika VIII (% normy) x 0,5
Poniższa tabela stanowi wytyczne dotyczące minimalnych poziomów czynnika VIII we krwi. W przypadku wymienionych zdarzeń krwotocznych aktywność czynnika VIII nie powinna spaść poniżej poziomu wskazanego (w % normy) w odpowiednim okresie.
W niektórych sytuacjach mogą być wymagane większe dawki niż te obliczone, szczególnie w przypadku obecności inhibitora o niskim mianie.
| Stopień krwawienia / Typ zabiegu chirurgicznego | Szczytowa aktywność czynnika AHF wymagana we krwi po infuzji (% normy lub j/dL osocza) | Częstotliwość infuzji |
| Stopień krwawienia Początkowy hemartroz; krwawienie mięśniowe lub jamy ustnej Rozszerzony hemartroz; krwawienie mięśniowe lub krwawiącego guza Krwawienia zagrażające życiu, takie jak krwotoki wewnątrzczaszkowe, krwawienia z gardła lub poważne krwawienia jamy brzusznej | 20 - 40 30 - 60 60 - 100 | Podawać co 12 - 24 godziny przez jeden do trzech dni, aż do ustąpienia epizodu krwawienia (na podstawie bólu) lub pełnego wyleczenia. Powtarzać infuzję co 12 - 24 godziny, zazwyczaj przez trzy dni lub dłużej, aż do ustąpienia bólu lub odzyskania funkcji. Powtarzać infuzję co 8 - 24 godziny, aż do ustąpienia zagrożenia. |
| Zabieg chirurgiczny Typ zabiegu Mały zabieg chirurgiczny, w tym ekstrakcje zębów Duży zabieg chirurgiczny | 30-60 80 -100 (przed i po zabiegu) | Jednorazowa infuzja, a także doustna terapia przeciwpłytkowa, w ciągu godziny przed zabiegiem, jest wystarczająca w około 70% przypadków. Co 24 godziny, przez co najmniej jeden dzień, aż do gojenia rany. Powtarzać infuzję co 8 - 24 godziny, w zależności od postępu gojenia rany. |
Stosowanie u dzieci
Recombinate może być stosowany zarówno u dorosłych, jak i u dzieci we wszystkich grupach wiekowych, w tym u noworodków. Zalecenia dotyczące dawkowania w leczeniu krwawień, wymienione powyżej, są takie same zarówno dla dorosłych, jak i dla dzieci. W przypadku profilaktyki krwawień (zapobiegania), w niektórych przypadkach mogą być konieczne krótsze odstępy między dawkami lub większe dawki w porównaniu z normalną dawką 20–40 j.m. czynnika VIII na kg masy ciała co 2–3 dni.
Monitorowanie przez lekarza
Lekarz wykona odpowiednie badania laboratoryjne, aby upewnić się, że osiągnięto odpowiedni poziom czynnika VIII. Jest to szczególnie ważne w przypadku większych zabiegów chirurgicznych.
Pacjenci z inhibitorami czynnika VIII
Jeśli poziom czynnika VIII we krwi nie osiąga oczekiwanych wartości lub jeśli krwawienie nie jest odpowiednio kontrolowane mimo zwiększenia dawki, należy podejrzewać obecność inhibitorów czynnika VIII. Obecność inhibitorów czynnika VIII zostanie sprawdzona przez lekarza.
W przypadku rozwoju inhibitorów czynnika VIII, do kontrolowania krwawienia może być konieczne zastosowanie większych ilości Recombinate. Jeśli dawka ta nie pozwala skontrolować krwawienia, lekarz może rozważyć zastosowanie innego produktu. Nie zwiększaj samodzielnie całkowitej dawki Recombinate w celu kontrolowania krwawienia bez wcześniejszej konsultacji z lekarzem.
Sposób i droga podania
Recombinate podaje się dożylnie po przygotowaniu roztworu za pomocą rozpuszczalnika zawartego w opakowaniu:
- przez zastrzyk wykonywany przez lekarza lub pielęgniarkę,
- przez wlew (infuzję) wykonywaną przez lekarza lub pielęgniarkę.
Szybkość podania należy dostosować do stanu pacjenta. Preparat może być podawany z maksymalną szybkością do 10 ml na minutę.
Częstotliwość podawania
Lekarz ustali i poinformuje o częstotliwości podawania Recombinate w zależności od skuteczności leczenia w Twoim indywidualnym przypadku.
Czas trwania leczenia
Zazwyczaj leczenie wspomagające Recombinate jest leczeniem na całe życie.
Jeśli podasz zbyt dużą dawkę Recombinate
- Nie odnotowano objawów przedawkowania rekombinowanym czynnikiem VIII krzepnięcia. W razie wątpliwości skontaktuj się z lekarzem.
Jeśli zapomnisz podać dawkę Recombinate
- Nie podawaj podwójnej dawki, aby nadrobić pominiętą dawkę.
- Natychmiast przejdź do kolejnej, regularnej dawki i kontynuuj podawanie w regularnych odstępach zgodnie z zaleceniem lekarza.
Jeśli przerwiesz leczenie Recombinate
Nie przerywaj stosowania Recombinate bez konsultacji z lekarzem ze względu na możliwość wystąpienia krwawień zagrażających życiu.
Jeśli masz jakiekolwiek wątpliwości dotyczące stosowania tego leku, skontaktuj się z lekarzem, farmaceutą lub pielęgniarką.
4. Możliwe działania niepożądane
Tak jak wszystkie leki, ten lek może powodować działania niepożądane, choć nie u wszystkich osób
one występują.
Podczas stosowania tego produktu obserwowano następujące działania niepożądane: nudności, wymioty,
ból brzucha, zawroty głowy, uczucie ogólnego zmęczenia, zawroty głowy, ogólne uczucie niedoboru, ból głowy,
przejściowe wysypki skórne (zaczerwienienie skóry), siniaki, reakcje w miejscu wstrzyknięcia, potliwość,
dreszcze, drżenie, gorączkę, ból nóg, zimne ręce i stopy, uczucie mrowienia w rękach i stopach, ból gardła,
infekcje uszu, niepowodzenie testów słuchowych, krwawienie z nosa i bladość.
Rzadko występowały zdarzenia niepożądane związane z nadwrażliwością, w tym: uogólnione pokrzywki i świąd
(wysypka skórna z silnym świądem i powstawaniem grudek), wysypka, trudności w oddychaniu, kaszel,
ból lub uczucie ściskania w klatce piersiowej, świsty podczas oddychania, niskie ciśnienie krwi (hipotensja),
utratę przytomności, przyspieszone bicie serca, ciężkie reakcje nadwrażliwościowe, które mogą powodować
trudności w połykaniu i/lub oddychaniu, obrzęk i zaczerwienienie twarzy i/lub rąk (anafilaksja).
W przypadku wystąpienia reakcji alergicznych lub anafilaktycznych należy natychmiast przerwać
wstrzykiwanie/infuzję i skontaktować się z lekarzem.
