Refacto AF
UcraniaContenido
INSTRUCCIONES para uso médico del medicamento ReFacto AF (ReFacto® AF)
Composición:
Principio activo: moroctocog alfa (factor de coagulación sanguínea VIII recombinante);
Cada vial contiene 250 UI, o 500 UI, o 1000 UI, o 2000 UI de moroctocog alfa;
Cada jeringa precargada contiene 3000 UI de moroctocog alfa;
Excipientes: sacarosa, cloruro de calcio dihidrato, L-histidina, polisorbato 80, cloruro de sodio.
Disolvente: cloruro de sodio, agua para inyección.
Forma farmacéutica.
Liofilizado para solución inyectable.
Principales propiedades físico-químicas:
Presentación – vial:
Liofilizado de color blanco, prácticamente libre de partículas extrañas visibles, humedad y defectos en el cierre del vial;
Disolvente: solución incolora y transparente, prácticamente libre de inclusiones visibles;
Presentación – jeringa precargada:
En la cámara superior: liofilizado de color blanco, prácticamente libre de partículas extrañas visibles, humedad y defectos en el cierre del recipiente;
En la cámara inferior: disolvente: solución incolora y transparente, prácticamente libre de inclusiones visibles.
Grupo farmacoterapéutico.
Agentes antihemorrágicos. Factor de coagulación sanguínea VIII. Código ATC B02BD02.
Propiedades farmacológicas.
Farmacodinámica.
ReFacto AF contiene factor de coagulación sanguínea recombinante VIII sin dominio B (moroctocog alfa). La actividad del medicamento en unidades internacionales (UI) se determina mediante un ensayo cromogénico según lo establecido en la Farmacopea Europea. La actividad específica del medicamento ReFacto AF es de 7.600–13.800 UI/mg de proteína.
El moroctocog alfa es una glicoproteína con una masa molecular aproximada de 170.000 Da, compuesta por 1438 aminoácidos, cuya secuencia es similar a la forma de 90 + 80 kDa del factor VIII (es decir, con el dominio B eliminado), y sus modificaciones postraduccionales son comparables a las presentes en la molécula obtenida a partir del plasma sanguíneo.
El moroctocog alfa se produce mediante tecnología de ADN recombinante utilizando células de ovario de hámster chino. El proceso de fabricación de ReFacto AF ha sido modificado de forma que se excluye la posibilidad de contaminación con proteínas exógenas de origen humano o animal durante el cultivo celular, la purificación o la producción del producto final; al mismo tiempo, el nombre del medicamento se modificó de ReFacto a ReFacto AF.
Las características funcionales de ReFacto AF son similares a las del factor VIII endógeno. La actividad del factor VIII está significativamente reducida en pacientes con hemofilia A, por lo que requieren terapia sustitutiva.
Tras la infusión del medicamento en pacientes con hemofilia, el factor VIII administrado se une al factor de von Willebrand que circula en la sangre.
El factor VIII activado actúa como cofactor del factor IX activado, acelerando la transformación del factor X en factor X activado. El factor X activado convierte la protrombina en trombina. La trombina, a su vez, convierte el fibrinógeno en fibrina, formándose así el coágulo. La hemofilia A es un trastorno hereditario ligado al sexo del sistema de coagulación, causado por niveles reducidos de factor VIII:C, que conduce a hemorragias abundantes en articulaciones, músculos o órganos internos. Las hemorragias pueden ocurrir espontáneamente o como consecuencia de traumatismos accidentales o quirúrgicos. Con la terapia sustitutiva, el nivel de factor VIII en el plasma sanguíneo aumenta, lo que permite corregir temporalmente esta deficiencia y reducir la tendencia a las hemorragias.
Eficacia clínica
Los datos mostrados en la Tabla 1 se obtuvieron en estudios del uso del medicamento ReFacto AF en pacientes previamente no tratados (PNT) y pacientes previamente tratados (PPT) menores de 12 años de edad.
Tabla 1
Dosificación y eficacia en pacientes pediátricos
| Indicador |
PLP |
PLP |
PNP |
| Dosificación según peso corporal (UI/kg) para infusión profilácticaa |
N = 14 (28, 51) |
N = 13 (21, 49) |
N = 22 (17, 161) |
| CTR total entre todos los sujetos, |
-- |
-- |
N = 23 (0,0; 39,5) |
| CTR total para sujetos que seguían régimen «según necesidad» al inicio del estudioc, |
N = 5 (1,6; 50,6) |
N = 9 (0,0; 46,6) |
-- |
| CTR total para sujetos que seguían régimen «profiláctico» al inicio del estudioc, |
N = 13 (0,0; 11,2) |
N = 9 (0,0; 13,0) |
-- |
| Dosificación según peso corporal (UI/kg) durante episodio hemorrágico para tratamiento de hemorragia, mediana (mín., máx.) |
N = 13 (28; 86) |
N = 14 (17; 229) |
N = 21 (11; 221) |
| % de hemorragias que fueron tratadas con éxito con ≤ 2 infusiones |
98,7 % |
98,8 % |
96,7 % |
a La dosificación y frecuencia de administración del medicamento Refacto AF en el estudio fueron determinadas por el médico según los estándares terapéuticos locales.
b Los sujetos en el estudio de pacientes no previamente tratados (PNT) no estaban obligados a seguir un tratamiento profiláctico continuo regular; sin embargo, excepto un sujeto (al que solo se administró tratamiento a demanda), la mayoría de los sujetos recibieron infusiones profilácticas regulares. Algunos sujetos comenzaron el estudio con infusiones a demanda y posteriormente pasaron a tratamiento profiláctico, mientras que otros solo tuvieron infusiones profilácticas esporádicas.
c Al inicio del estudio, los sujetos en el estudio de pacientes previamente tratados (PPT) declararon su régimen de tratamiento con factor VIII («profilaxis» o «a demanda»), pero no estaban obligados a mantener este régimen como condición para participar en el estudio. La dosificación y frecuencia de administración del medicamento Refacto AF en el estudio fue determinada por el médico según los estándares terapéuticos locales.
