Ganciclovir-Farmex
Ucraina
Indice
ISTRUZIONI PER L'USO MEDICINALE DEL MEDICINALE GANCICLOVIR-FARMEK (GANCICLOVIR-PHARMEX)
Composizione:
principio attivo: ganciclovir;
1 flaconcino contiene ganciclovir 500 mg (sotto forma di sale sodico di ganciclovir).
Forma farmaceutica. Liofilizzato per soluzione per infusione.
Principali proprietà fisico-chimiche: massa porosa liofilizzata o polvere di colore bianco o quasi bianco.
Gruppo farmacoterapeutico.
Agenti antivirali per uso sistemico. Nucleosidi e nucleotidi, esclusi gli inibitori della transcriptasi inversa. Ganciclovir.
Codice ATC J05A B06.
Proprietà farmacologiche.
Farmacodinamica.
Meccanismo d'azione
Ganciclovir è un analogo nucleosidico sintetico della 2’-desossiguanosina che inibisce la replicazione dei virus dell'herpes sia in vitro che in vivo. I virus umani sensibili al ganciclovir includono il citomegalovirus (CMV), i virus dell'herpes simplex di tipo 1 e 2 (HSV-1 e HSV-2), i virus dell'herpes umano di tipo 6, 7 e 8 (HHV-6, HHV-7, HHV-8), il virus di Epstein-Barr, il virus della varicella-zoster (Varicella zoster) e il virus dell'epatite B. Gli studi clinici sono stati limitati alla valutazione dell'efficacia del farmaco nei pazienti con infezione da citomegalovirus.
Nelle cellule infette da CMV, il ganciclovir viene fosforilato dalla proteina chinasi virale UL97 a ganciclovir monofosfato. La fosforilazione successiva avviene grazie all'azione di diverse chinasi cellulari, formando il ganciclovir trifosfato, che subisce un lento metabolismo intracellulare. È stato dimostrato che questo metabolismo avviene nelle cellule infette da citomegalovirus umano e dal virus dell'herpes simplex; dopo la scomparsa del ganciclovir dal liquido extracellulare, il tempo di dimezzamento intracellulare del farmaco è rispettivamente di 18 e 6–24 ore. Poiché la fosforilazione del ganciclovir dipende principalmente dall'azione della chinasi virale, essa avviene prevalentemente nelle cellule infette.
L'attività antivirale del ganciclovir è dovuta all'inibizione della sintesi del DNA virale attraverso: l'inibizione competitiva dell'incorporazione del desossiguanosina trifosfato nel DNA mediata dalla DNA polimerasi; l'incorporazione del trifosfato di ganciclovir nel DNA virale, che porta all'arresto dell'allungamento del DNA virale o ne limita fortemente la prosecuzione.
Attività antivirale
L'attività antivirale (IC50) del ganciclovir nei confronti del citomegalovirus, determinata in vitro, varia da 0,08 µM (0,02 µg/ml) a 14 µM (3,5 µg/ml).
Efficacia clinica e sicurezza
Resistenza virale
La possibilità di sviluppo di resistenza virale deve essere considerata nei pazienti che mostrano ripetutamente una scarsa risposta clinica o nei quali persiste l'eliminazione virale durante il trattamento.
La resistenza del citomegalovirus al ganciclovir può svilupparsi dopo un trattamento o una profilassi prolungata con ganciclovir a causa di mutazioni selettive nel gene della chinasi virale (UL97), responsabile della monofosforilazione del ganciclovir, e/o, più raramente, nel gene della polimerasi virale (UL54). I virus che presentano mutazioni nel gene UL97 sono resistenti solo al ganciclovir, mentre i virus con mutazioni in UL54 sono resistenti al ganciclovir ma possono mostrare resistenza crociata ad altri agenti antivirali il cui meccanismo d'azione è diretto contro la polimerasi virale, e viceversa.
Pediatria
In uno studio prospettico, 36 pazienti pediatrici (da 6 mesi a 16 anni) con grave immunodeficienza (pazienti con HIV e infezione da CMV) hanno ricevuto ganciclovir per via endovenosa alla dose di 5 mg/kg/die per 2 giorni, seguito da somministrazione orale di ganciclovir per un periodo medio di 32 settimane. Il ganciclovir si è dimostrato efficace e il profilo di tossicità è risultato simile a quello osservato negli adulti. La riduzione del rilascio di CMV, rilevata mediante coltura o reazione a catena della polimerasi (PCR), è stata associata all'uso del ganciclovir. La neutropenia è stata l'unica reazione avversa grave osservata durante lo studio; sebbene nessun bambino abbia richiesto l'interruzione del trattamento, a 4 di essi è stato necessario somministrare fattore stimolante le colonie granulocitiche (G-CSF) per mantenere il numero assoluto di neutrofili superiore a 400 cellule/mm³.
In uno studio retrospettivo, 122 bambini di età compresa tra 16 giorni e 18 anni (età media 2,5 anni), sottoposti a trapianto epatico, hanno ricevuto ganciclovir per almeno 14 giorni alla dose di 5 mg/kg per via endovenosa due volte al giorno, seguito da monitoraggio preventivo per CMV mediante PCR. Il rischio di sviluppare infezione da CMV è stato considerato elevato in 43 pazienti e normale in 79. L'infezione asintomatica da CMV è stata rilevata mediante PCR nel 34,4% dei pazienti e si è verificata più frequentemente nei pazienti ad alto rischio rispetto ai riceventi a rischio normale (58,1% contro 21,8%, p = 0,0001). L'infezione da CMV si è sviluppata in 12 pazienti (9,8%) (8 nel gruppo ad alto rischio contro 4 nel gruppo a rischio normale, p = 0,03). In 3 pazienti si è verificato un rigetto acuto entro 6 mesi dal rilevamento del CMV, mentre in 13 pazienti il rigetto è preceduto all'infezione da CMV. Non sono stati registrati decessi dovuti all'infezione da CMV. Nel complesso, il 38,5% dei pazienti non ha ricevuto farmaci antivirali dopo la profilassi postoperatoria iniziale.
In un'analisi retrospettiva, la sicurezza e l'efficacia del ganciclovir sono state confrontate con quelle del valganciclovir in 92 bambini di età compresa tra 7 mesi e 18 anni (età media 9 anni), sottoposti a trapianto renale e/o epatico. Tutti i bambini hanno ricevuto ganciclovir per via endovenosa alla dose di 5 mg/kg due volte al giorno per 2 settimane dopo il trapianto. I bambini trattati fino al 2004 hanno ricevuto ganciclovir per via orale in dosi da 30 mg/kg/dose fino a 1 g/dose, 3 volte al giorno (n = 41), mentre i bambini trattati dopo il 2004 hanno ricevuto valganciclovir fino a 900 mg una volta al giorno (n = 51). La frequenza complessiva di infezione da CMV è stata del 16% (15 su 92 pazienti). Il tempo di insorgenza dell'infezione da CMV è risultato comparabile in entrambi i gruppi.