U dzieci, które wcześniej nie były leczone lekami zawierającymi czynnik VIII, powstawanie przeciwciał
inhibitorycznych (patrz punkt 2) może być bardzo częste (więcej niż 1 pacjent na 10); jednak u
pacjentów, którzy wcześniej otrzymywali leczenie czynnikiem VIII (ponad 150 dni leczenia), ryzyko to jest
nieczęste (mniej niż 1 pacjent na 100). Jeśli do tego dojdzie, lek ten lub lek dziecka może przestać
właściwie działać i może wystąpić trwające krwawienie. W takiej sytuacji należy natychmiast skontaktować się
z lekarzem.
Zgłaszanie działań niepożądanych
Jeśli wystąpią u Państwa działania niepożądane, w tym te niewymienione w niniejszym ulotce, należy
skontaktować się z lekarzem. Można również zgłaszać działania niepożądane bezpośrednio za pośrednictwem
narodowego systemu: https://www.aifa.gov.it/content/segnalazioni-reazioni-avverse .
Zgłaszanie działań niepożądanych pozwala na dostarczenie dodatkowych informacji dotyczących bezpieczeństwa
stosowania tego leku.
5. Jak przechowywać Recombinate
- Lek należy przechowywać w miejscu niedostępnym dla dzieci.
- Przechowywać w lodówce (2 °C – 8 °C).
- Nie zamrażać.
- Przechowywać w opakowaniu zewnętrznym, aby chronić lek przed światłem.
- Nie należy stosować tego leku po dacie wygaśnięcia wskazanej na etykiecie i opakowaniu. Data wygaśnięcia znajduje się na opakowaniu po skrócie „Scad.”. Data ta odnosi się do ostatniego dnia danego miesiąca.
W okresie ważności produkt można przechowywać w temperaturze 15°C–25°C przed użyciem przez maksymalnie sześć miesięcy. Po przechowywaniu w temperaturze 15°C–25°C nie wolno ponownie umieszczać produktu w lodówce. Po odtworzeniu Recombinate należy podać w temperaturze pokojowej w ciągu trzech godzin.
Przechowywanie po odtworzeniu
- Ten produkt należy stosować tylko raz. Należy użyć produktu w ciągu trzech godzin od momentu odtworzenia.
- Nie chłodzić roztworu po odtworzeniu. Nie należy stosować Recombinate, jeśli roztwór zawiera osad lub jest mętny. Nie wyrzucać leków do kanalizacji ani do śmieci domowych. Zapytaj farmaceuty o sposób utylizacji leków, których już nie używasz. Pomoże to ochronić środowisko.
6. Skład opakowania i inne informacje
Skład Recombinate
- Substancją czynną jest octocog alfa, rekombinowany czynnik VIII krzepnięcia 25 IU/ml, 50 IU/ml lub 100 IU/ml. Produkt występuje w trzech dawkach: 250 IU, 500 IU lub 1000 IU (Jednostek Międzynarodowych) na fiolkę substancji czynnej.
- Pozostałe składniki to:
- dla proszku: albumina ludzka, chlorek sodu, histydyna, makrogol 3350, chlorek wapnia dwuwodny, kwas solny (do regulacji pH) i wodorotlenek sodu (do regulacji pH).
- dla rozpuszczalnika: woda do wstrzykiwań.
Wygląd zewnętrzny Recombinate i zawartość opakowania
Recombinate jest dostępne jako proszek i rozpuszczalnik do sporządzenia roztworu do wstrzykiwań – jest to sypka, biała lub lekko żółtawa masa. Po odtworzeniu roztwór jest klarowny, bezbarwny i pozbawiony zanieczyszczeń. Rozpuszczalnik (sterylna woda do wstrzykiwań) to klarowna, bezbarwna ciecz.
Opakowanie zawiera fiolkę z 250 IU lub 500 IU lub 1000 IU proszku, fiolkę z 10 ml rozpuszczalnika, urządzenie do odtworzenia (BAXJECT II), jednorazową strzykawkę plastikową, zestaw infuzyjny miniset, 2 waciki alkoholowe i 2 plastry opatrunkowe.
Zamiast BAXJECT II może zostać dostarczone urządzenie do odtworzenia z igłą, zawierające sterylną dwukierunkową igłę (do przeniesienia rozpuszczalnika do fiolki z Recombinate) oraz sterylną igłę filtracyjną (do przeniesienia odtworzonego roztworu do strzykawki).
Opakowanie jednostkowe
Właściciel pozwolenia na dopuszczenie do obrotu
Baxalta Innovations GmbH
Industriestrasse 67, A-1221 Wiedeń
Reprezentant w Włoszech:
Takeda Italia S.p.A.
Tel. +39 06 502601
Producent
Baxalta Belgium Manufacturing SA
Bd. René Branquart 80, B-7860 Lessines,
Belgia
Ten lek jest zarejestrowany w krajach członkowskich Europejskiego Obszaru Gospodarczego pod następującymi nazwami:
Belgia: Recombinate 250 (500, 1000) IU/10 ml
Recombinate 250 (500, 1000) IU/5 ml
Bułgaria: Recombinate 500 IU/5 ml
Cypr: Recombinate 250 (500, 1000) IU
Estonia: Recombinate 250 (500, 1000) IU/5 ml
Niemcy: Recombinate Antihämophilie Factor (rekombinant) 1000
Grecja: Recombinate 250 (500, 1000) IU
Litwa: Recombinate 250 (500, 1000) IU/10 ml
Malta: Recombinate 250 (500, 1000) IU
Holandia: Recombinate 250 (500, 1000) IE/10 ml
Recombinate 250 (500, 1000) IE/5 ml
Irlandia: Recombinate 250 (500, 1000) IU
Włochy: Recombinate 250 (500, 1000) IU/10 ml
Recombinate 250 (500, 1000) IU/5 ml
Łotwa: Recombinate 250 (500, 1000) IU/5 ml
Poniższe informacje przeznaczone są wyłącznie dla personelu medycznego:
STRESZCZENIE CHARAKTERYSTYKI PRODUKTU
1. NAZWA PRODUKTU LECZNICZEGO
Recombinate 250 IU/10 ml proszek i rozpuszczalnik do sporządzenia roztworu do wstrzykiwań
Recombinate 500 IU/10 ml proszek i rozpuszczalnik do sporządzenia roztworu do wstrzykiwań
Recombinate 1000 IU/10 ml proszek i rozpuszczalnik do sporządzenia roztworu do wstrzykiwań
2. SKŁAD JAKOŚCIOWY I ILOŚCIOWY
Octocog alfa 25 IU na ml roztworu odtworzonego
Po odtworzeniu: fiolka 10 ml zawiera 250 IU octocog alfa
Recombinate 250 IU/10 ml zawiera nominalnie 250 IU octocog alfa, rekombinowanego czynnika VIII
krzepnięcia, w każdej fiolce.