Abreviatura: TCR – tasa anual media de hemorragias.
Cabe señalar que no se comparan los valores de TCR al usar diferentes concentrados de factores de coagulación ni entre distintos estudios clínicos.
Inducción de la tolerancia inmunológica.
Se recopilaron datos sobre la inducción de la tolerancia inmunológica en pacientes con hemofilia A que desarrollaron inhibidores del factor VIII. En el estudio clínico fundamental con Refacto que incluyó a 25 pacientes previamente no tratados, se recopilaron datos sobre la inducción de la tolerancia inmunológica (15 con título alto, 10 con título bajo). De estos 25 pacientes, en 20 el título de inhibidores disminuyó a niveles < 0,6 UBI (unidades Bethesda), de los cuales 11 de los 15 inicialmente tenían títulos altos (≥ 5 UBI/ml) y 9 de los 10 tenían títulos bajos. Una disminución similar de los títulos se observó en 5 de 6 pacientes que tenían títulos bajos de inhibidores, pero no recibieron tratamiento mediante inducción de tolerancia inmunológica. No se realizaron observaciones de seguimiento a largo plazo.
Farmacocinética.
Las propiedades farmacocinéticas de Refacto, determinadas en un estudio cruzado de Refacto y un concentrado de factor VIII obtenido del plasma sanguíneo, con la participación de 18 pacientes previamente tratados (pacientes previamente tratados, PPT), se indican en la Tabla 2. Los análisis cuantitativos se realizaron mediante sustrato cromogénico (ver sección «Forma de administración y dosis»).
Tabla 2
| Evaluación de los parámetros farmacocinéticos de ReFacto en pacientes con hemofilia A |
|||
| Parámetro |
Media |
DE |
Mediana |
| AUCt (U·h/mL) |
19,9 |
4,9 |
19,9 |
| t½ (h) |
14,8 |
5,6 |
12,7 |
| CL (mL/h·kg) |
2,4 |
0,75 |
2,3 |
| tiempo medio de retención (h) |
20,2 |
7,4 |
18,0 |
| recuperación (U/dL de aumento del factor VIII:C por U/kg de factor VIII administrado) |
2,4 |
0,38 |
2,5 |
Abreviaciones: AUCt – área bajo la curva de concentración en plasma frente al tiempo desde cero hasta la última concentración medida; t½ – semivida; CL – aclaramiento; DE – desviación estándar.
En un estudio en el que se comparó la actividad de ReFactoAF, ReFacto y factor VIII en plasma mediante análisis cromogénico, se estableció la bioequivalencia entre ReFactoAF y ReFacto. La relación entre las medias geométricas de los parámetros farmacocinéticos estimados por mínimos cuadrados para ReFactoAF y ReFacto fue del 100,6 %, 99,5 % y 98,1 % para recuperación, AUCt y AUC∞ (área bajo la curva de concentración en plasma frente al tiempo desde cero hasta infinito), respectivamente. Los intervalos de confianza del 90 % correspondientes para la relación entre las medias geométricas de los parámetros farmacocinéticos de ReFactoAF y ReFacto se encontraron dentro del margen de bioequivalencia (80-125 %), lo que indica la bioequivalencia entre ReFactoAF y ReFacto.
En un estudio farmacocinético cruzado, los parámetros farmacocinéticos de ReFactoAF se determinaron en 25 pacientes (≥ 12 años) previamente tratados, al inicio del estudio y tras seis meses de administración repetida de ReFactoAF. La relación entre las medias geométricas de los parámetros farmacocinéticos estimados por mínimos cuadrados tras seis meses de tratamiento en comparación con los valores basales fue del 107 %, 100 % y 104 % para recuperación, AUCt y AUC∞, respectivamente. Los intervalos de confianza del 90 % correspondientes para la relación entre los parámetros farmacocinéticos tras seis meses de estudio en comparación con los valores basales se encontraron dentro del margen de equivalencia (80 % - 125 %). Esto indica la ausencia de cambios dependientes del tiempo en las propiedades farmacocinéticas de ReFactoAF.
En el mismo estudio, la actividad del medicamento ReFactoAF, un factor VIII recombinante de longitud completa (medicamento de comparación) y la actividad del factor VIII se midieron en muestras de plasma de pacientes (30 pacientes previamente tratados, ≥ 12 años) mediante un ensayo de coagulación en un solo paso. ReFactoAF demostró bioequivalencia farmacocinética con el medicamento de comparación, según el enfoque estándar para establecer bioequivalencia.
En pacientes previamente no tratados, los parámetros farmacocinéticos de ReFacto se evaluaron mediante análisis cromogénico. En estos pacientes (n = 59; mediana de edad 10 ± 8,3 meses), la recuperación media en la semana «0» fue de 1,5 ± 0,6 UI/dl por UI/kg (rango 0,2–2,8 UI/dl por UI/kg), lo cual es inferior a los valores obtenidos en pacientes previamente tratados, cuya recuperación media en la semana «0» fue de 2,4 ± 0,4 UI/dl por UI/kg (rango 1,1–3,8 UI/dl por UI/kg). En pacientes previamente no tratados, la recuperación media fue estable durante un período de dos años (5 visitas durante ese tiempo) y se mantuvo entre 1,5 y 1,8 UI/dl por UI/kg. Un estudio mediante modelo farmacocinético poblacional que incluyó datos de 44 pacientes previamente no tratados mostró que la semivida media predicha fue de 8,0 ± 2,2 horas.