In uno studio randomizzato controllato, 100 neonati (età ≤ 1 mese) con infezione congenita sintomatica da CMV con coinvolgimento del sistema nervoso centrale (SNC) hanno ricevuto ganciclovir 6 mg/kg per via endovenosa ogni 12 ore per 6 settimane oppure nessun trattamento. Dei 100 pazienti inclusi nello studio, 42 soddisfacevano tutti i criteri dello studio e avevano risultati audiometrici all'inizio e dopo 6 mesi di follow-up. Di questi, 25 hanno ricevuto ganciclovir e 17 non hanno ricevuto trattamento. In 21 su 25 pazienti trattati con ganciclovir, l'udito è migliorato o è rimasto normale rispetto al basale entro 6 mesi, rispetto a 10 su 17 pazienti del gruppo di controllo (84% e 59% rispettivamente, p = 0,06). In nessun paziente trattato con ganciclovir si è osservato un peggioramento dell'udito rispetto al basale entro 6 mesi, rispetto a 7 pazienti nel gruppo di controllo (p < 0,01). Dopo un anno, in 5 su 24 pazienti trattati con ganciclovir e in 13 su 19 pazienti del gruppo di controllo si è verificato un peggioramento dell'udito rispetto al basale (p < 0,01). Durante lo studio, la neutropenia è stata osservata in 29 su 46 pazienti trattati con ganciclovir rispetto a 9 su 43 del gruppo di controllo (p < 0,1). Sono stati registrati 9 decessi durante lo studio: 3 nel gruppo trattato con ganciclovir e 6 nel gruppo di controllo. Nessun decesso è stato correlato al farmaco in studio.
In uno studio randomizzato controllato di Fase III, 100 neonati di età compresa tra 3 e 33 giorni (età media 12 giorni) con grave infezione congenita sintomatica da CMV con coinvolgimento del SNC hanno ricevuto ganciclovir 6 mg/kg per via endovenosa due volte al giorno per 6 settimane (n = 48) oppure nessun trattamento antivirale (n = 52). Nei neonati trattati con ganciclovir, i risultati dello sviluppo neurologico sono migliorati a 6 e 12 mesi rispetto ai pazienti non trattati con farmaci antivirali. Sebbene i pazienti trattati con ganciclovir mostrassero un minore ritardo e migliori parametri neurologici normali, la maggior parte di loro era comunque in ritardo rispetto ai parametri di sviluppo normale all'età di 6 settimane, 6 mesi o 12 mesi. In questo studio la sicurezza non è stata valutata.
In uno studio retrospettivo è stato studiato l'effetto della terapia antivirale sulla sordità tardiva in neonati con infezione congenita da CMV (età 4–34 mesi, età media 10,3 ± 7,8 mesi, età mediana 8 mesi). Di 21 neonati con udito normale alla nascita, tutti hanno sviluppato sordità tardiva. Il trattamento antivirale consisteva in:
- somministrazione endovenosa di ganciclovir alla dose di 5 mg/kg al giorno per 6 settimane, seguita da somministrazione orale di valganciclovir alla dose di 17 mg/kg due volte al giorno per 6 settimane, poi una volta al giorno fino al raggiungimento dell'età di 1 anno
oppure
- somministrazione orale di valganciclovir alla dose di 17 mg/kg due volte al giorno per 12 settimane, poi una volta al giorno per 9 mesi.
In nessun bambino è stato necessario l'impianto cocleare e l'udito è migliorato nell'83% dei casi con sordità all'inizio. La neutropenia è stata l'unico effetto avverso riportato e in nessun paziente è stato necessario interrompere il trattamento.
Farmacocinetica.
L'esposizione sistemica (AUC0–∞) osservata in pazienti adulti sottoposti a trapianto epatico dopo un'infusione endovenosa singola di ganciclovir alla dose di 5 mg/kg per 1 ora è stata mediamente di 50,6 µg×h/ml (CV% 40). In questa popolazione di pazienti, la concentrazione massima in plasma (Cmax) è stata mediamente di 12,2 µg/ml (CV% 24).
Distribuzione
Il volume di distribuzione del ganciclovir dopo somministrazione endovenosa è correlato alla massa corporea. Il volume di distribuzione allo stato stazionario varia tra 0,54–0,87 l/kg. Il legame con le proteine plasmatiche è dell'1–2% alle concentrazioni di ganciclovir di 0,5 e 51 µg/ml. Il ganciclovir penetra nel liquido cerebrospinale, dove può raggiungere concentrazioni pari al 24–67% di quelle plasmatiche.
Metabolismo
Il ganciclovir non subisce in misura significativa metabolismo.
Eliminazione
Il ganciclovir viene eliminato principalmente attraverso l'escrezione renale mediante filtrazione glomerulare e secrezione tubulare attiva, in forma inalterata. In pazienti con funzionalità renale normale, oltre il 90% della dose endovenosa somministrata viene escreta in forma inalterata nelle urine entro 24 ore. La clearance sistemica media varia da 2,64 ± 0,38 ml/min/kg (n = 15) a 4,52 ± 2,79 ml/min/kg (n = 6), mentre la clearance renale varia da 2,57 ± 0,69 ml/min/kg (n = 15) a 3,48 ± 0,68 ml/min/kg (n = 20), corrispondente al 90–101% del ganciclovir somministrato. Il tempo di dimezzamento in soggetti senza insufficienza renale varia da 2,73 ± 1,29 (n = 6) a 3,98 ± 1,78 (n = 8) ore.
Linearità/non linearità
Il ganciclovir somministrato per via endovenosa mostra una farmacocinetica lineare nell'intervallo di dosi da 1,6 a 5,0 mg/kg.
Farmacocinetica in gruppi particolari di pazienti
Pazienti con insufficienza renale
La clearance totale del ganciclovir è linearmente correlata alla clearance della creatinina. In pazienti con compromissione renale lieve, moderata e grave, la clearance sistemica media è stata rispettivamente di 2,1, 1 e 0,3 ml/min/kg. Nei pazienti con compromissione renale, il tempo di dimezzamento è aumentato. Nei pazienti con compromissione renale grave, il tempo di dimezzamento aumenta di circa 10 volte (per le modalità di aggiustamento della dose nei pazienti con compromissione renale, vedere la sezione «Modalità di somministrazione e posologia»).
Pazienti in emodialisi
Un'emodialisi della durata di 4 ore riduce la concentrazione plasmatica di ganciclovir dopo somministrazione endovenosa e orale di circa il 50%.
Nel caso di emodialisi intermittente, i valori di clearance del ganciclovir variano da 42 a 92 ml/min, il tempo di dimezzamento del farmaco durante l'emodialisi è di 3,3–4,5 ore. La frazione di ganciclovir eliminata in una singola sessione di emodialisi varia dal 50 al 63%. Nel caso di dialisi continua, la clearance del ganciclovir è più bassa (4,0–29,6 ml/min), ma nel periodo successivo alla somministrazione viene eliminata una percentuale maggiore della dose assunta.
Pazienti con insufficienza epatica
La sicurezza e l'efficacia del ganciclovir non sono state studiate in pazienti con compromissione epatica. Non si prevede che la compromissione epatica influenzi la farmacocinetica del ganciclovir, poiché il farmaco viene eliminato attraverso i reni. Pertanto non sono necessarie raccomandazioni specifiche per la modifica della dose (vedere la sezione «Modalità di somministrazione e posologia»).