Produkt zawiera około 25 IU/ml octocog alfa, rekombinowanego czynnika VIII krzepnięcia, po odtworzeniu 10 ml wody do wstrzykiwań.
Octocog alfa 50 IU na ml roztworu odtworzonego
Po odtworzeniu: fiolka 10 ml zawiera 500 IU octocog alfa
Recombinate 500 IU/10 ml zawiera nominalnie 500 IU octocog alfa, rekombinowanego czynnika VIII
krzepnięcia, w każdej fiolce.
Produkt zawiera około 50 IU/ml octocog alfa, rekombinowanego czynnika VIII krzepnięcia, po odtworzeniu 10 ml wody do wstrzykiwań.
Octocog alfa 100 IU na ml roztworu odtworzonego
Po odtworzeniu: fiolka 10 ml zawiera 1000 IU octocog alfa
Recombinate 1000 IU/10 ml zawiera nominalnie 1000 IU octocog alfa, rekombinowanego czynnika VIII
krzepnięcia, w każdej fiolce.
Produkt zawiera około 100 IU/ml octocog alfa, rekombinowanego czynnika VIII krzepnięcia, po odtworzeniu 10 ml wody do wstrzykiwań.
Potencja jest określana za pomocą testu chromogenowego Europejskiej Farmakopei w odniesieniu do Standardu Mega FDA kalibrowanego według Standardu WHO. Aktywność specyficzna Recombinate wynosi około 4000–8000 IU/mg białka.
Recombinate zawiera rekombinowany czynnik VIII krzepnięcia (INN: octocog alfa). Octocog alfa (rekombinowany czynnik VIII krzepnięcia) jest białkiem oczyszczonym składającym się z 2332 aminokwasów. Ma sekwencję aminokwasów porównywalną do czynnika VIII oraz modyfikacje potranslacyjne podobne do cząsteczki pochodzącej z osocza. Rekombinowany czynnik VIII krzepnięcia jest glikoproteiną wyrażaną w komórkach ssaków, wytwarzaną metodami inżynierii biologicznej z linii komórkowej jajnika chomika chińskiego.
Substancje pomocnicze o znanym działaniu:
Każda fiolka zawiera 35 mg (1,5 mmol) sodu.
Pełna lista substancji pomocniczych znajduje się w punkcie 6.1.
3. POSTAĆ LEKU
Proszek i rozpuszczalnik do sporządzania roztworu do wstrzykiwania.
Krucha biała lub niemal biała proszek. Rozpuszczalnik (woda do sporządzeń iniekcyjnych) to klarowny, bezbarwny płyn.
4. INFORMACJE KLINICZNE
4.1 Wskazania terapeutyczne
Leczenie i profilaktyka krwawień u pacjentów z hemofilią A (wrodzony niedobór czynnika VIII).
Ten produkt nie zawiera czynnika von Willebranda i dlatego nie jest wskazany w chorobie von Willebranda.
Recombinate jest wskazany dla wszystkich grup wiekowych, od noworodków po dorosłych.
4.2 Dawkowanie i sposób podania
Leczenie powinno odbywać się pod nadzorem lekarza doświadczonych w leczeniu hemofilii.
Monitorowanie leczenia
Podczas terapii zaleca się odpowiednie oznaczanie poziomów czynnika VIII w celu ustalenia dawki do podania oraz częstotliwości powtarzania infuzji. Odpowiedź poszczególnych pacjentów na czynnik VIII może się różnić, co skutkuje różnymi okresami półtrwania i odzyskiem aktywności. Dawkowanie oparte na masie ciała może wymagać dostosowania u pacjentów z niedowagą lub nadwagą. Szczególnie w przypadku większych zabiegów chirurgicznych niezbędne jest dokładne monitorowanie terapii zastępczej za pomocą analizy czynności krzepnięcia (aktywność czynnika VIII w osoczu).
Dawkowanie
Dawka i długość trwania terapii zastępczej zależą od stopnia nasilenia niedoboru czynności hemostatycznej, lokalizacji i nasilenia krwawienia oraz stanu klinicznego pacjenta.
Leczenie powinno być prowadzone we współpracy z lekarzem specjalizującym się w zaburzeniach krzepnięcia oraz z laboratorium potrafiącym oznaczać stężenie AHF (czynnika VIII) w osoczu.
Podawaną liczbę jednostek czynnika VIII wyraża się w Jednostkach Międzynarodowych (J.M.), odnosząc się do obecnego standardu WHO dla produktów zawierających czynnik VIII. Aktywność czynnika VIII w osoczu wyrażana jest zarówno w procentach (w odniesieniu do osocza ludzkiego normalnego), jak i w Jednostkach Międzynarodowych (odnoszących się do międzynarodowego standardu dla czynnika VIII osoczkowego).
Jedna Jednostka Międzynarodowa (J.M.) aktywności czynnika VIII odpowiada ilości czynnika VIII zawartej w 1 ml normalnego osocza ludzkiego.
Terapia na żądanie
Oczekiwany wzrost in vivo poziomu szczytowego Recombinate wyrażony w J.M./dL osocza lub w % (procentach) wartości normalnej można obliczyć, mnożąc dawkę podaną na kg masy ciała (J.M./kg) przez 2.
Metoda obliczania jest przedstawiona w poniższych przykładach:
Oczekiwany wzrost % FVIII = liczba podanych jednostek × 2% / J.M. / kg
masa ciała (kg)
Przykład dla dorosłego o masie 70 kg: 1750 J.M. × 2% / J.M. / kg = ok. 50%
70 kg
lub
Wymagana dawka (J.M.) = masa ciała (kg) × oczekiwany wzrost % FVIII
2% / J.M. /kg
Przykład dla dziecka o masie 40 kg: 40 kg × 70% = 1400 J.M.
2% / J.M. /kg
Chociaż dawkę można oszacować za pomocą powyższego wzoru, zaleca się zdecydowanie przeprowadzenie, o ile to możliwe, odpowiednich badań laboratoryjnych, w tym oznaczania stężenia AHF w osoczu pacjenta w odpowiednich odstępach czasu, aby upewnić się, że osiągnięto i utrzymywane są odpowiednie poziomy AHF. Jeśli u pacjenta nie osiąga się pożądanego poziomu AHF w osoczu lub jeśli krwawienie nie ustępuje po podaniu odpowiedniej dawki, należy podejrzewać obecność inhibitora. Przy pomocy odpowiednich badań laboratoryjnych można wykazać i zmierzyć obecność inhibitora, wyrażając ją w Jednostkach Międzynarodowych AHF neutralizowanych przez 1 ml osocza (jednostki Bethesda) lub w całkowitej oszacowanej objętości osocza. Jeśli inhibitor występuje w ilości mniejszej niż 10 jednostek Bethesda na 1 ml, podanie dodatkowego AHF może zneutralizować inhibitor. W związku z tym podanie dodatkowych Jednostek Międzynarodowych AHF powinno prowadzić do oczekiwanego efektu. W takich przypadkach konieczne jest monitorowanie poziomów AHF za pomocą badań laboratoryjnych. Poziomy inhibitora przekraczające 10 jednostek Bethesda na 1 ml mogą uczynić niemożliwym lub nierealnym kontrolowanie hemostazy za pomocą AHF z powodu konieczności stosowania zbyt wysokich dawek.
