Refact AF
UcrainaIndice
ISTRUZIONI PER L'USO MEDICINALE DEL MEDICINALE ReFacto AF (ReFacto® AF)
Composizione:
principio attivo: moroctocog alfa (fattore della coagulazione del sangue VIII ricombinante);
1 flaconcino contiene 250 UI oppure 500 UI oppure 1000 UI oppure 2000 UI di moroctocog alfa;
1 siringa preriempita contiene 3000 UI di moroctocog alfa;
eccipienti: saccarosio, cloruro di calcio diidrato, L-istidina, polisorbato 80, cloruro di sodio.
solvente: cloruro di sodio, acqua per preparazioni iniettabili.
Forma farmaceutica.
Liofilizzato per soluzione iniettabile.
Principali caratteristiche fisico-chimiche:
forma farmaceutica – flaconcino:
liofilizzato di colore bianco, praticamente privo di particelle estranee visibili chiaramente, umidità e difetti di chiusura del flaconcino;
solvente: soluzione incolore e limpida, praticamente priva di inclusioni visibili;
forma farmaceutica – siringa preriempita:
nella camera superiore – liofilizzato di colore bianco, praticamente privo di particelle estranee visibili chiaramente, umidità e difetti di chiusura del contenitore;
nella camera inferiore – solvente: soluzione incolore e limpida, praticamente priva di inclusioni visibili.
Gruppo farmacoterapeutico.
Agenti emostatici. Fattore della coagulazione del sangue VIII. Codice ATC B02BD02.
Proprietà farmacologiche
Farmacodinamica
Refact AF contiene il fattore ricombinante della coagulazione del sangue VIII con dominio B rimosso (moroctocog alfa). L'attività del farmaco è espressa in unità internazionali (UI) e determinata mediante metodo cromogenico secondo la Farmacopea Europea. L'attività specifica del medicinale Refact AF è compresa tra 7.600 e 13.800 UI/mg di proteina.
Il moroctocog alfa è una glicoproteina con una massa molecolare approssimativa di 170.000 Da, costituita da 1438 amminoacidi, la cui sequenza è simile alla forma di 90 + 80 kDa del fattore VIII (cioè con il dominio B rimosso), e le modifiche post-traduzionali sono simili a quelle presenti nella molecola ottenuta dal plasma sanguigno.
Il moroctocog alfa è prodotto mediante tecnologia del DNA ricombinante utilizzando cellule ovariche di criceto cinese. Il processo produttivo di Refact AF è stato modificato in modo da escludere la possibilità di contaminazione con proteine esogene di origine umana o animale durante la coltivazione cellulare, la purificazione o la produzione del prodotto finito; contemporaneamente, il nome del farmaco è stato modificato da Refact a Refact AF.
Le caratteristiche funzionali di Refact AF sono simili a quelle del fattore VIII endogeno. L'attività del fattore VIII è notevolmente ridotta nei pazienti con emofilia A, che necessitano pertanto di una terapia sostitutiva.
Dopo somministrazione per infusione ai pazienti con emofilia, il fattore VIII somministrato si lega al fattore di von Willebrand circolante nel sangue.
Il fattore VIII attivato agisce come cofattore del fattore IX attivato, accelerando la trasformazione del fattore X in fattore X attivato. Il fattore X attivato converte la protrombina in trombina. La trombina a sua volta trasforma il fibrinogeno in fibrina, formando il trombo. L'emofilia A è un disturbo ereditario legato al sesso della coagulazione del sangue, determinato da livelli ridotti di fattore VIII:C, che porta a emorragie abbondanti nelle articolazioni, nei muscoli o negli organi interni. Le emorragie possono verificarsi spontaneamente o in seguito a traumi accidentali o chirurgici. Con la terapia sostitutiva, i livelli plasmatici di fattore VIII aumentano, consentendo di correggere temporaneamente la carenza di questo fattore e ridurre la tendenza alle emorragie.
Efficacia clinica
I dati riportati nella Tabella 1 sono stati ottenuti da studi sull'uso del medicinale Refact AF in pazienti precedentemente non trattati (PNT) e precedentemente trattati (PT), di età inferiore ai 12 anni.
Tabella 1
Dosaggio ed efficacia nell'uso in pazienti pediatrici
| Indicatore |
PLP |
PLP |
PNP |
| Dosaggio in base al peso corporeo (UI/kg) per infusione profilatticaa |
N = 14 (28, 51) |
N = 13 (21, 49) |
N = 22 (17, 161) |
| TRR totale tra tutti i soggetti, |
-- |
-- |
N = 23 (0,0; 39,5) |
| TRR totale nei soggetti che all'inizio dello studio seguivano una terapia “a richiesta”c, |
N = 5 (1,6; 50,6) |
N = 9 (0,0; 46,6) |
-- |
| TRR totale nei soggetti che all'inizio dello studio seguivano una terapia “profilattica”c, |
N = 13 (0,0; 11,2) |
N = 9 (0,0; 13,0) |
-- |
| Dosaggio in base al peso corporeo (UI/kg) durante episodi emorragici per il trattamento dell'emorragia, mediana (min., max.) |
N = 13 (28; 86) |
N = 14 (17; 229) |
N = 21 (11; 221) |
| % di emorragie trattate con successo con ≤ 2 infusioni |
98,7 % |
98,8 % |
96,7 % |
a Il dosaggio e la frequenza di somministrazione del medicinale Refact AF nello studio sono stati determinati dal medico sulla base delle linee guida terapeutiche locali.
b I soggetti nello studio PNP non erano tenuti a seguire un trattamento profilattico regolare e continuo; tuttavia, ad eccezione di un soggetto (a cui è stato somministrato esclusivamente il trattamento "a richiesta"), la maggior parte dei soggetti ha ricevuto infusioni profilattiche regolari. Alcuni soggetti hanno iniziato lo studio con infusioni "a richiesta" per poi passare a un trattamento profilattico, mentre altri hanno ricevuto infusioni profilattiche solo in modo sporadico.
c I soggetti nello studio PLP hanno indicato all'inizio dello studio il regime terapeutico con fattore VIII ("profilassi" o "a richiesta") e non erano tenuti a mantenere tale regime come condizione per partecipare allo studio. Il dosaggio e la frequenza di somministrazione del medicinale Refact AF nello studio sono stati determinati dal medico sulla base delle linee guida terapeutiche locali.