Pediatria
La farmacocinetica del ganciclovir somministrato per via endovenosa alla dose di 200 mg/m² è stata studiata in due studi che hanno coinvolto pazienti di età compresa tra 3 mesi e 16 anni dopo trapianto epatico (n = 18) e renale (n = 25), e valutata mediante un modello farmacocinetico di popolazione.
La clearance della creatinina (ClCr) è stata identificata come variabile indipendente statisticamente significativa (covariata) per la clearance del ganciclovir, mentre l'altezza del paziente è risultata una covariata statisticamente significativa per la clearance del ganciclovir, il volume di distribuzione allo stato stazionario e il volume di distribuzione periferico. Quando covariate come ClCr e altezza sono state aggiunte al modello, sono emerse evidenti differenze nella farmacocinetica del ganciclovir tra diverse fasce d'età, mentre età, sesso e tipo di trapianto d'organo non sono risultati covariate significative in queste popolazioni. Nella Tabella 1 sono riportati i parametri farmacocinetici calcolati per fasce d'età.
Tabella 1.
Parametri farmacocinetici del ganciclovir dopo somministrazione endovenosa calcolati sulla base della superficie corporea (200 mg/m²) in pazienti sottoposti a trapianto epatico o renale, espressi come mediana (valore minimo–massimo).
| |
età < 6 anni |
età da 6 a < 12 anni |
età da ≥ 12 a ≤ 16 anni |
| n=17 |
n=9 |
n=17 |
|
| Clearance (l/ora) |
4,23 (2,11–7,92) |
4,03 (1,88–7,8) |
7,53 (2,89–16,8) |
| Vcent (l) |
1,83 (0,45–5,05) |
6,48 (3,34–9,95) |
12,1 (3,6–18,4) |
| Vperiph (l) |
5,81 (2,9–11,5) |
16,4 (11,3–20,1) |
27 (10,6–39,3) |
| Vss (l) |
8,06 (3,35–16,6) |
22,1 (14,6–30,1) |
37,9 (16,5–57,2) |
| AUC0–24h (μg×h/ml) |
24,3 (14,1–38,9) |
40,4 (17,7–48,6) |
37,6 (19,2–80,2) |
| Cmax (μg/ml) |
12,1 (9,17–15) |
13,3 (4,73–15) |
12,4 (4,57–30,8) |
Inoltre, i parametri farmacocinetici del ganciclovir dopo somministrazione endovenosa secondo lo schema posologico approvato per gli adulti (5 mg/kg mediante infusione endovenosa per 1 ora) sono stati studiati in un piccolo gruppo di neonati e bambini di età compresa tra 9 mesi e 12 anni con funzione renale normale (n = 10, età media 3,1 anni). L’esposizione, definita come valore medio di AUC0–∞ al 1° giorno (n = 10) e AUC0–12 al 14° giorno (n = 7), è risultata pari a 19,4 ± 7,1 e 24,1 ± 14,6 μg×h/mL, con valori di Cmax di 7,59 ± 3,21 μg/mL (1° giorno) e 8,31 ± 4,9 μg/mL (14° giorno), rispettivamente. Utilizzando una posologia basata sul peso corporeo in questo studio, si è osservata una tendenza a un’esposizione inferiore nei bambini più piccoli. Nei bambini di età inferiore a 5 anni, i valori medi di AUC0–∞ al 1° giorno (n = 7) e AUC0–12h al 14° giorno (n = 4) sono stati pari a 17,7 ± 5,5 e 17,1 ± 7,5 μg×h/mL.
Lo schema posologico del ganciclovir per somministrazione endovenosa, calcolato in base al peso corporeo e alla funzione renale (3 × peso corporeo × clearance della creatinina), deriva dallo schema posologico del valganciclovir pediatrico e garantisce valori simili di esposizione al ganciclovir nei bambini dalla nascita fino ai 16 anni di età (vedere tabella 2).
Tabella 2.
Parametri di AUC0–24h* modellizzati (μg×h/mL) del ganciclovir nei bambini trattati con ganciclovir a una dose calcolata secondo la formula 3 × peso corporeo × clearance della creatinina, somministrata mediante infusione per 1 ora.
| Parametri |
età < 4 mesi |
età da ≥ 4 mesi a ≤ 2 anni |
età da > 2 a < 6 anni |
età da ≥ 6 a < 12 anni |
età da ≥ 12 a ≤ 16 anni |
Tutti i pazienti |
| Numero di bambini nel modello |
781 |
384 |
86 |
96 |
126 |
1 473 |
| Mediana |
55,6 |
56,9 |
54,4 |
51,3 |
51,4 |
55,4 |
| Valore medio |
57,1 |
58,0 |
55,1 |
52,6 |
51,8 |
56,4 |
| Min. |
24,9 |
24,3 |
16,5 |
23,9 |
22,6 |
16,5 |
| Max. |
124,1 |
133,0 |
105,7 |
115,2 |
94,1 |
133,0 |
| Pazienti con AUC < 40 μg × h/mL |
89 (11 %) |
38 (10 %) |
13 (15 %) |
23 (24 %) |
28 (22 %) |
191 (13 %) |
| Pazienti con AUC 40−60 μg × h/mL |
398 (51 %) |
195 (51 %) |
44 (51 %) |
41 (43 %) |
63 (50 %) |
741 (50 %) |
| Pazienti con AUC > 60 μg × h/mL |
294 (38 %) |
151 (39 %) |
29 (34 %) |
32 (33 %) |
35 (28 %) |
541 (37 %) |
| AUC — area sotto la curva «concentrazione plasmatica-tempo». BSA — superficie corporea. ClCr — clearance della creatinina. Max. — massimo. Min. — minimo. * La simulazione è stata effettuata utilizzando un modello farmacocinetico di popolazione validato e dati demografici di bambini trattati con valganciclovir o ganciclovir in studi clinici (n = 1 473 registrazioni di dati). |
||||||
Pazienti anziani
Non sono stati condotti studi su soggetti di età pari o superiore a 65 anni.
Caratteristiche cliniche.
Indicazioni
Ganciclovir-Farmex è indicato negli adulti e negli adolescenti ≥ 12 anni per:
- il trattamento delle infezioni da citomegalovirus (CMV) nei pazienti con immunodeficienza;
- la prevenzione delle infezioni da CMV mediante terapia preventiva nei pazienti sottoposti a immunosoppressione farmacologica (ad esempio dopo trapianto d'organo o chemioterapia antitumorale).
Ganciclovir-Farmex è indicato anche nei bambini dalla nascita per:
- la prevenzione delle infezioni da CMV mediante profilassi universale nei pazienti sottoposti a immunosoppressione farmacologica (ad esempio dopo trapianto d'organo o chemioterapia antitumorale).
Si raccomanda di seguire le linee guida ufficiali riguardo l'uso appropriato degli agenti antivirali.
Controindicazioni
Ipersensibilità al ganciclovir, al valganciclovir o ad uno qualsiasi degli eccipienti del medicinale.
Allattamento al seno.
Precauzioni particolari
Avvertenze nella preparazione della soluzione di ganciclovir
Poiché il ganciclovir è considerato potenzialmente teratogeno e cancerogeno per l'uomo, il medicinale deve essere maneggiato con cautela.