Poniższy schemat dawkowania podany w Tabeli I może być wykorzystany jako wskazówka dla dorosłych i dzieci. Podawaną ilość i częstotliwość infuzji należy zawsze dostosować do skuteczności klinicznej w poszczególnych przypadkach.
W zależności od sytuacji i uznania lekarza, Recombinate może być stosowany również w celu profilaktyki (krótko- lub długoterminowej) krwawień.
W przypadku poniższych zdarzeń krwotocznych aktywność czynnika VIII nie powinna spaść poniżej podanego poziomu aktywności plazmatycznej (w <% wartości normalnej>) w odpowiednim okresie. Poniższa tabela może być wykorzystana jako wskazówka do ustalania dawki w przypadku krwawień i zabiegów chirurgicznych:
Tabela I: Schemat dawkowania
| Krwawienie | ||
| Stopień krwawienia | Pik aktywności czynnika AHF wymaganej we krwi po infuzji (% normy lub UI/dL osocza) | Częstotliwość infuzji |
| Wcześniejsze wewnętrrzne krwawienie stawowe; krwawienie mięśniowe lub jamy ustnej | 20 - 40 | Rozpocząć infuzję co 12 - 24 godziny przez jeden do trzech dni, aż do ustąpienia epizodu krwawienia (na podstawie bólu) lub do pełnego wyleczenia |
| Rozleglejsze wewnętrrzne krwawienie stawowe; krwawienie mięśniowe lub hematoma | 30 - 60 | Powtarzać infuzję co 12 - 24 godziny, zazwyczaj przez trzy dni lub dłużej, aż do ustąpienia bólu i przywrócenia funkcji |
| Krwawienia zagrożone dla życia, takie jak krwawienie wewnątrzczaszkowe, krwawienie do gardła lub ciężkie krwawienie jamy brzusznej | 60 - 100 | Powtarzać infuzję co 8 - 24 godziny, aż do ustąpienia zagrożenia |
| Chirurgia | ||
| Typ zabiegu | ||
| Zabiegi mniejsze, w tym ekstrakcje zębów | 30-60 | Jednorazowa infuzja, a także doustna terapia przeciwpłytkowa w ciągu godziny przed lub po zabiegu wystarcza w około 70% przypadków. Następnie co 24 godziny przez co najmniej 1 dzień aż do gojenia rany. |
| Zabiegi większe | 80 -100 (przed i po zabiegu) | Powtarzać infuzję co 8 - 24 godziny, w zależności od postępu gojenia rany. |
Dane przedstawione reprezentują szczytowe poziomy aktywności AHF u pacjentów z oczekiwanym średniym okresem półtrwania Faktora VIII. Jeśli uznane za konieczne, szczytowe poziomy aktywności należy oznaczać w ciągu pół godziny po podaniu. U pacjentów z stosunkowo krótkim okresem półtrwania Faktora VIII może być konieczne zwiększenie dawki i/lub częstotliwości podawania.
Każdy fiolka Recombinate zawiera na etykiecie aktywność rekombinowanego Faktora Antyhemofilowego (Recombinate) wyrażoną w MI na fiolkę.
Oznaczenie miana odnosi się do Międzynarodowego Standardu WHO dla stężeń Faktora VIII:C. Przeprowadzone badania wykazały, że w celu dokładnego oznaczenia tych poziomów aktywności test aktywności należy przeprowadzać za pomocą próbek i pipet z tworzywa sztucznego oraz z wykorzystaniem podłoża zawierającego normalne poziomy Faktora von Willebranda.
Profilaktyka
W profilaktyce przeciwhemoragicznej długoterminowej u pacjentów z ciężką hemofilią A typowe dawki wynoszą od 20 do 40 MI Faktora VIII na kg masy ciała, podawane w odstępach 2–3 dni.
Pacjentów należy monitorować pod kątem rozwoju inhibitorów Faktora VIII. W przypadku nieosiągnięcia oczekiwanych poziomów aktywności Faktora VIII we krwi lub niemożliwości kontrolowania krwawienia odpowiednią dawką, należy przeprowadzić test w celu wykrycia ewentualnego obecności inhibitora Faktora VIII. U pacjentów z wysokimi poziomami inhibitora terapia oparta na Faktorze VIII może nie być skuteczna, dlatego należy rozważyć inne opcje terapeutyczne. Leczenie takich pacjentów powinno być prowadzone przez lekarzy doświadczonych w leczeniu pacjentów z hemofilią.
Zobacz również punkt 4.4.
Populacja pediatryczna
Recombinate może być stosowany u dzieci we wszystkich grupach wiekowych, w tym u noworodków (badania dotyczące bezpieczeństwa i skuteczności przeprowadzono zarówno u dzieci wcześniej leczonych, jak i u dzieci nieleczonych wcześniej: zobacz punkt 5.1). W leczeniu na żądanie dawkowanie u pacjentów pediatrycznych nie różni się od dawkowania u dorosłych. W profilaktyce długoterminowej krwawień u pacjentów z ciężką hemofilią A, w niektórych przypadkach mogą być konieczne krótsze odstępy między dawkami lub większe dawki w porównaniu z typową dawką 20–40 MI Faktora VIII na kg masy ciała podawaną w odstępach 2–3 dni.
Sposób podania
Preparat należy podawać dożylnie po odtworzeniu za pomocą rozpuszczalnika dostarczonego w zestawie (zobacz punkt 6.6). Odtworzonego produktu nie należy chłodzić. Zaleca się podawanie Recombinate w temperaturze pokojowej nie później niż w ciągu 3 godzin od odtworzenia. Szybkość podania powinna być dostosowana do komfortu pacjenta, maksymalnie do 10 ml/min. Przed i podczas podawania Recombinate należy monitorować puls pacjenta. W przypadku wystąpienia istotnego wzrostu tętna, zmniejszenie szybkości wlewu lub tymczasowe przerwanie wstrzykiwania zazwyczaj szybko powoduje ustąpienie objawów (zobacz punkty 4.4 i 4.8).
Instrukcje dotyczące odtworzenia leku przed podaniem znajdują się w punkcie 6.6.
4.3 Przeciwwskazania
Nadwrażliwość na substancję czynną lub którykolwiek z substancji pomocniczych wymienionych w punkcie 6.1. Znana reakcja alergiczna na białka wołowe, mysie lub chomikowe.
4.4 Szczególne ostrzeżenia i środki ostrożności stosowania
Śledzenie
W celu poprawy śledzenia leków biologicznych, nazwa i numer serii podanego leku powinny być wyraźnie zarejestrowane.