Abbreviazioni: TCR – tasso cumulativo annuo di emorragie.
È importante notare che il parametro TCR non è comparabile tra diversi concentrati di fattori della coagulazione né tra diversi studi clinici.
Induzione della tolleranza immunologica.
Sono stati raccolti dati sull'induzione della tolleranza immunologica in pazienti con emofilia A nei quali si sono sviluppati inibitori del fattore VIII. Nel contesto dello studio clinico principale con Refact AF, che ha coinvolto 25 pazienti mai trattati in precedenza, sono stati raccolti dati sull'induzione della tolleranza immunologica (15 con titolo elevato, 10 con titolo basso). Su questi 25 pazienti, in 20 il titolo degli inibitori si è ridotto a un livello < 0,6 UBT (unità di Bethesda), di cui 11 su 15 avevano inizialmente titoli elevati (≥ 5 UBT/ml) e 9 su 10 titoli bassi. Una riduzione simile dei titoli è stata osservata in 5 su 6 pazienti con titoli bassi di inibitori che non avevano ricevuto trattamento per l'induzione della tolleranza immunologica. Non sono stati effettuati follow-up a lungo termine.
Farmacocinetica.
Le proprietà farmacocinetiche di Refact AF, determinate in uno studio incrociato con Refact AF e un concentrato di fattore VIII ottenuto dal plasma, condotto su 18 pazienti precedentemente trattati (pazienti precedentemente esposti, PLP), sono riportate nella tabella 2. Le analisi quantitative sono state effettuate mediante substrato cromogeno (vedere sezione «Modalità di somministrazione e dosi»).
Tabella 2
| Valutazione dei parametri farmacocinetici di Refact AF in pazienti con emofilia A |
|||
| Parametro |
Media | DS |
Mediana |
| AUCt (MO·h/ml) |
19,9 |
4,9 |
19,9 |
| t½ (h) |
14,8 |
5,6 |
12,7 |
| CL (ml/h·kg) |
2,4 |
0,75 |
2,3 |
| tempo medio di permanenza (h) |
20,2 |
7,4 |
18,0 |
| recupero (MO/dl aumento del fattore VIII:C per MO/kg di fattore VIII somministrato) |
2,4 |
0,38 |
2,5 |
Abbreviazioni: AUCt – area sotto la curva «concentrazione nel plasma-tempo» dal tempo zero fino all'ultima concentrazione misurata; t½ – emivita; CL – clearance; CV – deviazione standard.
In uno studio in cui l'attività di Refacto AF, Refacto e del fattore VIII nel plasma sanguigno dei pazienti è stata confrontata mediante analisi cromogenica, è stata dimostrata la bioequivalenza tra Refacto AF e Refacto. Il rapporto tra le medie geometriche dei parametri farmacocinetici calcolati con il metodo dei minimi quadrati per Refacto AF e Refacto è risultato pari a 100,6%, 99,5% e 98,1% rispettivamente per il recupero, AUCt e AUC∞ (area sotto la curva «concentrazione nel plasma-tempo» dal tempo zero all'infinito). I corrispondenti intervalli di confidenza al 90% per il rapporto tra le medie geometriche dei parametri farmacocinetici di Refacto AF e Refacto rientravano nella finestra di bioequivalenza (80-125%), dimostrando la bioequivalenza tra Refacto AF e Refacto.
In uno studio farmacocinetico incrociato, i parametri farmacocinetici di Refacto AF sono stati determinati in 25 pazienti (≥ 12 anni) precedentemente trattati, all'inizio dello studio e dopo sei mesi di somministrazione ripetuta di Refacto AF. Il rapporto tra le medie geometriche dei parametri farmacocinetici calcolati con il metodo dei minimi quadrati dopo 6 mesi di trattamento rispetto ai valori basali è risultato pari a 107%, 100% e 104% rispettivamente per il recupero, AUCt e AUC∞. I corrispondenti intervalli di confidenza al 90% per il rapporto tra i parametri farmacocinetici dopo 6 mesi di studio rispetto ai valori basali erano entro i limiti della finestra di equivalenza (80-125%). Questo indica l'assenza di variazioni dipendenti dal tempo nelle proprietà farmacocinetiche di Refacto AF.
Nello stesso studio, l'attività di Refacto AF, di un fattore VIII ricombinante a lunghezza completa (farmaco di confronto) e del fattore VIII è stata misurata nei campioni di plasma di pazienti (30 pazienti precedentemente trattati, ≥ 12 anni) mediante un saggio di coagulazione in un unico passaggio. Refacto AF ha dimostrato bioequivalenza farmacocinetica rispetto al farmaco di confronto, applicando il criterio standard per stabilire la bioequivalenza.
Nei pazienti mai trattati in precedenza, i parametri farmacocinetici di Refacto sono stati valutati mediante analisi cromogenica. In questi pazienti (n = 59; età mediana 10 ± 8,3 mesi), il recupero medio alla settimana «0» era di 1,5 ± 0,6 UI/dl per UI/kg (intervallo 0,2–2,8 UI/dl per UI/kg), valore inferiore rispetto a quello osservato nei pazienti precedentemente trattati alla settimana «0», con un recupero medio di 2,4 ± 0,4 UI/dl per UI/kg (intervallo 1,1–3,8 UI/dl per UI/kg). Nei pazienti mai trattati in precedenza, il recupero medio è rimasto stabile durante il periodo di due anni (5 visite durante questo periodo), oscillando tra 1,5 e 1,8 UI/dl per UI/kg. Uno studio basato su un modello farmacocinetico popolazionale, che includeva i dati di 44 pazienti mai trattati in precedenza, ha mostrato un'emivita media prevista pari a 8,0 ± 2,2 ore.