È necessario evitare l'inalazione o il contatto diretto con la polvere contenuta nei flaconcini o con la soluzione ricostituita, nonché il contatto diretto con la pelle o le membrane mucose. La soluzione di ganciclovir è alcalina (pH circa 11). Si raccomanda di eseguire questa operazione indossando guanti in polietilene e occhiali protettivi.
In caso di contatto del ganciclovir con la pelle o le membrane mucose, lavare accuratamente con acqua e sapone; gli occhi devono essere sciacquati con acqua sterile o corrente, qualora non sia disponibile acqua sterile.
Interazioni con altri medicinali ed altre forme di interazione
Interazioni farmacocinetiche
Probenecid. L'associazione di probenecid con ganciclovir orale determina una riduzione statisticamente significativa della clearance renale del ganciclovir, con conseguente aumento statisticamente significativo dell'esposizione. Effetti simili sono attesi anche in caso di somministrazione concomitante di ganciclovir per via endovenosa e probenecid. Pertanto, i pazienti che assumono probenecid e ganciclovir devono essere attentamente monitorati per la tossicità del ganciclovir.
Didanosina. È stato dimostrato che l'assunzione concomitante di didanosina e ganciclovir determina un aumento costante delle concentrazioni plasmatiche di didanosina. Con la somministrazione endovenosa di ganciclovir alle dosi di 5–10 mg/kg al giorno, l'AUC della didanosina aumenta del 38–67%. Non si sono osservate variazioni clinicamente significative delle concentrazioni plasmatiche di ganciclovir. Tuttavia, considerando l'aumento delle concentrazioni plasmatiche di didanosina in presenza di ganciclovir, i pazienti devono essere attentamente monitorati per la tossicità della didanosina (vedere sezione «Precauzioni per l'uso»).
Altri agenti antiretrovirali. Gli isoenzimi del citocromo P450 non sono coinvolti nel metabolismo del ganciclovir. Di conseguenza, interazioni farmacocinetiche con inibitori della proteasi e inibitori non nucleosidici della trascrittasi inversa non sono attese.
Interazioni farmacodinamiche
Imipenem/cilastatina. Nei pazienti che ricevono contemporaneamente ganciclovir e imipenem/cilastatina sono state osservate convulsioni. Questi medicinali devono essere somministrati in combinazione con ganciclovir solo se i potenziali benefici superano il rischio (vedere sezione «Precauzioni per l'uso»).
Zidovudina. La zidovudina e il ganciclovir possono causare neutropenia e anemia. Un'interazione farmacodinamica può verificarsi durante l'uso concomitante di questi medicinali. Alcuni pazienti possono tollerare male il trattamento concomitante alle dosi complete di questi farmaci (vedere sezione «Precauzioni per l'uso»).
Altre possibili interazioni
È possibile un aumento della tossicità quando il ganciclovir viene somministrato contemporaneamente ad altri medicinali che possono avere effetto mielosoppressivo o causare insufficienza renale. Tra questi rientrano: agenti antibatterici (ad esempio dapsona, pentamidina, flucitosina, anfotericina B, trimetoprim/sulfametossazolo); immunosoppressori (ciclosporina, tacrolimus, micofenolato mofetile); agenti antineoplastici (ad esempio vincristina, vinblastina, doxorubicina e idrossiurea); nucleosidi (in particolare zidovudina, stavudina e didanosina), analoghi di nucleotidi (in particolare tenofovir, adefovir). Pertanto, questi medicinali devono essere somministrati contemporaneamente al ganciclovir solo se il beneficio terapeutico atteso supera il rischio (vedere sezione «Precauzioni per l'uso»).
Bambini
Gli studi sulle interazioni sono stati condotti esclusivamente negli adulti.
Caratteristiche particolari di impiego.
Ipersensibilità crociata
A causa della somiglianza della struttura chimica tra ganciclovir, aciclovir e penciclovir, possono verificarsi reazioni di ipersensibilità crociata con questi medicinali. Pertanto, si raccomanda cautela nell’uso di ganciclovir nei pazienti con nota ipersensibilità all’aciclovir o al penciclovir (o ai loro profarmaci, rispettivamente valganciclovir e famciclovir).
Mutagenicità, teratogenicità, cancerogenicità, fertilità e contraccezione
Prima di iniziare il trattamento con ganciclovir, i pazienti devono essere informati del rischio per il feto. Negli studi sugli animali, il ganciclovir ha mostrato effetti mutageni, teratogeni e cancerogeni, nonché inibizione della fertilità. Sulla base dei risultati degli studi clinici e preclinici, si ritiene probabile che il ganciclovir possa causare inibizione temporanea o irreversibile della spermatogenesi (vedi sezioni «Uso in gravidanza o durante l’allattamento» e «Effetti indesiderati»).
Il ganciclovir esercita un potenziale effetto teratogeno e cancerogeno e può causare malformazioni congenite e neoplasie maligne. Si raccomanda alle donne in età fertile di utilizzare metodi contraccettivi efficaci durante il trattamento con ganciclovir e per almeno 30 giorni dopo la fine del trattamento. Gli uomini devono utilizzare metodi contraccettivi barriera durante il trattamento e per almeno 90 giorni dopo la sua interruzione, a meno che non sia stato dimostrato che la partner non corre rischio di gravidanza (vedi sezioni «Uso in gravidanza o durante l’allattamento» e «Effetti indesiderati»).
L’uso di ganciclovir, specialmente nei bambini, richiede particolare cautela a causa della possibile cancerogenicità a lungo termine e del tossico effetto sulla funzione riproduttiva. I benefici del trattamento devono essere attentamente valutati caso per caso, considerando chiaramente i rischi.
Mielosoppressione
Il ganciclovir deve essere usato con cautela nei pazienti con citopenia ematologica in atto o in anamnesi correlata all’uso di medicinali, nonché nei pazienti sottoposti a radioterapia.
Nei pazienti trattati con ganciclovir sono stati osservati casi di grave leucopenia, neutropenia, anemia, trombocitopenia, pancitopenia e soppressione della funzione del midollo osseo. Il ganciclovir non deve essere somministrato se il numero assoluto di neutrofili è inferiore a 500 cellule/µL, il numero di piastrine è inferiore a 25.000/µL o il livello di emoglobina è inferiore a 8 g/dL (vedi sezioni «Modalità di somministrazione e posologia» e «Effetti indesiderati»).
Durante il trattamento si raccomanda il monitoraggio dell’emocromo completo, compreso il conteggio delle piastrine. Un monitoraggio ematologico più frequente può essere necessario nei pazienti con compromissione della funzionalità renale e nei neonati e lattanti (vedi sezione «Effetti indesiderati»). Nei primi 14 giorni di trattamento si raccomanda di effettuare il conteggio dei leucociti (preferibilmente con formula leucocitaria differenziata) ogni due giorni; nei pazienti con bassi livelli iniziali di neutrofili (<1000/µL), nei pazienti con precedente leucopenia durante terapie con altri agenti mielotossici e nei pazienti con compromissione renale, il monitoraggio deve essere effettuato giornalmente.
Nei pazienti che sviluppano grave leucopenia, neutropenia, anemia e/o trombocitopenia si raccomanda il trattamento con fattori ematopoietici di crescita e/o l’interruzione della terapia (vedi sezioni «Modalità di somministrazione e posologia» e «Effetti indesiderati»).