Nadwrażliwość
Zgłoszono przypadki ciężkich reakcji alergicznych po podaniu Recombinate. Pacjentów z znaną nadwrażliwością na białka mysie, wołowe lub chomikowe należy leczyć ostrożnie. Pacjentów należy poinformować o wczesnych objawach reakcji alergicznych, takich jak pokrzywka, uogólniona pokrzywka, uczucie ucisku w klatce piersiowej, świsty w oddychaniu, hipotensja oraz anafilaksja. W przypadku wystąpienia reakcji alergicznej lub anafilaktycznej należy natychmiast przerwać wstrzykiwanie lub wlew. W przypadku wstrząsu należy zastosować standardowe leczenie wstrząsu.
Inhibitory
Tworzenie się przeciwciał neutralizujących (inhibitorów) przeciwko Faktorowi VIII stanowi znaną komplikację w leczeniu pacjentów z hemofilią A. Inhibitory są zazwyczaj immunoglobulinami IgG skierowanymi przeciwko działaniu prokoagulacyjnemu Faktora VIII i są wyrażane w jednostkach Bethesda (UB) na ml osocza metodą zmodyfikowaną. Ryzyko rozwoju inhibitorów wiąże się z nasileniem choroby i czasem ekspozycji na Faktor VIII, jest większe w pierwszych 50 dniach ekspozycji, ale utrzymuje się przez całe życie, choć nie jest to zjawisko częste.
Znaczenie kliniczne rozwoju inhibitorów zależy od miana inhibitora: niskie miano inhibitorów, które występuje tymczasowo lub pozostaje stale niskie, ma mniejszy wpływ na ryzyko niewystarczającej odpowiedzi klinicznej niż wysokie miano inhibitorów.
Ogólnie rzecz biorąc, wszyscy pacjenci leczeni produktami zawierającymi Faktor VIII krzepnięcia powinni być dokładnie monitorowani pod kątem rozwoju inhibitorów za pomocą odpowiednich obserwacji klinicznych i badań laboratoryjnych. Jeśli nie uzyskuje się oczekiwanych poziomów aktywności Faktora VIII we krwi lub jeśli krwawienie nie jest kontrolowane odpowiednią dawką, należy przeprowadzić badanie w celu ustalenia obecności inhibitorów Faktora VIII. U pacjentów z wysokimi poziomami inhibitora terapia Faktorem VIII może nie być skuteczna, należy rozważyć inne opcje terapeutyczne. Opiekę nad tymi pacjentami powinny prowadzić lekarze doświadczeni w leczeniu hemofilii i inhibitorów Faktora VIII.
Zdarzenia kardiowaskularne
U pacjentów z istniejącymi czynnikami ryzyka kardiowaskularnego terapia zastępcza FVIII może zwiększać ryzyko kardiowaskularne.
Powikłania związane z cewnikiem
Jeśli konieczne jest użycie urządzenia do dostępu do żyły centralnej (CVAD), należy wziąć pod uwagę ryzyko powikłań związanych z CVAD, w tym infekcji lokalnych, bakteriemii i zakrzepicy w miejscu umiejscowienia cewnika.
Populacja pediatryczna
Ostrzeżenia i środki ostrożności stosowania u pacjentów pediatrycznych nie różnią się od tych stosowanych u dorosłych.
Ten lek zawiera 35 mg (1,5 mmol) sodu w każdej fiolce o pojemności 250 MI, 500 MI i 1000 MI, co odpowiada 1,8% maksymalnej zalecanej dziennej dawki sodu według WHO, wynoszącej 2 g sodu dla dorosłego. Należy to uwzględnić u pacjentów poddawanych diecie o obniżonej zawartości sodu.
4.5 Interakcje z innymi lekami i inne formy interakcji
Nie przeprowadzono badań interakcji.
4.6 Niepłodność, ciąża i karmienie piersią
Nie przeprowadzono badań rozrodczych na zwierzętach z użyciem Faktora VIII. Ze względu na rzadkość występowania hemofilii A u kobiet, nie są dostępne dane eksperymentalne dotyczące stosowania Faktora VIII w czasie ciąży lub karmienia piersią. Dlatego Faktor VIII należy podawać w czasie ciąży i karmienia piersią tylko wtedy, gdy wyraźnie wskazany.
4.7 Wpływ na zdolność prowadzenia pojazdów i obsługi maszyn
Nie zaobserwowano wpływu na zdolność prowadzenia pojazdów i obsługi maszyn.
4.8 Działania niepożądane
Tabela podsumowująca działania niepożądane
Poniższa tabela zawiera działania niepożądane zgłoszone w oparciu o doniesienia spontaniczne i badania kliniczne.
Poniższa tabela została sporządzona na podstawie klasyfikacji MedDRA według układów i narządów (SOC i Preferred Term).
Częstotliwość została oszacowana według następujących kryteriów: bardzo często (≥ 1/10), często (≥ 1/100, < 1/10), nieczęsto (≥ 1/1000, < 1/100), rzadko (≥ 1/10 000, < 1/1000), bardzo rzadko (< 1/10 000), nieznana (częstotliwość nie może być określona na podstawie dostępnych danych).
| Klasyfikacja wg układów i narządów według MedDRA | Częstość | Preferowany termin MedDRA |
| Infekcje i inwazje | niepowszechne 1 | Infekcja ucha 1 |
| Choroby układu krwiotwórczego i chłonnego | niepowszechne (PTPs)1bardzo powszechne (PUPs)1 | Inhibicja Faktora VIII 1 |
| Zaburzenia układu odpornościowego | nieznana | Reakcja anafilaktyczna Nadwrażliwość2 |
| Choroby układu nerwowego | niepowszechne | Zawroty głowy Tremor |
| nieznana | Utrata przytomności Omdlenie Bóle głowy Parestezja | |
| Choroby serca | nieznana | Cyanosis Tachykardia |
| Choroby układu naczyniowego | niepowszechne | Krwawienie z nosa Zarzewnica Siniaczki Hipotensja Bladość Odczucie zimna w kończynach |
| Choroby układu oddechowego, klatki piersiowej i śródpiersia | niepowszechne | Ból gardła i krtani |
| nieznana | Utrudnione oddychanie Kaszel Szumy w oddychaniu | |
| Choroby układu pokarmowego | niepowszechne | Nudności |
| nieznana | Wymioty Ból brzucha | |
| Choroby skóry i tkanki podskórnej | niepowszechne | Hyperhidrosis Swędzenie Wysypka Wysypka makularna i plamniczo-pęcherzykowa |
| nieznana | Światrwałość Ogólna pokrzywka Łuszczenie się skóry Rumień | |
| Choroby układu mięśniowo-szkieletowego i tkanki łącznej | niepowszechne | Ból kończyn |
| Choroby systemowe i stanów związanych z miejscem podania | powszechne | Dreszcze |
| niepowszechne | Odczucie zmęczenia Niedżuma | |
| nieznana | Nieprawidłowe wyniki testu stymulacji akustycznej | |
| Badania diagnostyczne 1 | niepowszechne | Anomalne wyniki testu stymulacji akustycznej |
Częstotliwość oparta jest na badaniach przeprowadzonych z użyciem wszystkich produktów zawierających czynnik VIII, które obejmowały pacjentów z ciężką hemofilią A. PTPs = pacjenci wcześniej leczeni, PUPs = pacjenci nieleczoni wcześniej.