En un estudio con ReFactoAF que incluyó a 19 pacientes previamente no tratados (PNT), el nivel de recuperación en 17 niños de entre 28 días y 2 años fue de 1,32 ± 0,65 UI/dl por UI/kg, y en 2 niños de entre 2 y < 6 años fue de 1,7 y 1,8 UI/dl por UI/kg. Excepto en los casos en que se detectaron inhibidores, el nivel medio de recuperación fue estable en el tiempo (6 visitas durante un período de dos años) y los valores individuales oscilaron entre 0 (en presencia de inhibidor) y 2,7 UI/dl por UI/kg. En la tabla 3 se muestran los parámetros farmacocinéticos observados de ReFactoAF tras la administración de una dosis de 50 UI/kg en un estudio realizado con 37 niños previamente tratados.
Tabla 3
| Valores medios ± DE de los parámetros farmacocinéticos del factor VIII tras la administración de una dosis única de 50 UI/kg en niños (hemofilia A) |
||
| Parámetro |
Número de sujetos |
Valor medioa ± DE |
| Recuperación, UI/dl por UI/kg Menores de 6 años de edad De 6 a 12 años de edad |
17 19 |
1,7 ± 0,4 2,1 ± 0,8 |
| Cmax, UI/mlb |
19 |
0,9 (45) |
| AUCinf, UI∙h/mlb |
14 |
9,9 (41) |
| t½, hb |
14 |
9,1 ± 1,9 |
| CL, ml/h/kgb |
14 |
4,4 (30) |
| Vss, ml/kgb |
14 |
56,4 (15) |
| a Valor medio geométrico (CV geométrico %) para todos los casos, excepto para el valor medio aritmético ± DE de la recuperación incremental y t½. b Solo pacientes de 6 a 12 años de edad. Abreviaturas: Cmax – concentración máxima observada en plasma; CV – coeficiente de variación; AUCinf – área bajo la curva de concentración en plasma frente al tiempo desde 0 hasta infinito; t½ – período de semivida; CL – aclaramiento; DE – desviación estándar; Vss – volumen de distribución en estado estacionario. |
||
Características clínicas.
Indicaciones.
Tratamiento y profilaxis de las hemorragias en pacientes con hemofilia A (deficiencia congénita del factor de coagulación sanguínea VIII).
Refacto AF está indicado para su uso en adultos y niños de cualquier edad, incluidos recién nacidos.
Refacto AF no contiene factor de von Willebrand y, por tanto, no está indicado para el tratamiento de la enfermedad de von Willebrand.
Contraindicaciones.
Hipersensibilidad al principio activo o a cualquiera de los excipientes.
Reacción alérgica conocida a las proteínas de hámster.
Interacción con otros medicamentos y otras formas de interacción.
No se han notificado interacciones entre el factor recombinante de coagulación sanguínea humana VIII y otros medicamentos.
Características de uso.
Seguimiento
Para mejorar el seguimiento de los medicamentos biológicos, se debe registrar claramente el nombre y el número de lote del medicamento administrado. Con este fin, los pacientes pueden pegar en su diario una de las etiquetas adhesivas desprendibles del frasco o de la jeringa precargada, para documentar el número de lote, o utilizarla para informar sobre cualquier efecto adverso.
Hipersensibilidad
ReFacto AF puede provocar reacciones de hipersensibilidad de tipo alérgico. El medicamento contiene pequeñas cantidades de proteínas de hámster. Se debe aconsejar a los pacientes que, ante la aparición de síntomas de hipersensibilidad, suspendan inmediatamente el uso del medicamento y consulten a su médico. Los pacientes deben informar sobre los primeros signos de reacciones de hipersensibilidad, incluyendo urticaria, urticaria generalizada, sensación de opresión en el pecho, sibilancias, hipotensión arterial y anafilaxia.
En caso de presentarse shock, se debe aplicar el tratamiento médico estándar para el estado de shock.
Inhibidores
La formación de anticuerpos neutralizantes (inhibidores) contra el factor VIII es una complicación conocida durante el tratamiento de personas con hemofilia A. Estos suelen ser inmunoglobulinas IgG dirigidas contra la actividad procoagulante del factor VIII. Su presencia en plasma se determina mediante un método cuantitativo modificado y se expresa en unidades de Bethesda (UB) por 1 ml. El riesgo de formación de inhibidores está relacionado con la gravedad de la enfermedad y con la exposición al factor VIII, siendo mayor durante los primeros 50 días de tratamiento, aunque el riesgo persiste durante toda la vida, aunque sea infrecuente.
La relevancia clínica de la formación de inhibidores dependerá de su título, siendo los inhibidores de bajo título menos propensos a causar una respuesta clínica insuficiente que los de alto título.
Se debe realizar un seguimiento cuidadoso de los pacientes que reciben tratamiento con factores recombinantes de coagulación VIII, mediante observación clínica adecuada y pruebas de laboratorio, para detectar la formación de inhibidores. Si no se alcanzan los niveles esperados de actividad del factor VIII en plasma o si la hemorragia no cesa con la dosis adecuada del medicamento, se deben realizar análisis para determinar la presencia de inhibidores del factor VIII. En pacientes con niveles altos de inhibidores, la terapia con factor VIII puede ser ineficaz, por lo que se debe considerar el uso de otros medicamentos. El tratamiento de estos pacientes debe ser supervisado por médicos con experiencia en el manejo de la hemofilia y la formación de inhibidores del factor VIII.
Notificación de falta de eficacia
En estudios clínicos y durante el uso poscomercialización de ReFacto, se han notificado casos de falta de eficacia del medicamento, principalmente en pacientes que lo utilizaban con fines profilácticos. La falta de eficacia se ha descrito como hemorragias en articulaciones diana, hemorragias en nuevas articulaciones o la sensación subjetiva del paciente de inicio de una nueva hemorragia. Al prescribir ReFacto AF, es fundamental individualizar la dosificación y monitorizar el nivel de factor en cada paciente para asegurar una respuesta terapéutica adecuada (ver sección «Reacciones adversas»).
Complicaciones cardiovasculares
En pacientes con factores de riesgo preexistentes para complicaciones cardiovasculares, el tratamiento sustitutivo con factor VIII puede aumentar el riesgo cardiovascular.