Compromissione della funzionalità renale
I pazienti con compromissione della funzionalità renale hanno un rischio aumentato di tossicità (in particolare ematologica). È necessario adeguare la dose del medicinale (vedi sezioni «Farmacocinetica in gruppi particolari di pazienti» e «Modalità di somministrazione e posologia»).
Uso con altri medicinali
Sono stati riportati casi di convulsioni in pazienti trattati con imipenem/cilastatina e ganciclovir; pertanto, il ganciclovir non deve essere somministrato contemporaneamente all’imipenem/cilastatina, a meno che i potenziali benefici terapeutici non superino il rischio (vedi sezione «Interazioni con altri medicinali ed altre forme di interazione»).
L’uso contemporaneo di ganciclovir con didanosina o con medicinali che esercitano effetti mielosoppressivi o nefrotossici deve essere attentamente monitorato, poiché ciò può portare allo sviluppo di tossicità additiva (vedi sezione «Interazioni con altri medicinali ed altre forme di interazione»).
Sostanze eccipienti
Questo medicinale nella formulazione da 500 mg contiene 2 mmol (45 mg) di sodio. Ciò deve essere tenuto in considerazione nei pazienti che devono controllare l’assunzione di sodio.
Uso in gravidanza o durante l’allattamento.
Fertilità
In uno studio clinico limitato su pazienti sottoposti a trapianto renale che avevano ricevuto ganciclovir per la prevenzione dell’infezione da CMV per un periodo fino a 200 giorni, è stato osservato un effetto del valganciclovir/ganciclovir sulla spermatogenesi, con riduzione della concentrazione e della motilità degli spermatozoi, rilevati dopo la fine del trattamento. Tale effetto è risultato reversibile. Circa 6 mesi dopo l’interruzione del ganciclovir, la concentrazione media degli spermatozoi e la motilità si sono ripristinate a livelli paragonabili a quelli osservati nei gruppi di controllo non trattati.
Negli studi sugli animali, il ganciclovir ha causato alterazioni della fertilità nei topi maschi e femmine, nonché inibizione della spermatogenesi e atrofia dei testicoli nei topi, nei ratti e nei cani, con dosi considerate clinicamente rilevanti.
Alla luce dei risultati degli studi clinici e preclinici, si ritiene probabile che il ganciclovir possa causare inibizione temporanea o irreversibile della spermatogenesi nell’uomo (vedi sezione «Caratteristiche particolari di impiego»).
Gravidanza
Non sono disponibili dati sulla sicurezza dell’uso di ganciclovir in gravidanza. Tuttavia, è noto che il ganciclovir attraversa rapidamente la barriera placentare. Negli studi sugli animali, l’uso di ganciclovir è stato associato a tossicità riproduttiva e teratogenicità. Pertanto, il ganciclovir non deve essere usato in gravidanza, eccetto nei casi in cui la necessità clinica del trattamento della paziente superi il potenziale rischio teratogeno per il feto.
Contraccezione
Alle donne in età fertile si raccomanda di utilizzare metodi contraccettivi efficaci durante il trattamento e per almeno 30 giorni dopo la fine del trattamento, poiché è possibile l’insorgenza di tossicità riproduttiva e teratogenicità. Ai pazienti di sesso maschile deve essere raccomandato di utilizzare metodi contraccettivi barriera durante il trattamento e per almeno 90 giorni dopo la fine del trattamento con ganciclovir, a meno che non sia stato dimostrato che la partner non corre rischio di gravidanza.
Allattamento
Non è noto se il ganciclovir passi nel latte materno nell’uomo, ma non si può escludere che possa passare nel latte e causare reazioni avverse gravi nel neonato. Dati ottenuti da studi sugli animali indicano che il ganciclovir viene escreto nel latte durante la lattazione nei ratti. Pertanto, l’allattamento al seno deve essere interrotto durante il trattamento con ganciclovir.
Capacità di guidare veicoli a motore e di usare macchinari.
L’uso di ganciclovir può influire in modo significativo sulla capacità di guidare veicoli a motore e di usare macchinari (vedi sezione «Effetti indesiderati»).
Modalità e dosi di somministrazione
La soluzione ottenuta diluendo la polvere sterile è destinata esclusivamente alla somministrazione endovenosa, preferibilmente mediante cannula di plastica e in una vena con adeguato flusso ematico. Non devono essere utilizzate iniezioni endovenose rapide o in bolo. La tossicità del ganciclovir può aumentare a causa di livelli eccessivi nel plasma sanguigno. Le iniezioni intramuscolari e sottocutanee del medicinale possono causare un grave irritazione dei tessuti a causa dell'elevato pH (9–11) della soluzione di Ganciclovir-Farmex.
Non si devono superare le dosi, la frequenza o la velocità di infusione raccomandate.
Trattamento delle infezioni da CMV
Adulti e adolescenti di età pari o superiore a 12 anni con funzionalità renale normale
Trattamento iniziale (induzione): infusione endovenosa di 5 mg/kg somministrata con velocità costante nell’arco di 1 ora, 2 volte al giorno (ogni 12 ore, 10 mg/kg/giorno) per 14–21 giorni.
Trattamento a lungo termine (di mantenimento): ai pazienti la cui funzione immunitaria non si è ripristinata e nei quali permane il rischio di recidiva della retinite da CMV, può essere somministrato un trattamento di mantenimento: 5 mg/kg mediante infusione endovenosa nell’arco di 1 ora, una volta al giorno per 7 giorni oppure 6 mg/kg una volta al giorno per 5 giorni alla settimana. La durata del trattamento di mantenimento viene stabilita individualmente.
Trattamento di malattia in evoluzione: a qualsiasi paziente con progressione dell’infezione da CMV, sia durante il trattamento di mantenimento che dopo l’interruzione del ganciclovir, il trattamento può essere ripreso secondo lo schema di induzione.
Bambini dalla nascita fino a 12 anni
Le informazioni attualmente disponibili sull’uso del medicinale nei bambini sono riportate nella sezione «Proprietà farmacologiche», tuttavia non è possibile fornire raccomandazioni relative al dosaggio.
Prevenzione delle infezioni da CMV mediante terapia preventiva
Adulti e adolescenti di età ≥ 12 anni con funzionalità renale normale
Regime di induzione: 5 mg/kg mediante infusione endovenosa nell’arco di 1 ora ogni 12 ore (10 mg/kg/giorno) per 7–14 giorni.
Regime di mantenimento: 5 mg/kg mediante infusione endovenosa nell’arco di 1 ora una volta al giorno per 7 giorni oppure 6 mg/kg una volta al giorno per 5 giorni alla settimana.
Bambini dalla nascita fino a 12 anni
Le informazioni attualmente disponibili sull’uso del medicinale nei bambini sono riportate nella sezione «Proprietà farmacologiche», tuttavia non è possibile fornire raccomandazioni relative al dosaggio.
Prevenzione delle infezioni da CMV mediante profilassi universale
Adulti e adolescenti di età pari o superiore a 16 anni: 5 mg/kg mediante infusione endovenosa nell’arco di 1 ora una volta al giorno per 7 giorni oppure 6 mg/kg una volta al giorno per 5 giorni alla settimana. La durata della profilassi viene stabilita in base al rischio di infezione da CMV.