Wczesne objawy reakcji nadwrażliwości to np. pokrzywka, duszność, kaszel, uczucie ucisku w klatce piersiowej, świsty podczas oddychania, anafilaksja, wysypka, hipotensja, świąd, dreszcze, rumień, gorączka, sinica, tachykardia, wymioty, omdlenia, ból głowy. Zaleca się ostrożność u pacjentów znanym z alergii na składniki preparatu (patrz punkty 4.3. i 4.4.).
Opis wybranych skutków niepożądanych
Może dojść do rozwoju przeciwciał neutralizujących (inhibitorów) u pacjentów z hemofilią A leczonych czynnikiem VIII, w tym Recombinate. Obecność inhibitorów może objawiać się niewystarczającą odpowiedzią kliniczną. W takich przypadkach zaleca się kontakt z wyspecjalizowanym ośrodkiem hemofilii.
Grupa pediatryczna
Podczas badań klinicznych nie stwierdzono różnic zależnych od wieku w zakresie skutków niepożądanych, z wyjątkiem rozwoju inhibitorów u dzieci wcześniej nieleczonych (PUPs).
Zgłaszanie podejrzewanych skutków niepożądanych
Zgłaszanie podejrzewanych skutków niepożądanych, które występują po dopuszczeniu do obrotu leku, jest ważne, ponieważ pozwala na ciągłe monitorowanie stosunku korzyści do ryzyka. Osoby pracujące w ochronie zdrowia powinny zgłaszać wszelkie podejrzewane skutki niepożądane za pośrednictwem strony https://www.aifa.gov.it/content/segnalazioni-reazioni-avverse .
4.9 Przedawkowanie
Nie znane są objawy przedawkowania.
5. WŁAŚCIWOŚCI FARMACOLOGICZNE
5.1 Właściwości farmakodynamiczne
Kategoria farmakoterapeutyczna: środki przeciwkrwawieniowe: czynnik VIII krzepnięcia krwi. Kod ATC: B02BD02.
Złożony preparat czynnika VIII / czynnika von Willebranda składa się z dwóch cząsteczek (czynnik VIII i czynnik von Willebranda) o różnych funkcjach fizjologicznych.
Po włączeniu do organizmu pacjenta z hemofilią, czynnik VIII wiąże się w obiegu z czynnikiem von Willebranda.
Aktywowany czynnik VIII działa jako kofaktor aktywowanego czynnika IX, przyspieszając przekształcanie czynnika X w aktywowany czynnik X; ten z kolei przekształca protrombinę w trombinę, która z kolei przekształca fibrynogen w fibrynę, prowadząc do powstania skrzepu. Hemofilia A to dziedziczny zaburzony krzepnięcia krwi związany z płcią, wynikający ze zmniejszonego poziomu czynnika VIII:C, powodujący obfite krwawienia do stawów, mięśni lub narządów wewnętrznych, zarówno samoistne, jak i przypadkowe lub spowodowane urazem chirurgicznym. Terapia zastępcza pozwala na zwiększenie stężenia czynnika VIII we krwi, umożliwiając tymczasową korektę niedoboru czynnika oraz korektę skłonności do krwawień.
Należy zwrócić uwagę, że wskaźnik rocznej liczby krwawień (ABR) nie jest porównywalny między różnymi preparatami czynnika VIII ani między różnymi badaniami klinicznymi.
Populacja pediatryczna
Recombinate został zbadany u 71 dzieci nigdy wcześniej nie leczonych (PUPs), których średni wiek w momencie pierwszej infuzji Recombinate wynosił 10 miesięcy (zakres: 2 dni – 50 miesięcy). Produkt okazał się dobrze tolerowany i nie był związany z istotnymi niepożądanymi odczynami krótkoterminowymi. Skuteczność kliniczna była porównywalna do innych cząsteczek FVIII o pełnej długości łańcucha zarówno w leczeniu krwawień ostrych, jak i w profilaktyce chirurgicznej (10 pacjentów przeszło zabiegi chirurgiczne). Długoterminowe badanie dalszego przebiegu u tych pacjentów wykazało częstość występowania niepożądanych odczynów związanych z produktem na poziomie 0,86/1000 infuzji, z których żaden nie był ciężki ani zagrażający życiu.
5.2 Właściwości farmakokinetyczne
Badania farmakokinetyczne przeprowadzone u 69 pacjentów wcześniej już leczonych wykazały, że średni okres półtrwania Recombinate krążącego we krwi wynosi 14,6 ± 4,9 godziny (n = 67), wartość ta nie różni się istotnie statystycznie od tej obserwowanej dla HemofilM, Czynnika Przeciwhemofilowego (Ludzkiego) uzyskanego z osocza (pdAHF), którego średni okres półtrwania wynosi 14,7 ± 5,1 godziny (n = 61). Rzeczywiste odzyskanie stężenia względem wartości wyjściowej po podaniu dawki 50 IU/kg po infuzji Recombinate wyniosło 123,9 ± 47,7 IU/dL (n = 23), co jest istotnie wyższe niż rzeczywiste odzyskanie stężenia względem wartości wyjściowej obserwowane po podaniu HemofilM, wynoszące 101,7 ± 31,6 IU/dL (n = 61). Jednakże obliczony stosunek odzyskania rzeczywistego do oczekiwanego (czyli wzrost aktywności czynnika VIII o 2% na 1 IU rAHF/kg masy ciała) dla Recombinate (121,2 ± 48,9%) jest podobny do tego dla HemofilM (123,4 ± 16,4%).
Łącznie przeprowadzono 494 badania odzyskania u 68 pacjentów nigdy wcześniej nie leczonych.
Dwustu dwanaście badań odzyskania przeprowadzono podczas leczenia epizodów krwawień, przy czym średnie rzeczywiste odzyskanie ± SD wyniosło 70,0 ± 37,9 IU/dL (N = 208, cztery odzyskania pominięto w analizie, ponieważ wartości były poza zakresem odniesienia). Wysoka zmienność wynika z szerokiego zakresu rzeczywiście podanych dawek, od 13,8 do 103,2 IU/kg (średnia ± SD 36,0 ± 16,2, mediana 30,2 IU/kg). Uwzględniając zmienne dawki, obliczono stosunek rzeczywistego odzyskania do przewidywanego, który wyniósł średnią 1,0 ± 0,3.