In uno studio sull'uso di Refacto AF coinvolgente 19 pazienti mai trattati in precedenza (PTP), il livello di recupero è stato di 1,32 ± 0,65 UI/dl per UI/kg in 17 bambini di età compresa tra 28 giorni e 2 anni e di 1,7 e 1,8 UI/dl per UI/kg in 2 bambini di età compresa tra 2 e < 6 anni. A parte i casi in cui sono stati rilevati inibitori, il livello medio di recupero è rimasto stabile nel tempo (6 visite durante un periodo di 2 anni), con valori individuali compresi tra 0 (in presenza di inibitore) e 2,7 UI/dl per UI/kg. Nella Tabella 3 sono riportati i parametri farmacocinetici osservati per Refacto AF dopo somministrazione di una dose di 50 UI/kg in uno studio condotto su 37 bambini precedentemente trattati.
Tabella 3
| Valori medi ± DS dei parametri farmacocinetici del fattore VIII dopo una singola dose di 50 UI/kg in bambini (PSP) |
||
| Parametro |
Numero di soggetti |
Valore medioa ± DS |
| Risultato, UI/dl per UI/kg Di età inferiore a 6 anni Di età compresa tra 6 e 12 anni |
17 19 |
1,7 ± 0,4 2,1 ± 0,8 |
| Cmax, UI/mlb |
19 |
0,9 (45) |
| AUCinf, UI∙ora/mlb |
14 |
9,9 (41) |
| t½, orab |
14 |
9,1 ± 1,9 |
| CL, ml/ora/kgb |
14 |
4,4 (30) |
| Vss, ml/kgb |
14 |
56,4 (15) |
| a Media geometrica (CV% geometrico) per tutti i parametri, eccetto la media aritmetica ± DS per il recupero incrementale e t½. b Solo pazienti di età compresa tra 6 e 12 anni. Abbreviazioni: Cmax – concentrazione plasmatica massima osservata; CV – coefficiente di variazione; AUCinf – area sotto la curva concentrazione plasmatica-tempo da 0 all’infinito; t½ – emivita; CL – clearance; DS – deviazione standard; Vss – volume di distribuzione allo stato stazionario. |
||
Caratteristiche cliniche.
Indicazioni.
Trattamento e profilassi delle emorragie nei pazienti con emofilia A (deficit congenito del fattore della coagulazione del sangue VIII).
Refact AF è indicato per l'uso in adulti e bambini di tutte le età, inclusi neonati.
Refact AF non contiene il fattore di von Willebrand e pertanto non è indicato per il trattamento della malattia di von Willebrand.
Controindicazioni.
Ipersensibilità al principio attivo o a qualsiasi componente del prodotto.
Reazione allergica nota alle proteine del criceto.
Interazioni con altri medicinali e altre forme di interazione.
Non sono state riportate interazioni tra il fattore della coagulazione umano ricombinante VIII e altri farmaci.
Caratteristiche di impiego.
Tracciabilità
Per migliorare il tracciamento dei medicinali biologici, è necessario registrare chiaramente il nome e il numero di lotto del medicinale somministrato. A tale scopo, i pazienti possono applicare una delle etichette autoadesive staccabili prelevate dal flaconcino o dalla siringa preriempita sul proprio diario personale per documentare il numero di lotto, oppure utilizzarla per segnalare eventuali effetti indesiderati.
Ipersensibilità
Refact AF può causare reazioni di ipersensibilità di tipo allergico. Il medicinale contiene tracce di proteine di criceto. I pazienti devono essere informati che, in caso di comparsa di sintomi di ipersensibilità, devono interrompere immediatamente l'uso del medicinale e consultare un medico. I pazienti devono segnalare tempestivamente i segni iniziali di reazioni di ipersensibilità, inclusi orticaria, orticaria generalizzata, sensazione di oppressione al torace, respiro sibilante, ipotensione arteriosa e anafilassi.
In caso di insorgenza di shock, deve essere attuato il trattamento medico standard per lo stato di shock.
Inibitori
La formazione di anticorpi neutralizzanti (inibitori) del fattore VIII è una complicanza nota che può verificarsi nel trattamento di pazienti con emofilia A. Tali anticorpi sono generalmente immunoglobuline IgG dirette contro l'attività procoagulante del fattore VIII. La loro presenza nel plasma viene determinata mediante un metodo quantitativo modificato e espressa in unità di Bethesda (UB) per 1 ml. Il rischio di formazione di inibitori è correlato alla gravità della malattia e all'esposizione al fattore VIII, ed è massimo durante i primi 50 giorni di trattamento, ma permane per tutta la vita, sebbene con frequenza rara.
L'importanza clinica della formazione di inibitori dipende dal loro titolo: gli inibitori a basso titolo comportano un rischio minore di risposta clinica inadeguata rispetto a quelli ad alto titolo.
È necessario effettuare un attento monitoraggio clinico e di laboratorio dei pazienti in trattamento con fattori ricombinanti della coagulazione VIII per rilevare l'eventuale formazione di inibitori. Se non si raggiungono i livelli attesi di attività del fattore VIII nel plasma o se il sanguinamento non si arresta nonostante la somministrazione della dose appropriata del medicinale, devono essere effettuati test per determinare la presenza di inibitori del fattore VIII. Nei pazienti con livelli elevati di inibitori, la terapia con fattore VIII può risultare inefficace e si deve considerare l'uso di altri medicinali. Il trattamento di tali pazienti deve essere effettuato da medici esperti nella gestione dell'emofilia e della formazione di inibitori del fattore VIII.
Segnalazione di inefficacia
Negli studi clinici e durante l'uso post-marketing di Refact AF sono stati segnalati casi di inefficacia del medicinale, soprattutto nei pazienti che utilizzavano il prodotto a scopo profilattico. L'inefficacia è stata descritta come emorragie nei giunti articolari target, emorragie in nuovi giunti articolari o percezione soggettiva da parte del paziente dell'inizio di una nuova emorragia. Quando si prescrive Refact AF, è importante personalizzare il dosaggio e monitorare il livello del fattore in ciascun paziente per garantire una risposta terapeutica adeguata (vedere sezione «Effetti indesiderati»).
Complicanze cardiovascolari
Nei pazienti con fattori di rischio preesistenti per complicanze cardiovascolari, il trattamento sostitutivo con fattore VIII può aumentare i rischi cardiovascolari.