Complicaciones relacionadas con el catéter
Si es necesario utilizar un dispositivo de acceso venoso central, se debe considerar el riesgo de complicaciones asociadas con su uso, incluyendo infecciones locales, bacteriemia y trombosis del catéter (ver sección «Reacciones adversas»).
Contenido de sodio
Tras la disolución del polvo, un frasco o una jeringa precargada contienen 1,27 mmol (o 29 mg) de sodio, lo que equivale al 1,5 % de la ingesta diaria máxima recomendada por la OMS para un adulto (2 g). Dependiendo del peso corporal del paciente y de la dosis de ReFacto AF, los pacientes pueden utilizar varios frascos o jeringas precargadas. Esta información debe tenerse en cuenta en pacientes que siguen una dieta con restricción de sodio.
Uso durante el embarazo o la lactancia.
No se han realizado estudios sobre el efecto del factor VIII en la función reproductiva en animales, por lo que no existen datos sobre su impacto en la fertilidad. Debido a que la hemofilia A es rara en mujeres, no hay experiencia sobre el uso del factor VIII durante el embarazo o la lactancia. Por tanto, el factor VIII solo debe administrarse durante el embarazo o la lactancia si es estrictamente necesario.
Capacidad para afectar la velocidad de reacción al conducir vehículos o manejar maquinaria.
ReFacto AF no afecta la capacidad para conducir vehículos ni para trabajar con maquinaria.
Vía de administración y dosis.
El tratamiento debe iniciarse bajo la supervisión de un médico con experiencia en el tratamiento de la hemofilia A.
Control del tratamiento.
Para determinar la dosis necesaria durante el curso del tratamiento y la frecuencia de las infusiones repetidas, se recomienda realizar estudios del nivel del factor VIII. La respuesta a la administración del factor VIII puede variar entre diferentes pacientes, manifestándose en distintos niveles de recuperación y diferentes períodos de semivida. La dosis calculada según el peso corporal del paciente puede requerir ajustes en pacientes con peso excesivo o insuficiente. En particular, en caso de intervenciones quirúrgicas importantes, es obligatorio un monitoreo cuidadoso de la terapia sustitutiva mediante análisis de coagulación (actividad del factor VIII en plasma).
Para el monitoreo del nivel de actividad del factor VIII en los pacientes durante el tratamiento con el medicamento ReFacto AF, se recomienda utilizar el método de análisis cromogénico. Si para determinar la actividad del factor VIII en muestras de sangre de los pacientes se utiliza el tiempo de tromboplastina parcial activado in vitro (TTPa), calculado mediante un ensayo en una sola etapa de actividad coagulante, los resultados de la actividad del factor VIII pueden verse afectados tanto por los tipos de reactivos como por los patrones utilizados en la cuantificación. Asimismo, pueden existir diferencias significativas entre los resultados cuantitativos obtenidos mediante el método cromogénico y aquellos determinados mediante el TTPa mediante ensayo en una sola etapa de actividad coagulante. Habitualmente, los resultados obtenidos mediante el ensayo en una sola etapa de actividad coagulante son un 20-50 % más bajos que los resultados cuantitativos obtenidos mediante el método cromogénico. Para corregir esta discrepancia, puede utilizarse un patrón de laboratorio de ReFacto AF (ver sección «Farmacocinética»). Esto es especialmente importante cuando se utilizan otros laboratorios y/o reactivos.
Dosificación.
La dosis y la duración de la terapia sustitutiva dependen del grado de deficiencia del factor VIII, la localización y gravedad de la hemorragia, así como del estado clínico del paciente. Las dosis empleadas deben ajustarse según la respuesta clínica del paciente. En caso de presencia de inhibidores, puede ser necesario administrar dosis más altas del medicamento o prescribir un tratamiento específico adecuado.
La cantidad de unidades de factor VIII se expresa en UI (unidades internacionales), referidas al estándar activo de la OMS para productos que contienen factor VIII. La actividad del factor VIII en plasma se expresa en porcentaje (en relación con su contenido en plasma humano normal) o en unidades internacionales (en relación con el estándar internacional para factor VIII en plasma). 1 UI de actividad del factor VIII equivale a la cantidad de factor VIII presente en 1 ml de plasma humano normal.
La actividad de otro producto de moroctocog alfa (Xyntha), registrado fuera de Ucrania, se determina mediante un estándar de actividad de fabricación calibrado frente al estándar internacional de la OMS, utilizando un ensayo en una sola etapa de actividad coagulante. Debido a las diferencias entre los métodos utilizados para determinar la actividad de los productos Xyntha y ReFacto AF, 1 UI del producto Xyntha (calibrado mediante ensayo en una sola etapa) equivale aproximadamente a 1,38 UI del producto ReFacto AF (calibrado mediante ensayo cromogénico). Si se prescribe ReFacto AF a un paciente que habitualmente recibe tratamiento con Xyntha, el médico puede considerar la posibilidad de ajustar la dosis según los niveles de recuperación del factor VIII.
A los pacientes con hemofilia A se les debe recomendar llevar consigo una cantidad suficiente del medicamento factor VIII, de acuerdo con el régimen de tratamiento vigente, por si fuera necesario durante un viaje. Se debe aconsejar a los pacientes consultar con su médico antes de viajar.
Tratamiento a demanda.
El cálculo de la dosis necesaria de factor VIII se basa en datos empíricos que indican que 1 UI de factor VIII por kg de peso corporal aumenta la actividad del factor VIII en plasma sanguíneo en 2 UI/dl. La dosis necesaria se calcula mediante la siguiente fórmula:
cantidad necesaria de unidades (UI) =
peso corporal (kg) × incremento deseado del factor VIII (%, o UI/dl) × 0,5 (UI/kg por UI/dl),
donde 0,5 UI/kg por UI/dl es el valor recíproco de la recuperación, que es el valor habitualmente observado tras la infusión de factor VIII.