Łącznie 68 badań odzyskania przeprowadzono, gdy pacjenci otrzymywali kolejne infuzje w ramach długotrwałego leczenia istniejących już epizodów krwawień. Rzeczywisty poziom odzyskania FVIII został skorygowany o poziom FVIII przed infuzją. Średnie rzeczywiste odzyskanie ± SD wyniosło 88,6 ± 38,2 IU/dL (N = 66, dwa odzyskania pominięto w analizie, ponieważ wartości były poza zakresem odniesienia). Ponownie, szeroki zakres rzeczywiście podanych dawek, od 18,5 do 85,7 IU/kg (średnia ± SD 38,6 ± 15,9, mediana 32,1 IU/kg), spowodował znaczącą zmienność obserwowanych poziomów odzyskania. Średni stosunek ± SD rzeczywistego do przewidywanego odzyskania wyniósł 1,0 ± 0,3, mediana 1,0.
Łącznie 214 badań odzyskania przeprowadzono, gdy pacjenci byli już ustabilizowani, przy średnim rzeczywistym odzyskaniu 71,6 ± 29,7 IU/dL (N = 209, pięć odzyskań pominięto w analizie, ponieważ wartości były poza zakresem odniesienia). Dawki podane wahały się od 10,4 do 68,1 IU/kg (średnia ± SD 38,0 ± 12,7, mediana 36,1 IU/kg). Średni stosunek ± SD rzeczywistego do przewidywanego odzyskania wyniósł 1,0 ± 0,3.
5.3 Dane przedkliniczne dotyczące bezpieczeństwa
Recombinate działa jak endogenny czynnik VIII. Dawkowanie wielokrotnie wyższe niż zalecane u ludzi na kilogram masy ciała nie wykazało efektów toksycznych w badaniach na zwierzętach laboratoryjnych. Recombinate został poddany badaniom mutagenności zarówno in vitro przy znacznie wyższych dawkach niż stężenie plazmatyczne AHF, jak i in vivo przy dawkach do 10-krotnie wyższych niż maksymalna dawka sugerowana w klinice, bez wywoływania mutacji odwracalnych, aberracji chromosomowych ani wzrostu liczby mikrojąder w policromatycznych erytrocytach szpiku kostnego.
Ponieważ doświadczenie kliniczne nie dostarcza żadnych dowodów na działanie rakotwórcze i mutagenne, nie uznano za konieczne przeprowadzanie długoterminowych badań oceniających potencjalną rakotwórczość u zwierząt.
6. DANE FARMACEUTYCZNE
6.1 Wykaz substancji pomocniczych
Proszek:
Albumina humana
Natrium chloridum
Histidina
Macrogol 3350
Calcium chloridum dihydricum
Kwas chlorowodorowy (do regulacji pH)
Natrium hydroxidum (do regulacji pH)
Roztwórnik:
Woda do preparatów do wstrzykiwań
6.2 Niekompatybilności
W przypadku braku badań zgodności, tego leku nie wolno mieszać z innymi lekami.
Należy stosować wyłącznie zestaw do wlewu dołączony do opakowania, ponieważ leczenie może nie powieść się z powodu adsorpcji czynnika VIII krzepnięcia krwi ludzkiej na wewnętrznych powierzchniach niektórych urządzeń do wlewu.
6.3 Okres ważności
3 lata. Po rekonstytucji preparat Recombinate nie należy chłodzić i należy podać w ciągu trzech godzin.
6.4 Szczególne środki ostrożności dotyczące przechowywania
Przechowywać w lodówce (2°C – 8°C).
Nie zamrażać.
Przechowywać w opakowaniu zewnętrznym, aby chronić lek przed światłem.
W okresie ważności lek może być przechowywany do sześciu miesięcy w temperaturze 15°C – 25°C przed użyciem.
Nie wolno ponownie chłodzić po przechowywaniu w temperaturze 15–25°C.
Warunki przechowywania po rekonstytucji leku podano w punkcie 6.3.
6.5 Rodzaj i zawartość opakowania
Jedno opakowanie zawiera fiolkę z proszkiem, fiolkę z 10 ml rozcieńczalnika (obie z szkła typu I z korkami gumowymi) oraz urządzenie do rekonstytucji (BAXJECT II) + jednorazową strzykawkę plastikową sterylną + minizestaw do wlewu sterylny + 2 waty nasączone alkoholem + dwa plasterki.
Zamiast BAXJECT II opakowanie może zawierać urządzenie z igłą do rekonstytucji, zawierające sterylną dwukierunkową igłę (do przeniesienia rozcieńczalnika do fiolki Recombinate) oraz sterylną igłę filtracyjną (do przeniesienia roztworu po rekonstytucji do strzykawki).
Opakowanie jednostkowe.
6.6 Szczególne środki ostrożności dotyczące usuwania i manipulacji
Preparat należy podawać dożylnie po rekonstytucji za pomocą sterylnej wody do preparatów do wstrzykiwań zawartej w opakowaniu. Należy użyć jednorazowej plastikowej strzykawki zawartej w opakowaniu.
- Użyć w ciągu 3 godzin od momentu rekonstytucji.
- Nie chłodzić po rekonstytucji.
- Nieużywany lek oraz odpady pochodzące z tego leku należy usunąć zgodnie z obowiązującymi przepisami lokalnymi.
- Roztwór powinien być klarowny lub lekko mleczny. Nie należy stosować roztworów mętnych lub zawierających osad. Przed podaniem należy wizualnie sprawdzić, czy odtworzone preparaty nie zawierają cząstek obcych lub nieprawidłowego zabarwienia.
- Nie należy stosować, jeśli produkt, jego sterylny system barierowy lub opakowanie są uszkodzone lub wykazują jakiekolwiek oznaki degradacji.