Bambini dalla nascita fino a ≤ 16 anni
Le informazioni attualmente disponibili sull’uso del medicinale nei bambini sono riportate nella sezione «Proprietà farmacologiche», tuttavia non è possibile fornire raccomandazioni relative al dosaggio.
La singola dose giornaliera raccomandata di ganciclovir, somministrata mediante infusione endovenosa nell’arco di un’ora, viene calcolata in base alla SSM, determinata con la formula di Mosteller (Mostellar), e alla clearance della creatinina (ClCr), determinata con la formula di Schwartz (Schwartz). Le formule di calcolo sono riportate di seguito. La durata della profilassi universale deve essere stabilita individualmente in considerazione del rischio di sviluppo di infezione da CMV.
Dose pediatrica (mg) = 3 × SSM × ClCr (vedere le formule della SSM di Mosteller e della ClCr di Schwartz riportate di seguito).
Se la ClCr calcolata con la formula di Schwartz supera i 150 ml/min/1,73 m², il valore massimo da utilizzare nella formula è 150 ml/min/1,73 m².
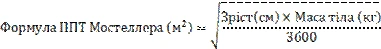
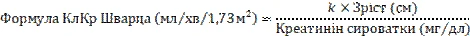
k = 0,33 per pazienti di età < 1 anno con basso peso corporeo alla nascita,
0,45 per pazienti di età < 2 anni,
0,55 per ragazzi di età compresa tra 2 e < 13 anni e ragazze di età compresa tra 2 e 16 anni,
0,7 per ragazzi di età compresa tra 13 e 16 anni.
Per i pazienti di età pari o superiore a 16 anni deve essere utilizzato il dosaggio raccomandato per gli adulti.
I valori di k indicati si basano sulla determinazione della creatinina sierica con il metodo di Jaffe (Jaffe) e potrebbero richiedere un aggiustamento qualora si utilizzino metodi enzimatici.
Si raccomanda di rivedere regolarmente i livelli sierici di creatinina, l’altezza e il peso corporeo del paziente e di modificare la dose se necessario.
Raccomandazioni particolari per il dosaggio
Pazienti con compromissione della funzionalità renale
I bambini (dalla nascita fino a 16 anni) con compromissione della funzionalità renale che ricevono una dose profilattica di ganciclovir calcolata con la formula: 3 × SSM × ClCr, non richiedono ulteriore aggiustamento della dose, poiché tale dose è già corretta in base alla ClCr.
Ai pazienti di età pari o superiore a 12 anni con compromissione della funzionalità renale che ricevono terapia preventiva e trattamento dell’infezione da CMV con una dose calcolata in base al peso corporeo, la dose di ganciclovir deve essere aggiustata in base alla ClCr, come indicato nella tabella 3.
Tabella 3
Aggiustamento della dose per pazienti con compromissione della funzionalità renale
| Clearance della creatinina (ml/min) |
Dose di induzione |
Dose di mantenimento |
| > 70 |
5 mg/kg ogni 12 ore |
5 mg/kg al giorno |
| 50–69 |
2,5 mg/kg ogni 12 ore |
2,5 mg/kg al giorno |
| 25–49 |
2,5 mg/kg al giorno |
1,25 mg/kg al giorno |
| 10–24 |
1,25 mg/kg al giorno |
0,625 mg/kg al giorno |
| < 10 |
1,25 mg/kg 3 volte/settimana dopo emodialisi |
0,625 mg/kg 3 volte/settimana dopo emodialisi |
La clearance della creatinina (ml/min) può essere calcolata sulla base della creatinina sierica secondo la seguente formula:
(140 – età [anni] × massa corporea [kg])
Negli uomini ClCr = 72 × 0,011 × creatinina sierica [µmol/l]
Nelle donne ClCr = 0,85 × valore calcolato per gli uomini.
Poiché nei pazienti con alterata funzionalità renale è raccomandata una correzione della dose, è necessario monitorare i livelli di creatinina sierica o la clearance della creatinina calcolata.
Pazienti con alterata funzionalità epatica
L’efficacia e la sicurezza dell’uso di ganciclovir non sono state studiate nei pazienti con alterata funzionalità epatica (vedere il paragrafo «Proprietà farmacologiche»).
Pazienti con grave leucopenia, marcata neutropenia, anemia, trombocitopenia e pancitopenia (vedere il paragrafo «Particolari avvertenze e precauzioni per l’uso» prima dell’inizio della terapia)
Se durante il trattamento con ganciclovir si verifica una marcata riduzione delle cellule ematiche, si dovrà considerare la possibilità di trattare con fattori di crescita ematopoietici e/o interrompere il trattamento (vedere i paragrafi «Particolari avvertenze e precauzioni per l’uso» e «Effetti indesiderati»).
Anziani
L’efficacia e la sicurezza dell’uso di ganciclovir negli anziani non sono state studiate. Poiché negli anziani la funzionalità renale è spesso ridotta, il ganciclovir deve essere somministrato con attenzione e tenendo rigorosamente conto della funzionalità renale (vedere il paragrafo «Proprietà farmacologiche»).
Bambini
L’esperienza nell’uso del ganciclovir nei bambini di età inferiore ai 12 anni è limitata (vedere i paragrafi «Farmacocinetica», «Particolari avvertenze e precauzioni per l’uso»). Gli effetti indesiderati osservati sono stati simili a quelli riscontrati negli adulti. Tuttavia, l’uso del ganciclovir nei bambini richiede particolare cautela a causa del potenziale effetto cancerogeno a lungo termine e della tossicità riproduttiva. Il beneficio terapeutico deve superare i rischi. Il ganciclovir non è indicato per il trattamento delle infezioni congenite e neonatali da CMV.
Modalità di somministrazione
Attenzione!
Il ganciclovir deve essere somministrato per infusione endovenosa nell’arco di 1 ora, con una concentrazione non superiore a 10 mg/ml. Non deve essere somministrato mediante iniezione endovenosa rapida o bolus, poiché la tossicità del ganciclovir può aumentare a causa di un eccessivo livello plasmatico del farmaco.
Non deve essere somministrato per via intramuscolare o sottocutanea, poiché ciò potrebbe causare un grave irritazione dei tessuti a causa del pH elevato (~11) della soluzione di ganciclovir (vedere il paragrafo «Effetti indesiderati»).
Non devono essere superate la dose raccomandata, la frequenza o la velocità di infusione.
Ganciclovir-Farmex è un liofilizzato (polvere) per soluzione per infusione. Dopo la ricostituzione, il medicinale Ganciclovir-Farmex è una soluzione incolore fino a giallo chiaro, quasi priva di particelle visibili.
L’infusione deve essere effettuata in una vena con adeguato flusso ematico, preferibilmente attraverso un catetere in plastica.
Preparazione del concentrato ricostituito
La ricostituzione deve essere effettuata in condizioni asettiche.
- Rimuovere il tappo per accedere alla parte centrale del tappo di gomma. Aspirare 10 ml di acqua per preparazioni iniettabili in una siringa e trasferirli lentamente nel flacone attraverso il centro del tappo di gomma. Non utilizzare acqua per preparazioni iniettabili contenente agenti antibatterici (parabeni, paraidrossibenzoati), poiché non compatibili con il medicinale Ganciclovir-Farmex.