La dosis del medicamento prescrito y la frecuencia de administración deben determinarse siempre individualmente, considerando la eficacia clínica del producto.
En caso de presentarse manifestaciones hemorrágicas, la actividad del factor VIII en plasma sanguíneo no debe descender por debajo de los niveles (en % o en UI/dl) indicados en la tabla 4. Esta tabla puede utilizarse como guía para la dosificación del factor VIII en casos de hemorragia y durante intervenciones quirúrgicas.
Tabla 4
| Gravedad del sangrado / tipo de intervención quirúrgica |
Nivel necesario del factor VIII (% del normal o UI/dl de plasma) |
Frecuencia de administración (horas) / duración del tratamiento (días) |
| Sangrado |
||
| Signos iniciales de hemartrosis, hemorragias musculares o sangrado en la cavidad bucal |
20–40 |
Repetir la administración cada 12–24 horas durante al menos 1 día hasta la cesación del sangrado, evidenciada por la ausencia de dolor o signos de cicatrización |
| Hemartrosis y hemorragias musculares de moderada gravedad o hematomas |
30–60 |
Repetir la administración cada 12–24 horas durante 3–4 días o más, hasta la desaparición del dolor y recuperación de la movilidad de las extremidades |
| Hemorragias que amenazan la vida |
60–100 |
Repetir la administración cada 8–24 horas hasta la desaparición del riesgo vital |
| Intervenciones quirúrgicas |
||
| Intervenciones quirúrgicas menores, incluida la extracción dental |
30–60 |
Repetir la administración cada 24 horas, durante al menos 1 día, hasta la cicatrización |
| Intervenciones quirúrgicas mayores |
80–100 (antes y después de la intervención quirúrgica) |
Repetir la administración cada 8–24 horas hasta una adecuada cicatrización de la herida; posteriormente continuar el tratamiento durante al menos 7 días para mantener la actividad del factor VIII en un nivel del 30–60 % (UI/dl) |
Prevención
Para la prevención prolongada de hemorragias en pacientes con hemofilia A grave, normalmente se administran 20-40 UI del factor VIII por kg de peso corporal cada 2-3 días. En algunos casos, especialmente en pacientes jóvenes, puede ser necesario aumentar la dosis o la frecuencia de administración del medicamento.
Pacientes pediátricos
Al tratar a niños pequeños (menores de 6 años) con Refacto AF, puede ser necesario utilizar dosis más altas en comparación con los pacientes adultos y los niños mayores (ver sección «Farmacocinética»).
Pacientes de edad avanzada
Los pacientes de 65 años o más no fueron incluidos en los estudios clínicos. En general, la dosis para pacientes de edad avanzada debe ajustarse individualmente.
Pacientes con insuficiencia renal o hepática
No se ha estudiado el ajuste de la dosis en pacientes con insuficiencia renal o hepática durante los estudios clínicos.
Vía de administración
Uso intravenoso.
Refacto AF se administra mediante infusión intravenosa durante varios minutos tras disolverse en el diluyente adecuado. La velocidad de administración dependerá del nivel de comodidad del paciente. Se recomienda formación adecuada a las personas que administren el medicamento y que no sean personal sanitario.
Presentación – frasco
El polvo liofilizado se disuelve en el diluyente adecuado (solución de cloruro sódico al 0,9 %) que se incluye en una jeringa precargada, utilizando un adaptador estéril. Tras añadir el diluyente, el frasco debe agitarse suavemente hasta que el polvo se disuelva completamente.
Información más detallada sobre la preparación y administración de la solución se proporciona a continuación.
Tras la reconstitución, la solución resultante se aspira nuevamente en la jeringa. La solución debe ser transparente o ligeramente opalescente e incolora. La solución debe desecharse si se observan partículas extrañas o cambios en el color.
Presentación – jeringa precargada:
El polvo liofilizado en la cámara superior de la jeringa precargada debe disolverse con el diluyente (solución de cloruro sódico, 9 mg/ml (0,9 %)) que se encuentra en la cámara inferior. La jeringa precargada debe agitarse suavemente hasta que todo el polvo se disuelva completamente. Información más detallada sobre la preparación y administración de la solución se proporciona a continuación.
Tras la reconstitución, la solución será transparente o ligeramente opalescente e incolora. La solución debe desecharse si se observan inclusiones mecánicas visibles o cambios en el color.
El medicamento Refacto AF contiene polisorbato 80 tras la reconstitución, que puede acelerar la extracción de ftalato de di-(2-etilhexilo) del cloruro de polivinilo (PVC). Esta característica debe tenerse en cuenta durante la preparación y administración del medicamento, incluido el tiempo de almacenamiento en un recipiente de PVC tras la preparación de la solución. Es importante seguir estrictamente las recomendaciones indicadas en la sección «Condiciones de conservación».
Los restos no utilizados del medicamento o los materiales empleados para la disolución y administración deben desecharse de acuerdo con los requisitos locales.
Presentación – frasco
Preparación de la solución:
- Deje que la temperatura del liofilizado y del diluyente en la jeringa precargada alcance la temperatura ambiente.
- Retire la tapa de plástico tipo flip-top del frasco de Refacto AF para exponer la parte central del tapón de goma.
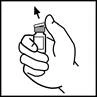
- Limpie la parte superior del frasco con una gasa con alcohol incluida en el envase o use otro antiséptico y deje secar. Tras la limpieza, no toque el tapón de goma ni permita que entre en contacto con ninguna superficie.
- Retire la cubierta protectora del adaptador al frasco. No retire el adaptador del envase.
- Coloque el frasco sobre una superficie plana. Sosteniendo el envase del adaptador, coloque el adaptador sobre el frasco. Presione firmemente hasta que haga clic y quede fijado en la parte superior del frasco, y la punta del adaptador atraviese el tapón del frasco.
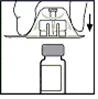
- Retire el envase del adaptador y desecharlo.