| Rekonstytucja: należy stosować technikę bezpyłową | |
| Rekonstytucja za pomocą BAXJECT II | Rekonstytucja za pomocą igieł |
| 1. Przygotować Recombinate (proszek) oraz Wodę do wstrzykiwań (rozpuszczalnik) w temperaturze 15°C–25°C. 2. Usunąć osłony z butelek zawierających liofilizat i rozpuszczalnik. 3. Zdezynfekować przekłady za pomocą waty nasączonej alkoholem. Postawić butelki na płaskiej powierzchni. 4. Otworzyć opakowanie urządzenia BAXJECT II, usuwając papierową osłonę, nie dotykając wnętrza (Rys. a). Nie wyjmować urządzenia z opakowania. 5. Odwrócić pudełko, wsunąć przez przekładkę butelki z rozpuszczalnikiem przezroczysty końcówkę z plastiku. Uchwycić brzeg pudełka i wyjąć je, zwalniając urządzenie BAXJECT II (Rys. b). Nie usuwać niebieskiej osłony z urządzenia BAXJECT II. 6. Trzymając BAXJECT II podłączony do butelki z rozpuszczalnikiem, odwrócić układ tak, aby butelka z rozpuszczalnikiem znalazła się w górnej części urządzenia. | 1. Przygotować Recombinate (proszek) oraz Wodę do wstrzykiwań (rozpuszczalnik) w temperaturze 15°C–25°C. 2. Usunąć osłony z butelek zawierających liofilizat i rozpuszczalnik. 3. Zdezynfekować przekłady za pomocą waty nasączonej alkoholem. Postawić butelki na płaskiej powierzchni. 4. Usunąć osłonę z jednego końca dwustronnej igły i wsunąć odsłonięty koniec igły przez przekładkę butelki z rozpuszczalnikiem. 5. Usunąć osłonę z drugiego końca dwustronnej igły. Odwrócić butelkę z rozpuszczalnikiem nad butelką z Recombinate trzymaną pionowo i szybko wsunąć wolny koniec igły w środek przekładki butelki z Recombinate. Rozpuszczalnik przepłynie do butelki z liofilizatem dzięki działaniu próżni. 6. Rozłączyć obie butelki, wyciągając igłę z przekładki butelki z rozpuszczalnikiem, |
Wsunąć białą plastikową końcówkę przez przekładkę butelki z Recombinate. Rozpuszczalnik zostanie wessany do butelki z liofilizatem pod wpływem próżni (Rys. c). 7. Delikatnie wymieszać zawartość aż do całkowitego rozpuszczenia. Upewnić się, że Recombinate jest całkowicie rozpuszczony, w przeciwnym razie substancja czynna nie przejdzie przez filtr urządzenia. Produkt rozpuszcza się szybko (zwykle w mniej niż minutę). Rys. a Rys. b Rys. c    | następnie usunąć igłę z butelki z Recombinate. Delikatnie wymieszać zawartość aż do całkowitego rozpuszczenia. Upewnić się, że Recombinate jest całkowicie rozpuszczony, w przeciwnym razie substancja czynna pozostanie na filtrze igły. |
| Podanie: należy stosować technikę bezpyłową | |
Zaleca się rozpoczęcie podania nie później niż trzy godziny po rekonstytucji. Nie należy chłodzić odtworzonego produktu. Przed podaniem leki do stosowania parenteralnego należy poddać wizualnej kontroli pod kątem obecności cząstek obcych lub nieprawidłowego zabarwienia, o ile pozwala na to roztwór i opakowanie. Dla Recombinate dopuszczalne jest zabarwienie od bezbarwnego do lekko żółtawego. 1. Usunąć niebieską osłonę z urządzenia BAXJECT II. NIE WCIĄGAJ POWIETRZA DO STRZYKAWKI. Podłączyć strzykawkę do BAXJECT II (Rys. d). 2. Odwrócić układ (tak aby butelka z koncentratem znalazła się w górnej części urządzenia). Wciągnąć koncentrat do strzykawki powoli wyciągając tłoczek (Rys. e). 3. Odłączyć strzykawkę. 4. Podłączyć zestaw do podania do strzykawki. Podawać dożylnie. Preparat można podawać z prędkością do 10 ml na minutę. Przed i podczas podawania Recombinate należy kontrolować tętno pacjenta. W przypadku istotnego wzrostu tętna, zmniejszenie szybkości wlewu lub tymczasowe przerwanie iniekcji zwykle szybko powoduje ustąpienie objawów (patrz punkty 4.4 i 4.8). Rys. d Rys. e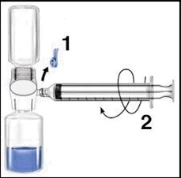 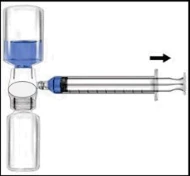 | Zaleca się rozpoczęcie podania nie później niż trzy godziny po rekonstytucji. Nie należy chłodzić odtworzonego produktu. Przed podaniem leki do stosowania parenteralnego należy poddać wizualnej kontroli pod kątem obecności cząstek obcych lub nieprawidłowego zabarwienia, o ile pozwala na to roztwór i opakowanie. Dla Recombinate dopuszczalne jest zabarwienie od bezbarwnego do lekko żółtawego. 1. Podłączyć igłę z filtrem do jednorazowej strzykawki i wyciągnąć tłoczek, aby wprowadzić powietrze do strzykawki. 2. Wprowadzić igłę z filtrem do odtworzonego Recombinate. 3. Wprowadzić powietrze do butelki, a następnie wciągnąć odtworzony materiał do strzykawki. 4. Usunąć i wyrzucić igłę z filtrem. Podłączyć zestaw do podania do strzykawki. Podawać dożylnie. Preparat można podawać z prędkością do 10 ml na minutę. Przed i podczas podawania Recombinate należy kontrolować tętno pacjenta. W przypadku istotnego wzrostu tętna, zmniejszenie szybkości wlewu lub tymczasowe przerwanie iniekcji zwykle szybko powoduje ustąpienie objawów (patrz punkty 4.4 i 4.8). 5. Do wciągnięcia zawartości każdej butelki odtworzonego Recombinate należy użyć nowej, nieużywanej igły z filtrem. |
7. WŁAŚCICIEL AUTORIZACJI DO WPUSZCZENIA NA RYNEK
Baxalta Innovations GmbH - Industriestrasse 67, A-1221 Vienna
8. NUMER W ZEZWOLENIA NA WPUSZCZENIE DO OBROTU
Zezwolenie AIC nr 028687010 – „250 JEDN./10 ml proszek i rozpuszczalnik do sporządzenia roztworu do wstrzykiwań” 1 fiolka proszek + 1 fiolka rozpuszczalnika z urządzeniem do rekonstytucji bez igły
Zezwolenie AIC nr 028687022 – „500 JEDN./10 ml proszek i rozpuszczalnik do sporządzenia roztworu do wstrzykiwań” 1 fiolka proszek + 1 fiolka rozpuszczalnika z urządzeniem do rekonstytucji bez igły
Zezwolenie AIC nr 028687034 – „1000 JEDN./10 ml proszek i rozpuszczalnik do sporządzenia roztworu do wstrzykiwań” 1 fiolka proszek + 1 fiolka rozpuszczalnika z urządzeniem do rekonstytucji bez igły
Zezwolenie AIC nr 028687109 – „250 JEDN./10 ml proszek i rozpuszczalnik do sporządzenia roztworu do wstrzykiwań” 1 fiolka proszek + 1 fiolka rozpuszczalnika z urządzeniem do rekonstytucji z dwugrotową igłą + igła filtracyjna
Zezwolenie AIC nr 028687111 – „500 JEDN./10 ml proszek i rozpuszczalnik do sporządzenia roztworu do wstrzykiwań” 1 fiolka proszek + 1 fiolka rozpuszczalnika z urządzeniem do rekonstytucji z dwugrotową igłą + igła filtracyjna
Zezwolenie AIC nr 028687123 – „1000 JEDN./10 ml proszek i rozpuszczalnik do sporządzenia roztworu do wstrzykiwań” 1 fiolka proszek + 1 fiolka rozpuszczalnika z urządzeniem do rekonstytucji z dwugrotową igłą + igła filtracyjna
9. DATA PIERWSZEGO POZWOLENIA / ODNOWIENIA POZWOLENIA
Data pierwszego pozwolenia: 12 maja 1993
Data najnowszego odnowienia: 17 maja 2010