Complicanze associate al catetere
Qualora sia necessario utilizzare un dispositivo per l'accesso venoso centrale, si devono considerare i rischi di complicanze legate al suo utilizzo, inclusi infezioni locali, batteriemia e trombosi del catetere (vedere sezione «Effetti indesiderati»).
Contenuto di sodio
Dopo ricostituzione, un flaconcino o una siringa preriempita contengono 1,27 mmol (o 29 mg) di sodio, pari al 1,5% della dose giornaliera massima raccomandata dall'OMS per un adulto (2 g). A seconda del peso corporeo del paziente e della dose di Refact AF, i pazienti possono dover utilizzare più flaconcini o siringhe preriempite. Tale informazione deve essere presa in considerazione nei pazienti che seguono una dieta a basso contenuto di sodio.
Uso in gravidanza o durante l'allattamento.
Non sono stati condotti studi sull'effetto del fattore VIII sulla funzione riproduttiva negli animali, pertanto non sono disponibili dati sul suo effetto sulla fertilità. Poiché l'emofilia A è rara nelle donne, non esiste esperienza sull'uso del fattore VIII durante la gravidanza e l'allattamento. Pertanto, il fattore VIII deve essere utilizzato durante la gravidanza e l'allattamento al seno solo in caso di effettiva necessità.
Capacità di guidare veicoli o di usare macchinari.
Refact AF non influenza la capacità di guidare veicoli o di usare macchinari.
Modalità e dosaggio di somministrazione.
Il trattamento deve essere iniziato sotto la supervisione di un medico esperto nel trattamento dell'emofilia A.
Monitoraggio del trattamento.
Per determinare la dose necessaria durante il corso del trattamento e la frequenza delle infusioni ripetute, si raccomanda di effettuare analisi del livello del fattore VIII. La risposta al fattore VIII può variare tra i diversi pazienti, manifestandosi con diversi livelli di recupero e diversi periodi di emivita. La dose calcolata in base al peso corporeo del paziente potrebbe richiedere aggiustamenti nei pazienti con peso corporeo eccessivo o insufficiente. In particolare, in caso di interventi chirurgici importanti, è obbligatorio un accurato monitoraggio della terapia sostitutiva mediante analisi della coagulazione (attività del fattore VIII nel plasma).
Per il monitoraggio del livello di attività del fattore VIII nei pazienti durante il trattamento con Refact AF, si raccomanda l'uso del metodo cromogenico. Se per determinare l'attività del fattore VIII nei campioni di sangue dei pazienti si utilizza il tempo di tromboplastina parziale attivato in vitro (aPTT), calcolato mediante analisi in una sola fase dell'attività coagulativa, i risultati dell'attività del fattore VIII possono essere influenzati sia dal tipo di reagenti che dagli standard utilizzati nella quantificazione. Inoltre, possono sussistere differenze significative tra i risultati della quantificazione ottenuti con il metodo cromogenico e quelli ottenuti con l'aPTT mediante analisi in una sola fase dell'attività coagulativa. Di solito, i risultati ottenuti con l'analisi in una sola fase sono inferiori del 20–50% rispetto ai risultati della quantificazione ottenuti con il metodo cromogenico. Per correggere questa discrepanza, può essere utilizzato lo standard di laboratorio Refact AF (vedere la sezione «Farmacocinetica»). Questo è particolarmente importante quando si utilizzano laboratori e/o reagenti diversi.
Dosaggio.
Il dosaggio e la durata della terapia sostitutiva dipendono dal grado di carenza del fattore VIII, dalla localizzazione e dalla gravità dell'emorragia, nonché dallo stato clinico del paziente. Le dosi utilizzate devono essere adattate in base alla risposta clinica del paziente. In presenza di inibitori, potrebbe essere necessario l'uso di dosi più elevate del medicinale o la prescrizione di un trattamento specifico appropriato.
La quantità di unità di fattore VIII somministrata viene espressa in UI legate allo standard internazionale dell'OMS per i preparati contenenti fattore VIII. L'attività del fattore VIII nel plasma sanguigno viene espressa in percentuale (rispetto al suo contenuto nel plasma umano normale) oppure in unità internazionali (rispetto allo standard internazionale per il fattore VIII nel plasma). 1 UI di attività del fattore VIII equivale alla quantità di fattore VIII presente in 1 ml di plasma umano normale.
L'attività di un altro medicinale, moroctocog alfa (Xyntha), registrato al di fuori dell'Ucraina, viene determinata mediante uno standard produttivo calibrato rispetto allo standard internazionale dell'OMS utilizzando un'analisi in una sola fase dell'attività coagulativa. A causa delle differenze tra i metodi utilizzati per determinare l'attività dei preparati Xyntha e Refact AF, 1 UI del medicinale Xyntha (calibrato mediante analisi in una sola fase) equivale approssimativamente a 1,38 UI del medicinale Refact AF (calibrato mediante analisi cromogenica). Se un paziente che normalmente riceve il trattamento con Xyntha viene trattato con Refact AF, il medico potrebbe prendere in considerazione la possibilità di aggiustare la dose in base ai valori di recupero del livello del fattore VIII.
Ai pazienti con emofilia A si raccomanda di avere sempre a disposizione una quantità sufficiente di fattore VIII in conformità allo schema terapeutico in vigore, nel caso in cui il trattamento fosse necessario durante un viaggio. Si consiglia ai pazienti di consultare il medico prima di intraprendere un viaggio.
Trattamento su richiesta.
Il calcolo della dose necessaria di fattore VIII si basa su dati empirici secondo cui 1 UI di fattore VIII per 1 kg di peso corporeo aumenta l'attività del fattore VIII nel plasma sanguigno di 2 UI/dl. La dose necessaria viene calcolata secondo la seguente formula:
quantità necessaria di unità (UI) =
peso corporeo (kg) × aumento desiderato del fattore VIII (%, oppure UI/dl) × 0,5 (UI/kg per UI/dl),
dove 0,5 UI/kg per UI/dl rappresenta il reciproco del recupero, che di solito si osserva dopo l'infusione del fattore VIII.
La dose del medicinale prescritto e la frequenza di somministrazione devono sempre essere determinate caso per caso in base all'efficacia clinica del medicinale.