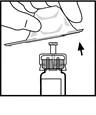
- Conecte el émbolo a la jeringa con diluyente: inserte el émbolo en la abertura del tope de la jeringa, presione y gire el émbolo hasta que quede firmemente fijado en el tope.
- Rompa la tapa protectora de plástico con indicador de primera apertura de la jeringa con diluyente, rompiendo la perforación de la tapa. Esto se hace agitando la tapa hasta que la perforación se rompa. No toque la superficie interior de la tapa ni la punta de la jeringa. Puede ser necesario volver a colocar la tapa (si el Refacto AF preparado no se va a usar inmediatamente), por lo que debe guardarse con la abertura hacia arriba.
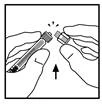
- Coloque el frasco sobre una superficie plana. Conecte la jeringa con diluyente al adaptador del frasco insertando la punta de la jeringa en la abertura del adaptador, presionando y girando la jeringa en sentido horario hasta lograr una conexión segura.
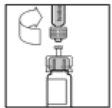
- Presione lentamente el émbolo para inyectar todo el diluyente en el frasco de Refacto AF.
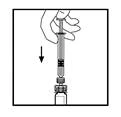
- Manteniendo la jeringa conectada al adaptador, agite suavemente el frasco hasta que el polvo se disuelva completamente.
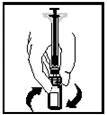
- Antes de la administración, la solución preparada debe inspeccionarse visualmente para detectar partículas extrañas. La solución resultante debe ser transparente o ligeramente opalescente e incolora.
Si se utilizan más de un frasco de Refacto AF por infusión, cada frasco debe prepararse según las instrucciones anteriores. A continuación, se debe retirar la jeringa del diluyente, dejando el adaptador del frasco en su lugar, y se puede utilizar una jeringa grande con conector tipo Luer para extraer la solución de cada frasco.
- Asegurándose de que el émbolo permanezca completamente introducido en el cilindro de la jeringa, invierta el frasco. Lentamente, aspire toda la solución a través del adaptador del frasco hacia la jeringa.
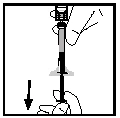
- Desconecte la jeringa del adaptador del frasco, tirando y girando suavemente la jeringa en sentido antihorario. Deseche el frasco con el adaptador conectado.
Si la solución no se va a usar inmediatamente, la tapa de la jeringa debe colocarse cuidadosamente de nuevo. No toque la punta de la jeringa ni la superficie interior de la tapa.
La solución preparada debe usarse inmediatamente o dentro de las 3 horas posteriores a su preparación. La solución preparada puede conservarse a una temperatura no superior a 25 °C hasta su uso.
Administración (infusión intravenosa)
Refacto AF debe administrarse utilizando el conjunto de infusión suministrado y la jeringa precargada con diluyente suministrada, o una jeringa plástica estéril desechable con conector tipo Luer.
- Conecte la jeringa al conector tipo Luer del catéter del conjunto de infusión.
- Coloque una banda elástica y prepare el sitio de inyección limpiando bien la piel con una gasa con alcohol suministrada con el kit.
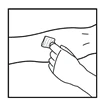
- Introduzca la aguja del catéter del conjunto de infusión en la vena y retire la banda elástica. Elimine el aire del catéter del conjunto de infusión tirando de la jeringa. El medicamento preparado debe administrarse por vía intravenosa durante varios minutos. El médico puede modificar la velocidad de infusión recomendada para hacer el procedimiento más cómodo para el paciente.
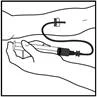
Presentación – jeringa precargada
Preparación de la solución:
- Lleve la jeringa precargada a temperatura ambiente.
- Saque el kit para la jeringa precargada de Refacto AF y colóquelo sobre una superficie limpia, asegurándose de tener todos los componentes necesarios.
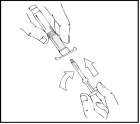
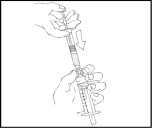
- Sujete el émbolo como se muestra en el esquema siguiente. Enrosque firmemente el émbolo en la abertura del soporte para los dedos de la jeringa precargada de Refacto AF presionando y girando en sentido horario hasta que note resistencia (aproximadamente 2 giros).
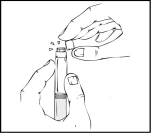
Durante todo el proceso de preparación de la solución, la jeringa precargada de Refacto AF debe mantenerse verticalmente (el polvo blanco debe estar sobre la solución transparente) para evitar posibles fugas.
- Manteniendo la jeringa precargada en posición vertical, rompa la tapa protectora blanca con indicador de primera apertura, doblándola de derecha a izquierda (o con movimientos suaves oscilantes), para romper la perforación de la tapa y ver el tapón de goma gris de la punta de la jeringa precargada de Refacto AF.
- Saque de su envase la tapa protectora estéril azul ventilada.
Manteniendo constantemente la jeringa precargada de Refacto AF en posición vertical, retire el tapón de goma gris y sustitúyalo por la tapa protectora azul ventilada. Esta tapa protectora tiene pequeñas aberturas que permiten la salida de aire, evitando así una presión excesiva. No toque el extremo abierto de la jeringa ni la tapa protectora azul.
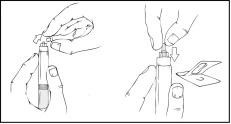
- Con cuidado y lentamente, empuje el émbolo presionando hasta que los dos émbolos internos de la jeringa precargada se encuentren y todo el diluyente pase a la cámara superior que contiene el polvo de Refacto AF.
Nota: Para evitar fugas de líquido por la punta de la jeringa, no presione el émbolo con fuerza excesiva.
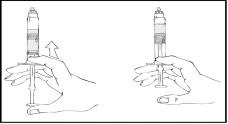
- Manteniendo la jeringa Refacto AF en posición vertical, agítela suavemente hasta que el polvo se disuelva.