- Agitare delicatamente il flacone per assicurare un completo bagnamento del liofilizzato.
- Ruotare delicatamente il flacone in movimenti circolari per alcuni minuti, fino a ottenere una soluzione completamente ricostituita.
- Esaminare attentamente la soluzione ricostituita per verificare la presenza di particelle meccaniche prima della diluizione con un diluente compatibile. La soluzione ricostituita del medicinale Ganciclovir-Farmex può essere incolore fino a giallo chiaro.
Preparazione della soluzione finale diluita per infusione
Aspirare dal flacone il volume necessario, calcolato in base al peso corporeo del paziente, e diluirlo successivamente in 100 ml di un appropriato diluente per infusione. Non è raccomandato utilizzare soluzioni per infusione con concentrazione superiore a 10 mg/ml. I seguenti diluenti per infusione sono compatibili con il ganciclovir: soluzione allo 0,9 % di cloruro di sodio; soluzione al 5 % di destrosio; soluzione di Ringer; soluzione di Ringer lattato. Non mescolare Ganciclovir-Farmex con altri medicinali per somministrazione endovenosa.
La soluzione diluita non deve essere somministrata per via endovenosa in un tempo superiore a 1 ora, come indicato nel paragrafo «Modalità di somministrazione» sopra.
La soluzione ricostituita con acqua per preparazioni iniettabili ha dimostrato stabilità chimica e fisica per 12 ore se conservata a 25 °C. La soluzione ricostituita non deve essere conservata in frigorifero né congelata.
Dal punto di vista microbiologico, la soluzione ricostituita deve essere utilizzata immediatamente; in caso contrario, la responsabilità per il tempo e le condizioni di conservazione prima dell’uso ricade sull’utilizzatore.
La soluzione diluita per infusione ha dimostrato stabilità chimica e fisica per 24 ore a una temperatura di 2–8 °C. La soluzione pronta per l’infusione non deve essere congelata. Dal punto di vista microbiologico, la soluzione pronta per l’infusione del medicinale Ganciclovir-Farmex deve essere utilizzata immediatamente; in caso contrario, la responsabilità per il tempo e le condizioni di conservazione prima dell’uso ricade sull’utilizzatore. In ogni caso, il periodo di validità non deve superare le 24 ore a una temperatura di 2–8 °C, qualora la ricostituzione e la diluizione siano state effettuate in condizioni asettiche controllate e validate.
Il medicinale non utilizzato o i rifiuti derivanti da tale medicinale devono essere smaltiti in conformità ai requisiti locali.
Bambini.
L’esperienza nell’uso del ganciclovir nei bambini di età inferiore ai 12 anni è limitata (vedere i paragrafi «Proprietà farmacologiche», «Indicazioni», «Interazioni con altri medicinali ed altre forme di interazione», «Particolari avvertenze e precauzioni per l’uso», «Modalità di somministrazione e dosi», «Sovradosaggio» ed «Effetti indesiderati»).
Sovradosaggio.
Sintomi
Sono stati riportati casi di sovradosaggio di ganciclovir (alcuni con esito fatale) durante studi clinici e nell’uso post-commercializzazione. La maggior parte dei casi non ha comportato effetti indesiderati o ha incluso uno o più effetti indesiderati elencati di seguito:
- tossicità ematologica: mielosoppressione, in particolare pancitopenia, leucopenia, neutropenia, granulocitopenia, insufficienza del midollo osseo;
- epatotossicità: epatite, alterazione della funzionalità epatica;
- tossicità renale: peggioramento dell’ematuria in pazienti con preesistente alterazione della funzionalità renale, insufficienza renale acuta, aumento dei livelli di creatinina;
- tossicità gastrointestinale: dolore addominale, diarrea, vomito;
- neurotossicità: tremore generalizzato, convulsioni.
Trattamento
Il ganciclovir viene eliminato durante l’emodialisi; pertanto, l’emodialisi può essere utilizzata per ridurre i livelli plasmatici del farmaco nei pazienti a cui è stata somministrata una dose eccessiva (vedere il paragrafo «Farmacocinetica»).
Pazienti particolari
Insufficienza renale: si prevede che un sovradosaggio di ganciclovir possa aumentare la tossicità renale nei pazienti con insufficienza renale (vedere il paragrafo «Particolari avvertenze e precauzioni per l’uso»).
Bambini: non sono disponibili informazioni specifiche.
Effetti indesiderati.
Profilo generale di sicurezza
Nota: il valganciclovir è una forma inattiva (profarmaco) del ganciclovir e pertanto gli effetti indesiderati associati all’uso del valganciclovir possono essere previsti anche con l’uso del ganciclovir. Il ganciclovir per somministrazione orale non è più prodotto, ma gli effetti indesiderati riportati durante il suo utilizzo possono essere osservati anche nei pazienti che ricevono ganciclovir per via endovenosa. Pertanto, l’elenco include gli effetti indesiderati osservati durante la somministrazione endovenosa o orale di ganciclovir o valganciclovir.
Nei pazienti trattati con ganciclovir/valganciclovir, gli effetti indesiderati più gravi e comuni sono stati neutropenia, anemia e trombocitopenia. Altri effetti indesiderati sono riportati di seguito.
La frequenza degli effetti indesiderati riportati di seguito è stata determinata sulla base dei dati aggregati del gruppo di pazienti con infezione da HIV (n = 1704) che ricevevano terapia di mantenimento con ganciclovir o valganciclovir. Fanno eccezione agranulocitosi, granulocitopenia e reazione anafilattica, i cui dati di frequenza provengono dal periodo post-marketing. Gli effetti indesiderati sono elencati in base ai sistemi organici secondo MedDRA [Dizionario Medico per le attività regolatorie], e la loro frequenza è classificata nelle seguenti categorie: molto frequente (≥ 1/10), frequente (≥ 1/100, < 1/10), non frequente (≥ 1/1000, < 1/100), raro (≥ 1/10.000, < 1/1000).
Il profilo generale di sicurezza di ganciclovir/valganciclovir è paragonabile nelle popolazioni di pazienti con infezione da HIV e nei pazienti sottoposti a trapianto d’organo, con l’eccezione che il distacco della retina è stato riportato solo nei pazienti con infezione da HIV affetti da retinite da CMV. Tuttavia, esistono alcune differenze nella frequenza di alcune reazioni. L’uso endovenoso di ganciclovir è associato a un rischio minore di diarrea rispetto alla somministrazione orale di valganciclovir. Aumento della temperatura corporea, infezioni da Candida, depressione, neutropenia grave (ANC [numero assoluto di neutrofili] < 500/µL) e reazioni cutanee si verificano più frequentemente nei pazienti con infezione da HIV. Disfunzione renale e epatica si osservano più spesso nei pazienti sottoposti a trapianto d’organo.
Infezioni e infestazioni: molto frequente — infezioni da Candida, in particolare candidosi orale, infezioni delle vie respiratorie superiori; frequente — sepsi, influenza, cellulite, infezione delle vie urinarie.
Sistema emolinfopoietico: molto frequente — neutropenia, anemia; frequente — trombocitopenia, leucopenia, pancitopenia; non frequente — insufficienza del midollo osseo; raro — agranulocitosi*, anemia aplastica, granulocitopenia*.