In caso di emorragia, l'attività del fattore VIII nel plasma sanguigno non deve scendere al di sotto dei livelli (in % o in UI/dl) indicati nella tabella 4. Tale tabella può essere utilizzata come guida per il dosaggio del fattore VIII in caso di emorragia e di intervento chirurgico.
Tabella 4
| Gravità dell’emorragia/ tipo di intervento chirurgico |
Livello necessario del fattore VIII (% del normale, o UI/dl di plasma) |
Frequenza di somministrazione (ore)/ durata della terapia (giorni) |
| Emorragia |
||
| Primi segni di emartrosi, emorragie muscolari o emorragie orali |
20–40 |
Ripetere la somministrazione ogni 12–24 ore per almeno 1 giorno fino alla cessazione dell’emorragia, indicata dall’assenza di dolore o dalla presenza di guarigione |
| Emartrosi, emorragie muscolari di grado moderato o ematomi |
30–60 |
Ripetere la somministrazione ogni 12–24 ore per 3–4 giorni o più fino alla scomparsa del dolore e al ripristino della mobilità articolare |
| Emorragie che mettono in pericolo la vita |
60–100 |
Ripetere la somministrazione ogni 8–24 ore fino alla scomparsa del pericolo per la vita |
| Interventi chirurgici |
||
| Interventi chirurgici minori, inclusa l’estrazione dentaria |
30–60 |
Ripetere la somministrazione ogni 24 ore per almeno 1 giorno fino alla guarigione |
| Interventi chirurgici maggiori |
80–100 (prima e dopo l’intervento chirurgico) |
Ripetere la somministrazione ogni 8–24 ore fino a un adeguato rimarginamento della ferita, quindi continuare la terapia per almeno 7 giorni per mantenere l’attività del fattore VIII al livello del 30–60 % (UI/dl) |
Prevenzione
Per la prevenzione prolungata delle emorragie nei pazienti con emofilia A grave, si utilizzano solitamente 20–40 UI di fattore VIII per kg di peso corporeo ogni 2–3 giorni. In alcuni casi, specialmente nei pazienti più giovani, può rendersi necessario aumentare la dose o la frequenza di somministrazione del medicinale.
Pediatria
Quando si tratta bambini più piccoli (di età inferiore ai 6 anni) con Refact AF, può essere necessario utilizzare dosi più elevate rispetto a quelle impiegate negli adulti e nei bambini più grandi (vedere la sezione «Farmacocinetica»).
Pazienti anziani
I pazienti di età pari o superiore a 65 anni non sono stati inclusi negli studi clinici. In generale, la dose nei pazienti anziani deve essere adattata individualmente.
Pazienti con insufficienza renale o epatica
La necessità di aggiustamento della dose nei pazienti con insufficienza renale o epatica non è stata studiata negli studi clinici.
Modalità di somministrazione
Per via endovenosa.
Refact AF viene somministrato per infusione endovenosa lenta per alcuni minuti dopo la ricostituzione con il solvente appropriato. La velocità di somministrazione deve essere determinata in base al comfort del paziente. Si raccomanda un'adeguata formazione per le persone che somministrano il medicinale ma che non sono operatori sanitari.
Forma farmaceutica – flacone
Il prodotto liofilizzato viene ricostituito con il solvente appropriato (soluzione di sodio cloruro 0,9 %) fornito in una siringa preriempita, utilizzando un adattatore sterile. Dopo l’aggiunta del solvente, il flacone deve essere ruotato delicatamente fino a completa dissoluzione della polvere.
Ulteriori informazioni dettagliate sulla preparazione e sull’uso della soluzione sono riportate di seguito.
Dopo la ricostituzione, la soluzione ottenuta viene aspirata nuovamente nella siringa. La soluzione deve essere limpida o leggermente opalescente e incolore. La soluzione deve essere scartata se si osservano particelle estranee o un cambiamento di colore.
Forma farmaceuticа – siringa preriempita:
La polvere liofilizzata nella camera superiore della siringa preriempita deve essere ricostituita con il solvente (soluzione di sodio cloruro 9 mg/ml (0,9 %)) contenuto nella camera inferiore. La siringa preriempita deve essere ruotata delicatamente finché tutta la polvere non si sarà completamente sciolta. Ulteriori informazioni dettagliate sulla preparazione e sull’uso della soluzione sono riportate di seguito.
Dopo la ricostituzione, la soluzione sarà limpida o leggermente opalescente e incolore. La soluzione deve essere scartata se si osservano inclusioni meccaniche visibili o un cambiamento di colore.
Il medicinale Refact AF, dopo la ricostituzione, contiene polisorbato 80, che può accelerare l’estrazione del di-(2-etilesil)ftalato dal polivinilcloruro (PVC). Tale caratteristica deve essere tenuta in considerazione durante la preparazione e la somministrazione del medicinale, compreso il tempo di conservazione in contenitori in PVC dopo la ricostituzione della soluzione. È fondamentale seguire attentamente le raccomandazioni riportate nella sezione «Condizioni di conservazione».
Eventuali residui non utilizzati del medicinale o i materiali impiegati per la ricostituzione e la somministrazione devono essere smaltiti in conformità con i requisiti locali.
Forma farmaceutica – flacone
Preparazione della soluzione:
- Portare il liofilizzato e il solvente nella siringa preriempita alla temperatura ambiente.
- Rimuovere il tappo di plastica a chiusura rapida dal flacone Refact AF per esporre la parte centrale del tappo in gomma.
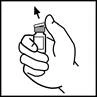
- Disinfettare la parte superiore del flacone con un tampone alcolico incluso nella confezione o con un altro disinfettante e lasciare asciugare. Dopo la pulizia, non toccare il tappo in gomma né farlo venire a contatto con alcuna superficie.
- Sollevare il rivestimento protettivo della confezione in plastica trasparente dell’adattatore per il flacone. Non rimuovere l’adattatore dalla confezione.
- Posizionare il flacone su una superficie piana. Tenendo la confezione con l’adattatore, posizionare l’adattatore sul flacone. Premere con forza fino a quando l’adattatore si inserisce con un clic nella parte superiore del flacone e la punta penetra attraverso il tappo.
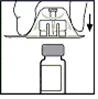
- Rimuovere la confezione dall’adattatore e smaltirla.