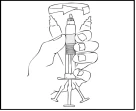
Inspeccione visualmente la solución para detectar partículas extrañas o cambios de color. La solución debe ser transparente, ligeramente opalescente e incolora. Si la solución en la jeringa precargada contiene partículas extrañas o ha cambiado de color, no debe usarse.
- Continuando a mantener la jeringa precargada de Refacto AF en posición vertical, empuje lentamente el émbolo hasta que la mayor parte, pero no todo, el aire sea expulsado de la cámara (superior).
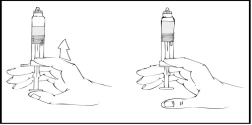
Refacto AF debe administrarse dentro de las 3 horas posteriores a la preparación de la solución o a la retirada del tapón de goma gris de la jeringa precargada.
Si no va a usar inmediatamente la solución de Refacto AF, la jeringa debe mantenerse en posición vertical con la tapa protectora azul en la jeringa precargada hasta que esté listo para realizar la infusión. La solución preparada puede conservarse a temperatura ambiente durante 3 horas. Si no se usa dentro de las 3 horas, la solución debe desecharse.
Administración (infusión intravenosa)
Refacto AF debe administrarse utilizando el conjunto de infusión suministrado.
- Retire la tapa protectora azul y conecte firmemente el conjunto de infusión a la jeringa precargada de Refacto AF.
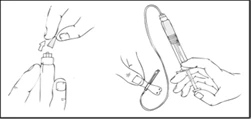
- Coloque una banda elástica y prepare el sitio de inyección limpiando bien la piel con una gasa con alcohol suministrada con el kit.
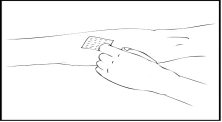
- Retire la tapa protectora de la aguja e introduzca la aguja del catéter del conjunto de infusión en la vena. Retire la banda elástica. La solución preparada del medicamento Refacto AF debe administrarse por vía intravenosa durante varios minutos. El médico puede modificar la velocidad de infusión recomendada para hacer el procedimiento más cómodo para usted. No intente realizar la infusión sin la formación adecuada.
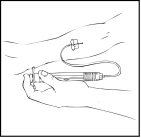
La solución preparada de Refacto AF no debe administrarse a través del mismo conjunto de infusión o recipiente que otros medicamentos.
- Tras la infusión de Refacto AF, retire y deseche el conjunto de infusión. La cantidad de medicamento que queda en el conjunto de infusión no afecta al tratamiento. Información adicional sobre la administración de múltiples jeringas de Refacto AF con jeringas de 10 cm³ o mayores del tipo Luer Lock (jeringas de 10 cm³ o mayores del tipo Luer Lock no incluidas en el envase)
- Prepare la solución en todas las jeringas de Refacto AF según las instrucciones de preparación de la solución indicadas anteriormente.
Manteniendo la jeringa de Refacto AF llena en posición vertical, empuje lentamente el émbolo hasta que la mayor parte, pero no todo, el aire sea expulsado de la jeringa.
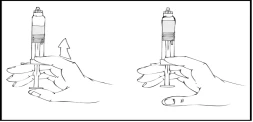
- Abra el conector para jeringas del tipo Luer Lock (no incluido en el envase).
- Conecte una jeringa estéril de 10 cm³ o mayor del tipo Luer Lock a uno de los orificios (puertos) del conector para jeringas, y la jeringa de Refacto AF al otro puerto abierto en el lado opuesto.
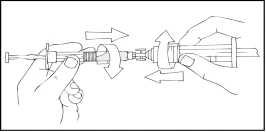
- Manteniendo la jeringa de Refacto AF en la parte superior, presione lentamente el émbolo hasta que el contenido pase a la jeringa vacía de 10 cm³ o mayor del tipo Luer Lock.
- Retire la jeringa vacía de Refacto AF y repita los pasos 3 y 4 con otras jeringas adicionales con solución preparada.
- Retire el conector para jeringas de la jeringa de 10 cm³ o mayor del tipo Luer Lock y conecte el conjunto de infusión, como se describe anteriormente en las instrucciones de administración.
Los restos no utilizados de la solución, los frascos vacíos y las agujas y jeringas usadas deben desecharse en un contenedor adecuado para residuos médicos, ya que estos materiales pueden causar daño a otras personas si no se eliminan adecuadamente.
Pacientes pediátricos
Refacto AF puede administrarse a niños de cualquier edad, incluyendo recién nacidos, como se indica en la sección «Posología y forma de administración».
Sobredosificación
No se han notificado síntomas de sobredosificación con el uso de medicamentos con factor VIII recombinante.
Reacciones adversas.
Resumen del perfil de seguridad
La hipersensibilidad y reacciones alérgicas (que pueden incluir angioedema, sensación de ardor y hormigueo en el lugar de la infusión, escalofríos, sofocos, urticaria generalizada, dolor de cabeza, urticaria, hipotensión arterial, letargo, náuseas, excitación, taquicardia, opresión en el pecho, zumbidos en los oídos, vómitos, sibilancias) con el uso de ReFacto AF son raras y en algunos casos pueden progresar a una anafilaxia grave, incluyendo shock (ver sección «Precauciones de uso»).
ReFacto AF puede contener trazas de proteína de hámster. Muy raramente se ha observado la formación de anticuerpos contra la proteína de hámster, pero no se han presentado complicaciones clínicas. En un estudio con ReFact0, en 20 de 113 pacientes previamente tratados (PPT) se observó un aumento del título de anticuerpos contra la proteína de ovario de hámster chino, sin efecto clínico aparente.
La formación de anticuerpos neutralizantes (inhibidores) puede ocurrir en pacientes con hemofilia A que reciben tratamiento con factor VIII, incluyendo ReFacto AF. La aparición de inhibidores puede manifestarse como una respuesta clínica insuficiente. En tales casos, se recomienda consultar a un centro especializado en el tratamiento de la hemofilia.