Sistema immunitario: frequente — ipersensibilità; non frequente — reazioni anafilattiche*.
Disturbi del metabolismo e della nutrizione: molto frequente — riduzione dell’appetito; frequente — perdita di peso.
Disturbi psichiatrici: frequente — depressione, confusione mentale, ansia; non frequente — eccitazione, disturbi psicotici, alterazioni del pensiero, allucinazioni.
Sistema nervoso: molto frequente — cefalea; frequente — insonnia, disgeusia (alterazione del gusto), ipoestesia, parestesia, neuropatia periferica, convulsioni, vertigini; non frequente — tremore.
Organi della vista: frequente — edema della cornea, distacco della retina, corpi mobili nel vitreo, dolore oculare, disturbi della vista, congiuntivite.
Organi dell’udito e dell’equilibrio: frequente — dolore all’orecchio; non frequente — sordità.
Sistema cardiaco: non frequente — aritmia.
Disturbi vascolari: frequente — ipotensione arteriosa.
Apparato respiratorio, torace e mediastino: molto frequente — dispnea, tosse.
Apparato gastrointestinale: molto frequente — diarrea, nausea, vomito, dolore addominale; frequente — dolore addominale superiore, costipazione, meteorismo, disfagia, dispepsia, sensazione di distensione addominale, ulcere orali, pancreatite.
Sistema epatobiliare: frequente — aumento dei livelli ematici di fosfatasi alcalina e AST [aspartato aminotransferasi], alterazione della funzionalità epatica, aumento dei livelli ematici di ALT [alanina aminotransferasi].
Tessuto cutaneo e sottocutaneo: molto frequente — dermatite; frequente — sudorazione notturna, prurito, eruzioni cutanee, alopecia; non frequente — secchezza cutanea, orticaria.
Apparato muscoloscheletrico e connettivo: frequente — mialgia, dolore articolare, dolore alla schiena, crampi muscolari.
Renale e sistema urinario: frequente — riduzione del clearance renale della creatinina, alterazione della funzionalità renale, aumento dei livelli ematici di creatinina; non frequente — ematuria, insufficienza renale.
Sistema riproduttivo: non frequente — sterilità maschile.
Disturbi generali: molto frequente — astenia, debolezza; frequente — dolore, brividi, malessere, astenia, reazioni nel sito di iniezione; non frequente — dolore toracico.
* I dati sulla frequenza di questi effetti indesiderati provengono dal periodo post-marketing; la frequenza di tutti gli altri effetti indesiderati è stata determinata sulla base di studi clinici.
Descrizione di singoli effetti indesiderati
Neutropenia. Il rischio di sviluppare neutropenia non può essere previsto sulla base del numero di neutrofili prima del trattamento. La neutropenia si manifesta solitamente durante la prima o la seconda settimana di terapia induttiva e dopo un dosaggio cumulativo ≤ 200 mg/kg. Il numero di cellule di solito si normalizza entro 2–5 giorni dall’interruzione del farmaco o dalla riduzione della dose (vedere sezione «Informazioni importanti sull’uso del medicinale»).
Neutropenia grave. La neutropenia grave è stata riportata più spesso nei pazienti con infezione da HIV (14%) in terapia di mantenimento con valganciclovir, ganciclovir orale o endovenoso (n = 1704), rispetto ai pazienti sottoposti a trapianto d’organo che ricevevano valganciclovir o ganciclovir per via orale. Nei pazienti che ricevevano valganciclovir o ganciclovir orale entro il 100° giorno dal trapianto, la frequenza di neutropenia grave era rispettivamente del 5% e del 3%, mentre nei pazienti che ricevevano valganciclovir entro il 200° giorno dal trapianto, la frequenza di neutropenia grave era del 10%.
Trombocitopenia. I pazienti con un basso numero iniziale di piastrine (< 100.000/µL) presentano un rischio aumentato di sviluppare trombocitopenia. I pazienti con immunosoppressione iatrogena dovuta al trattamento con farmaci immunosoppressori hanno un rischio maggiore di sviluppare trombocitopenia rispetto ai pazienti con AIDS (vedere sezione «Informazioni importanti sull’uso del medicinale»). La trombocitopenia grave può essere associata a emorragie potenzialmente letali.
Convulsioni. Sono state osservate convulsioni in pazienti che assumevano contemporaneamente imipenem-cilastatina e ganciclovir (vedere sezioni «Interazioni con altri medicinali ed altre forme di interazione» e «Informazioni importanti sull’uso del medicinale»).
Distacco della retina. Questo effetto indesiderato è stato riportato solo negli studi condotti su pazienti con infezione da HIV che ricevevano ganciclovir per il trattamento della retinite da CMV.
Reazioni nel sito di iniezione. Nei pazienti trattati con ganciclovir, si verificano frequentemente reazioni nel sito di iniezione. Il ganciclovir deve essere somministrato secondo le raccomandazioni riportate nella sezione «Modalità di somministrazione e posologia» al fine di ridurre il rischio di irritazione locale dei tessuti.
Bambini
Non sono stati condotti studi ufficiali sulla sicurezza dell’uso di ganciclovir nei bambini di età ≤ 12 anni; tuttavia, in base all’esperienza con valganciclovir, forma inattiva (profarmaco) di ganciclovir, il profilo generale di sicurezza della forma attiva è simile nei pazienti pediatrici e adulti. La neutropenia si verifica più frequentemente nei bambini, ma non vi è correlazione tra l’insorgenza di neutropenia e infezioni nei bambini. Il rischio più elevato di sviluppare citopenia nei neonati e nei lattanti richiede un attento monitoraggio del conteggio ematico in queste fasce d’età.
I dati sull’uso di valganciclovir o ganciclovir nei neonati o nei lattanti con HIV/AIDS o con infezione congenita sintomatica da CMV sono limitati, ma il profilo di sicurezza corrisponde al noto profilo di sicurezza di valganciclovir/ganciclovir.
La segnalazione degli effetti indesiderati dopo l’autorizzazione del medicinale è di fondamentale importanza. Permette di monitorare il rapporto beneficio/rischio del medicinale. I professionisti sanitari, i farmacisti, i pazienti o i loro rappresentanti legali devono segnalare tutti i casi sospetti di effetti indesiderati e l’inefficacia del medicinale attraverso il Sistema informatizzato automatizzato di farmacovigilanza al seguente indirizzo: https://aisf.dec.gov.ua.
Durata della conservazione. 3 anni.
Condizioni di conservazione.
Conservare nella confezione originale a una temperatura non superiore a 25 °C.
Conservare fuori dalla portata dei bambini.
Incompatibilità.
Il ganciclovir non deve essere mescolato con altri farmaci somministrati per via endovenosa. Il ganciclovir precipita in soluzioni contenenti parabeni.
Confezione.
500 mg in flacone, 1 flacone in blister, 1 blister in astuccio di cartone.
Categoria di vendita. Sotto prescrizione medica.
Produttore.
S.R.L. «FARMEKS GRUP».
Sede del produttore e indirizzo del luogo di esercizio dell’attività.
Ucraina, 08301, Oblast’ di Kiev, città di Boryspil’, via Ševčenka, 100