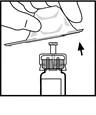
- Collegare lo stantuffo alla siringa con solvente: inserire lo stantuffo nell’apertura della chiusura della siringa, premere e ruotare fino a quando si inserisce saldamente nella chiusura.
- Rimuovere il cappuccio protettivo con indicatore di prima apertura dalla siringa con solvente, rompendo la perforazione del cappuccio. Ciò si ottiene agitando il cappuccio finché la perforazione non si rompe. Non toccare la superficie interna del cappuccio né la punta della siringa. Poiché potrebbe essere necessario riapplicare il cappuccio (se Refact AF non viene utilizzato immediatamente), posizionarlo con l’apertura rivolta verso l’alto.
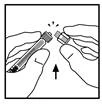
- Posizionare il flacone su una superficie piana. Collegare la siringa con solvente all’adattatore del flacone inserendo la punta della siringa nell’apertura dell’adattatore, premendo con forza e ruotando in senso orario fino a ottenere un collegamento sicuro.
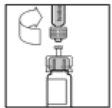
- Premere lentamente lo stantuffo per iniettare tutto il solvente nel flacone di Refact AF.
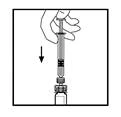
- Mantenendo la siringa collegata all’adattatore, ruotare delicatamente il flacone fino a completa dissoluzione della polvere.
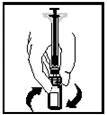
- Prima della somministrazione, la soluzione preparata deve essere ispezionata visivamente per verificare la presenza di particelle estranee. La soluzione ottenuta deve essere limpida o leggermente opalescente e incolore.
Se si utilizzano più flaconi di Refact AF per un’infusione, ogni flacone deve essere preparato secondo le istruzioni precedenti. Successivamente, rimuovere la siringa del solvente, lasciando l’adattatore sul flacone, e utilizzare una singola siringa più grande con bloccaggio a tenone per aspirare la soluzione da ciascun flacone.
- Una volta verificato che lo stantuffo sia completamente inserito nel cilindro della siringa, capovolgere il flacone. Aspirare lentamente tutta la soluzione attraverso l’adattatore del flacone nella siringa.
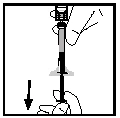
- Scollegare la siringa dall’adattatore del flacone, staccandola con cura e ruotando in senso antiorario. Smaltire il flacone con l’adattatore collegato.
Se la soluzione non viene utilizzata immediatamente, il cappuccio della siringa deve essere riposizionato con attenzione. Non toccare la punta della siringa né la superficie interna del cappuccio.
La soluzione preparata deve essere utilizzata immediatamente o entro 3 ore dalla ricostituzione. Prima della somministrazione, la soluzione può essere conservata a una temperatura non superiore a 25 °C.
Somministrazione (infusione endovenosa)
Refact AF deve essere somministrato utilizzando il set per infusione fornito e la siringa preriempita con solvente fornita, oppure una singola siringa sterile monouso in plastica con bloccaggio a tenone.
- Collegare la siringa al connettore a tenone del catetere del set per infusione.
- Applicare un laccio emostatico e preparare l’area di iniezione pulendo accuratamente la cute con un tampone alcolico fornito nel kit.
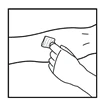
- Inserire l’ago del catetere del set per infusione in una vena e rimuovere il laccio emostatico. Rimuovere l’aria dal catetere del set per infusione aspirando con la siringa. Il medicinale preparato deve essere somministrato per via endovenosa in pochi minuti. Il medico può modificare la velocità raccomandata dell’infusione per rendere la procedura più confortevole per il paziente.
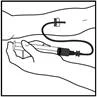
Forma farmaceutica – siringa preriempita
Preparazione della soluzione:
- Portare la siringa preriempita alla temperatura ambiente.
- Preparare il kit per la siringa preriempita Refact AF e posizionarlo su una superficie pulita, assicurandosi di avere tutti i componenti necessari.
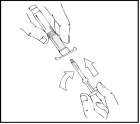
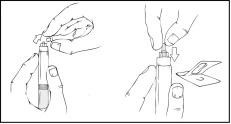
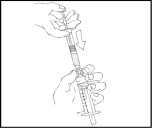
- Tenere lo stantuffo come mostrato nello schema seguente. Inserire saldamente lo stantuffo nell’apertura del supporto per le dita della siringa preriempita Refact AF premendo e ruotando in senso orario fino a quando si sente resistenza (circa 2 giri).
Durante l’intero processo di preparazione della soluzione, la siringa preriempita Refact AF deve essere mantenuta in posizione verticale (la polvere bianca deve trovarsi sopra il solvente trasparente) per evitare fuoriuscite.
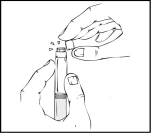
- Tenendo la siringa preriempita in posizione verticale, rimuovere il cappuccio protettivo bianco con indicatore di prima apertura, staccandolo da destra verso sinistra (o con movimenti oscillatori delicati), per rompere la perforazione del cappuccio e visualizzare il cappuccio in gomma grigio sulla punta della siringa preriempita Refact AF.
- Estrarre dalla confezione il cappuccio protettivo sterile blu valvolato.
Mantenendo costantemente la siringa preriempita Refact AF in posizione verticale, rimuovere il cappuccio in gomma grigio e sostituirlo con il cappuccio protettivo blu valvolato. Questo cappuccio protettivo presenta piccoli fori che permettono all’aria di uscire, prevenendo un’eccessiva pressione. Non toccare l’estremità aperta della siringa né il cappuccio protettivo blu.
- Con attenzione e lentamente spingere lo stantuffo, premendo fino a quando i due stantuffi all’interno della siringa preriempita si incontrano e tutto il solvente passa nella camera superiore contenente la polvere di Refact AF.
Nota: Per evitare fuoriuscite di liquido dalla punta della siringa, non premere lo stantuffo con forza eccessiva.
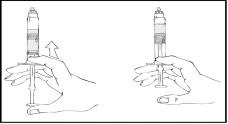
- Tenendo la siringa Refact AF in posizione verticale, ruotarla delicatamente fino a completa dissoluzione della polvere.