Lista de reacciones adversas
La información que se presenta a continuación se clasifica según el sistema de clasificación por órganos y sistemas del Diccionario Médico para la Actividad Regulatoria (MedDRA). Las reacciones adversas se clasifican según su frecuencia de aparición: muy frecuentes (≥ 1/10); frecuentes (≥ 1/100 a < 1/10) y poco frecuentes (≥ 1/1000 a < 1/100). A continuación se indican las reacciones adversas observadas en estudios clínicos con ReFacto o ReFacto AF. La frecuencia se indica considerando todos los eventos adversos espontáneos ocurridos durante el uso del medicamento en todos los estudios clínicos con 715 pacientes.
Dentro de cada grupo, los efectos adversos se enumeran en orden decreciente de gravedad.
Trastornos de la sangre y del sistema linfático:
muy frecuentes: inhibición del factor VIII (en pacientes previamente no tratados)*;
poco frecuentes: inhibición del factor VIII (en pacientes previamente tratados)*.
Sistema inmunológico:
poco frecuentes: reacciones anafilácticas.
Trastornos del metabolismo y de la nutrición:
frecuentes: disminución del apetito.
Sistema nervioso:
muy frecuentes: cefalea;
frecuentes: vértigo;
poco frecuentes: neuropatía periférica, somnolencia, disgeusia.
Órgano cardiaco:
poco frecuentes: angina de pecho, taquicardia, palpitaciones.
Vascular:
frecuentes: hemorragia/hematoma;
poco frecuentes: hipotensión, tromboflebitis, hiperemia facial.
Órganos respiratorio, torácico y mediastínico:
muy frecuentes: tos;
poco frecuentes: disnea.
Trastornos gastrointestinales:
frecuentes: diarrea, náuseas, dolor abdominal, vómitos.
Piel y tejido subcutáneo:
frecuentes: urticaria, prurito, erupción cutánea;
poco frecuentes: hiperhidrosis.
Sistema músculo-esquelético y tejido conjuntivo:
muy frecuentes: artralgia;
frecuentes: mialgia.
Trastornos generales y condiciones en el sitio de administración:
muy frecuentes: fiebre;
frecuentes: escalofríos, complicaciones relacionadas con catéter venoso permanente;
poco frecuentes: astenia, dolor, inflamación y otras reacciones en el sitio de administración.
Análisis de laboratorio:
frecuentes: prueba positiva para anticuerpos; prueba positiva para anticuerpos contra el factor VIII;
poco frecuentes: aumento de los niveles de aspartato aminotransferasa, alanina aminotransferasa, bilirrubina en sangre, creatinfosfocinasa en sangre.
* La frecuencia se indica basándose en estudios de todos los factores VIII que incluyeron pacientes con hemofilia A grave. PPT: pacientes previamente tratados; PNT: pacientes no tratados previamente.
Pediatría
Se ha notificado un caso de quiste en un paciente de 11 años y un caso de alteración de la conciencia en un paciente de 13 años; el desarrollo de estos estados podría estar relacionado con el tratamiento con ReFacto AF.
La seguridad de ReFacto AF se evaluó en estudios con adultos, niños y adolescentes previamente tratados (n = 18, de 12 a 16 años en el estudio y n = 49, de 7 a 16 años en un estudio adicional), y se observó una tendencia al aumento de la frecuencia de eventos adversos en niños de 7 a 16 años en comparación con adultos.
Adicionalmente, se obtuvieron datos sobre seguridad en estudios con pacientes previamente tratados (n = 18 menores de 6 años y n = 19 de 6 a 12 años) o no tratados previamente (n = 23 menores de 6 años). Los datos obtenidos indican que los perfiles de seguridad son similares a los observados en pacientes adultos.
Notificación de reacciones adversas sospechosas
Es importante notificar las reacciones adversas sospechosas tras la autorización del medicamento. Esto permite continuar con la vigilancia del balance beneficio-riesgo del medicamento. Los profesionales sanitarios deben informar sobre cualquier reacción adversa sospechosa de acuerdo con los requisitos locales.
Periodo de validez.
Forma farmacéutica – frasco: para el polvo liofilizado – 3 años; para el disolvente – 5 años.
Forma farmacéutica – jeringa precargada: 3 años.
Condiciones de almacenamiento.
Conservar en el envase original a una temperatura de entre 2 y 8 °C. No congelar, para evitar dañar la jeringa precargada.
El medicamento puede conservarse durante un máximo de 3 meses a una temperatura no superior a 25 °C dentro del periodo de validez. No se puede volver a colocar en el refrigerador una vez que se ha almacenado a temperatura ambiente.
La solución preparada debe utilizarse inmediatamente o bien dentro de las 3 horas siguientes a la preparación de la solución (y/o retirada de la tapa gris del conector, en el caso de la forma farmacéutica de jeringa precargada), siempre que se conserve a una temperatura no superior a 25 °C.
Mantener fuera del alcance de los niños.
Incompatibilidades.
Dado que no se han realizado estudios de compatibilidad con este medicamento, no debe mezclarse con otros medicamentos, incluyendo otros líquidos para infusión.
Para la administración de la solución debe utilizarse el conjunto para infusión suministrado, ya que el factor de coagulación VIII puede adsorberse en las superficies internas de otros equipos de infusión.
Envase.
Forma farmacéutica – frasco:
1 frasco con liofilizado, 1 jeringa precargada con disolvente, 1 adaptador para frasco, 1 sistema para infusión, 2 torundas con alcohol, 1 apósito adhesivo y 1 compresa de gasa en una caja de cartón.
Forma farmacéutica – jeringa precargada:
1 jeringa precargada con liofilizado en la cámara superior y disolvente (4 ml) en la cámara inferior, 1 émbolo, 1 sistema para infusión, 2 torundas con alcohol, 1 apósito adhesivo, 1 compresa de gasa y 1 tapa en una caja de cartón.
Categoría de dispensación. Bajo receta.
Fabricante.
Wyeth Farma S.A.
Fecha de la última revisión.