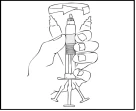
Controllare visivamente la soluzione per verificare la presenza di particelle estranee o cambiamenti di colore. La soluzione deve essere limpida, leggermente opalescente e incolore. Se la soluzione nella siringa preriempita contiene particelle estranee o ha cambiato colore, non deve essere utilizzata.
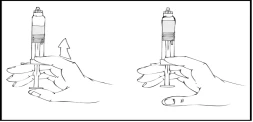
- Continuando a mantenere la siringa preriempita Refact AF in posizione verticale, spingere lentamente lo stantuffo finché la maggior parte, ma non tutta, dell’aria viene espulsa dalla (camera superiore).
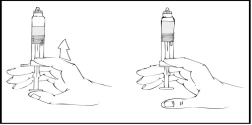
Refact AF deve essere somministrato entro 3 ore dalla ricostituzione della soluzione o dalla rimozione del cappuccio in gomma grigio dalla siringa preriempita.
Se non si utilizza immediatamente la soluzione Refact AF, conservare la siringa in posizione verticale con il cappuccio protettivo blu sulla siringa preriempita fino al momento dell’infusione. La soluzione preparata può essere conservata a temperatura ambiente per 3 ore. Se non utilizzata entro 3 ore, la soluzione deve essere scartata.
Somministrazione (infusione endovenosa)
Refact AF deve essere somministrato utilizzando il set per infusione fornito.
- Rimuovere il cappuccio protettivo blu e collegare saldamente il set per infusione alla siringa preriempita Refact AF.
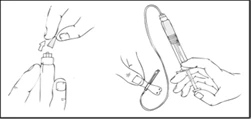
- Applicare un laccio emostatico e preparare l’area di iniezione pulendo accuratamente la cute con un tampone alcolico fornito nel kit.
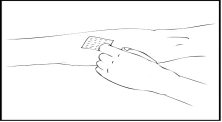
- Rimuovere il cappuccio protettivo dall’ago e inserire l’ago del catetere del set per infusione in una vena. Rimuovere il laccio emostatico. La soluzione preparata di Refact AF deve essere somministrata per via endovenosa in pochi minuti. Il medico può modificare la velocità raccomandata dell’infusione per rendere la procedura più confortevole. Non tentare di effettuare l’infusione autonomamente senza un’adeguata formazione.
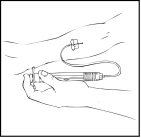
La soluzione preparata di Refact AF non deve essere somministrata attraverso lo stesso set per infusione o contenitore insieme ad altri medicinali.
- Dopo l’infusione di Refact AF, rimuovere e smaltire il set per infusione. La quantità di medicinale rimasta nel set per infusione non influisce sul trattamento. Informazioni aggiuntive sull’uso di più siringhe Refact AF con siringhe da 10 cm³ o di dimensioni superiori del tipo a tenone (siringhe da 10 cm³ o di dimensioni superiori del tipo a tenone non incluse nel kit)
- Preparare la soluzione in tutte le siringhe Refact AF seguendo le istruzioni per la preparazione della soluzione riportate sopra.
Tenendo la siringa Refact AF piena in posizione verticale, spingere lentamente lo stantuffo finché la maggior parte, ma non tutta, dell’aria viene espulsa dalla siringa.
- Aprire la confezione del connettore per siringhe a tenone (non incluso nel kit).
- Collegare una siringa sterile da 10 cm³ o di dimensioni superiori del tipo a tenone a un’apertura (porta) del connettore per siringhe e la siringa Refact AF all’altra porta aperta sul lato opposto.
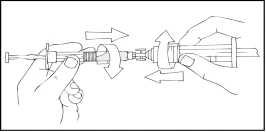
- Tenendo la siringa Refact AF in alto, premere lentamente lo stantuffo finché il contenuto non passa nella siringa vuota da 10 cm³ o di dimensioni superiori del tipo a tenone.
- Rimuovere la siringa preriempita Refact AF vuota e ripetere le procedure 3 e 4 con altre siringhe aggiuntive contenenti soluzione preparata.
- Rimuovere il connettore per siringhe dalla siringa da 10 cm³ o di dimensioni superiori del tipo a tenone e collegare il set per infusione come descritto in precedenza nell’istruzione per la somministrazione.
I residui non utilizzati del medicinale, i flaconi vuoti e gli aghi e siringhe usati devono essere smaltiti in un apposito contenitore per rifiuti sanitari, poiché questi materiali possono rappresentare un pericolo per gli altri se non smaltiti correttamente.
Pediatria
Refact AF può essere utilizzato in bambini di qualsiasi età, inclusi i neonati, come indicato nella sezione «Modalità di somministrazione e dosi».
Sovradosaggio
Non sono stati riportati sintomi di sovradosaggio con l’uso di medicinali contenenti fattore VIII ricombinante.
Effetti indesiderati.
Riassunto del profilo di sicurezza
Reazioni di ipersensibilità e allergiche (che possono includere angioedema, sensazione di calore e formicolio nel sito di infusione, brividi, vampate di calore, orticaria generalizzata, cefalea, orticaria, ipotensione arteriosa, letargia, nausea, agitazione, tachicardia, senso di oppressione toracica, ronzio nelle orecchie, vomito, respiro sibilante) si verificano raramente con l’uso di Refact AF e in alcuni casi possono progredire fino a una grave anafilassi, inclusa la reazione anafilattica con shock (vedere il paragrafo "Avvertenze particolari e precauzioni di impiego").
Refact AF può contenere tracce di proteine di criceto. È stato osservato molto raramente lo sviluppo di anticorpi contro le proteine di criceto, senza tuttavia complicazioni cliniche. In uno studio con Refact, in 20 su 113 pazienti precedentemente trattati (PPT) è stato osservato un aumento del titolo di anticorpi contro le proteine delle ovaie del criceto cinese, senza effetti clinici visibili.
La formazione di anticorpi neutralizzanti (inibitori) può verificarsi in pazienti con emofilia A trattati con fattore VIII, incluso Refact AF. La comparsa di inibitori può manifestarsi con una risposta clinica inadeguata. In tali casi si raccomanda di rivolgersi a un centro specializzato nel trattamento dell’emofilia.
Elenco degli effetti indesiderati
Le informazioni riportate di seguito sono classificate per sistemi e organi secondo il dizionario medico per l’attività regolatoria (MedDRA). Gli effetti indesiderati sono classificati in base alla frequenza di insorgenza: molto frequenti (≥ 1/10); frequenti (da ≥ 1/100 a < 1/10); non frequenti (da ≥ 1/1000 a < 1/100). Di seguito sono riportati gli effetti indesiderati osservati negli studi clinici con Refact o Refact AF. La frequenza si riferisce a tutti gli eventi avversi casuali verificatisi durante l’uso del medicinale in tutti gli studi clinici che hanno coinvolto 715 pazienti.
In ciascuna categoria, gli effetti indesiderati sono elencati in ordine decrescente di gravità.
Sistema emolinfopoietico:
molto frequenti: inibizione del fattore VIII (in pazienti precedentemente non trattati)*;
non frequenti: inibizione del fattare VIII (in pazienti precedentemente trattati)*.
Sistema immunitario: non frequenti: reazioni anafilattiche.
Disturbi del metabolismo e della nutrizione: frequenti: riduzione dell’appetito.
Sistema nervoso: molto frequenti: cefalea;
frequenti: vertigini;
non frequenti: neuropatia periferica, sonnolenza, disgeusia.
Organo cardiaco: non frequenti: angina pectoris, tachicardia, palpitazioni.
Sistema vascolare: frequenti: emorragia/ematoma;
non frequenti: ipotensione, tromboflebite, iperemia facciale.
Sistema respiratorio, torace e mediastino: molto frequenti: tosse;
non frequenti: dispnea.
Apparato gastrointestinale: frequenti: diarrea, nausea, dolore addominale, vomito.
Pelle e tessuto sottocutaneo: frequenti: orticaria, prurito, eruzione cutanea;
non frequenti: iperidrosi.
Sistema osteomuscolare e connettivo: molto frequenti: artralgia;
frequenti: mialgia.
Disturbi generali e condizioni in sede di somministrazione: molto frequenti: aumento della temperatura corporea;
frequenti: brividi, complicanze legate al catetere venoso permanente;
non frequenti: astenia, dolore, infiammazione e altre reazioni in sede di somministrazione.
Esami di laboratorio:
frequenti: test positivo per anticorpi; test positivo per anticorpi anti-fattore VIII;
non frequenti: aumento dei livelli ematici di aspartato aminotrasferasi, alanina aminotrasferasi, bilirubina, creatinfosfochinasi.
* La frequenza si basa su studi condotti con tutti i fattori VIII, inclusi pazienti con emofilia A grave. PPT: pazienti precedentemente trattati; PNT: pazienti non trattati precedentemente.
Pediatria
È stato riportato un caso di cisti in un paziente di 11 anni e un caso di offuscamento della coscienza in un paziente di 13 anni; lo sviluppo di questi eventi potrebbe essere correlato al trattamento con Refact AF.
La sicurezza di Refact AF è stata valutata in studi condotti su adulti, bambini e adolescenti precedentemente trattati (n = 18, età compresa tra 12 e 16 anni nello studio principale e n = 49, età compresa tra 7 e 16 anni in uno studio aggiuntivo), rilevando una tendenza all’aumento della frequenza degli eventi avversi nei bambini di età compresa tra 7 e 16 anni rispetto agli adulti.
Ulteriori dati sulla sicurezza sono stati ottenuti in studi su pazienti precedentemente trattati (n = 18, età inferiore a 6 anni e n = 19, età compresa tra 6 e 12 anni) o non trattati precedentemente (n = 23, età inferiore a 6 anni). I dati ottenuti indicano una similarità del profilo di sicurezza rispetto a quello osservato nei pazienti adulti.
Segnalazione delle reazioni avverse sospette
È importante segnalare tempestivamente le reazioni avverse sospette dopo l’autorizzazione del medicinale. Questo permette di continuare a monitorare il rapporto beneficio/rischio del medicinale. I professionisti sanitari devono segnalare qualsiasi reazione avversa sospetta secondo le procedure locali previste.
Periodo di validità.
Forma farmaceutica – flaconcino: per il prodotto in polvere – 3 anni, per il solvente – 5 anni.
Forma farmaceutica – siringa preriempita: 3 anni.
Condizioni di conservazione.
Conservare nella confezione originale a una temperatura compresa tra 2 e 8 °C. Non congelare, per evitare danni alla siringa preriempita.
Il medicinale, entro il periodo di validità, può essere conservato a una temperatura non superiore a 25 °C per un massimo di 3 mesi. Una volta conservato a temperatura ambiente, il medicinale non può essere riportato in frigorifero.
La soluzione ricostituita deve essere utilizzata immediatamente o entro 3 ore dalla ricostituzione (e/o rimozione del tappo grigio dalla punta, nel caso della forma farmaceutica siringa preriempita), purché conservata a una temperatura non superiore a 25 °C.
Conservare fuori dalla portata dei bambini.
Incompatibilità.
Poiché non sono stati condotti studi di compatibilità con questo medicinale, non deve essere mescolato con altri medicinali, inclusi altri soluzioni per infusione.
Per l’infusione della soluzione deve essere utilizzato il set per infusione fornito nel kit, poiché il fattore VIII della coagulazione può adsorbirsi sulle superfici interne di altri dispositivi per infusione.
Confezione.
Forma farmaceutica – flaconcino:
1 flaconcino con liofilizzato, 1 siringa preriempita con solvente, 1 adattatore per flaconcino, 1 set per infusione, 2 tamponi alcolici, 1 cerotto e 1 compressa di garza, il tutto contenuto in una scatola di cartone.
Forma farmaceutica – siringa preriempita:
1 siringa preriempita contenente liofilizzato nella camera superiore e solvente da 4 ml nella camera inferiore, 1 stantuffo, 1 set per infusione, 2 tamponi alcolici, 1 cerotto, 1 compressa di garza e 1 tappo, il tutto contenuto in una scatola di cartone.
Categoria di prescrivibilità. Sotto prescrizione medica.
Produttore.
Wyeth Farma S.A.
Data dell’ultima revisione.