Firmagon
Ucraina
Indice
ISTRUZIONI PER L'USO MEDICO DEL MEDICINALE FIRMAGON (FIRMAGON®)
Composizione:
Principio attivo: degarelix;
1 flaconcino contiene degarelix (come acetato) 80 mg o 120 mg;
dopo ricostituzione la concentrazione è pari a 40 mg/ml o 20 mg/ml;
Eccipienti: mannitolo (E 421);
1 siringa preriempita con solvente contiene 4,2 ml o 3,0 ml di acqua per preparazioni iniettabili.
Forma farmaceutica. Polvere per soluzione iniettabile.
Principali caratteristiche fisico-chimiche:
polvere: liofilizzato bianco o quasi bianco, sotto forma di cake;
solvente: liquido trasparente incolore.
Categoria farmacoterapeutica. Antagonisti ormonali e sostanze analoghe.
Codice ATC L02B X02.
Proprietà farmacologiche
Farmacodinamica
Degarelix è un antagonista selettivo dell'ormone di rilascio delle gonadotropine (GnRH) che si lega in modo competitivo e reversibile ai recettori ipofisari dell'ormone di rilascio delle gonadotropine (GnRH), riducendo rapidamente il rilascio delle gonadotropine, ormone luteinizzante (LH) e ormone follicolo-stimolante (FSH), e quindi riduce la secrezione di testosterone da parte dei testicoli. Il carcinoma della prostata è sensibile agli androgeni e risponde al trattamento che elimina la fonte di androgeni. A differenza degli agonisti del GnRH, gli inibitori del GnRH non inducono un rilascio di LH con conseguente picco di testosterone/stimolazione della crescita tumorale, né un potenziale peggioramento dei sintomi all'inizio della terapia.
Una somministrazione singola di 240 mg di Firmagon seguita da una dose di mantenimento mensile di 80 mg riduce rapidamente i livelli di LH, FSH e, di conseguenza, di testosterone. La concentrazione di diidrotestosterone (DHT) nel siero sanguigno si riduce in modo analogo a quella di testosterone.
Firmagon è efficace nel raggiungere e mantenere la soppressione della secrezione di testosterone al di sotto del livello di castrazione farmacologica di 0,5 ng/ml. La dose di mantenimento di 80 mg al mese determina una soppressione sostenuta della secrezione di testosterone nel 97% dei pazienti per almeno un anno.
Non sono state osservate fluttuazioni del testosterone dopo iniezioni ripetute durante il trattamento con Firmagon. Il livello medio di testosterone dopo un anno di trattamento è stato di 0,087 ng/ml (intervallo interquartile 0,06–0,15), N=167.
Risultati dello studio di conferma di fase III
L'efficacia e la sicurezza di degarelix sono state valutate in uno studio aperto, multicentrico, randomizzato, a gruppi paralleli, con controllo attivo. Lo studio ha confrontato l'efficacia e la sicurezza di due diversi regimi di dosaggio mensili di degarelix con dose iniziale di 240 mg (40 mg/ml) seguita da somministrazioni sottocutanee mensili di 160 mg (40 mg/ml) o 80 mg (20 mg/ml), rispetto all'amministrazione intramuscolare mensile di 7,5 mg di leuprorelina in pazienti affetti da cancro alla prostata che richiedevano terapia antiandrogenica. Complessivamente, 620 pazienti sono stati randomizzati in uno dei tre gruppi di trattamento, di cui 504 (81%) hanno completato lo studio. Nel gruppo di trattamento con degarelix 240/80 mg, 41 pazienti (20%) hanno abbandonato lo studio rispetto ai 32 (16%) del gruppo trattato con leuprorelina.
Dei 610 pazienti trattati:
- 31% aveva un cancro alla prostata localizzato;
- 29% aveva un cancro alla prostata localmente avanzato;
- 20% aveva un cancro alla prostata metastatico;
- 7% aveva uno stato metastatico non definito;
- 13% aveva anamnestici di intervento chirurgico o radioterapia curativa e un attuale aumento del livello dell'antigene prostatico specifico (PSA).
I dati basali personali erano simili in tutti i gruppi. L'età media era di 74 anni (range da 47 a 98 anni). L'obiettivo primario era dimostrare l'efficacia di degarelix nel raggiungere e mantenere la soppressione del testosterone a un livello inferiore a 0,5 ng/ml per 12 mesi di trattamento.
È stata selezionata la dose di mantenimento efficace più bassa, pari a 80 mg di degarelix.
Raggiungimento del livello di testosterone (T) nel siero sanguigno ≤ 0,5 ng/ml
Firmagon è efficace nel raggiungere una rapida soppressione del testosterone, vedere tabella 1.
Tabella 1
Percentuale di pazienti che hanno raggiunto un livello di T ≤ 0,5 ng/ml dopo l'inizio del trattamento
| Periodo di trattamento |
Degarelix 240/80 mg |
Leuprolide 7,5 mg |
| Giorno 1 |
52 % |
0 % |
| Giorno 3 |
96 % |
0 % |
| Giorno 7 |
99 % |
1 % |
| Giorno 14 |
100 % |
18 % |
| Giorno 28 |
100 % |
100 % |
Prevenzione delle fluttuazioni del testosterone
Per fluttuazione si intende un livello di testosterone superiore al valore basale di almeno il 15% durante le prime 2 settimane.
Nessun paziente trattato con degarelix ha presentato fluttuazioni del testosterone; si è osservata una riduzione media del livello di testosterone del 94% al terzo giorno. Nella maggior parte dei pazienti trattati con leuprolide, si sono verificate fluttuazioni del testosterone; si è osservato un aumento medio del livello di testosterone del 65% al terzo giorno. Tale differenza è risultata statisticamente significativa (p < 0,001).
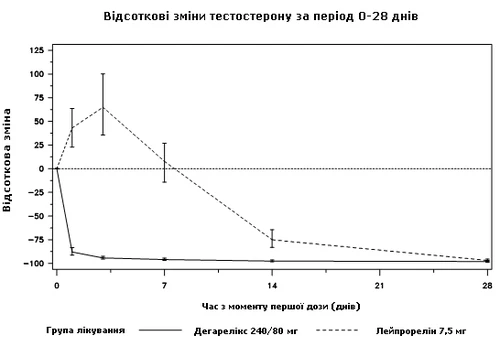
Diagramma 1. Variazione percentuale del testosterone rispetto ai livelli basali nei gruppi di trattamento fino al giorno 28 (mediana con intervalli interquartili)
Il criterio primario di valutazione dello studio era il livello di soppressione del testosterone dopo un anno di trattamento con degarelix o leuprolide. Non è stato dimostrato alcun vantaggio clinico di degarelix rispetto a leuprolide e antiandrogeno nella fase iniziale del trattamento.
Ripristino del testosterone
È stato condotto uno studio su pazienti con aumento del PSA dopo terapia locale (principalmente prostatectomia radicale e radioterapia), ai quali è stato somministrato Firmagon per sette mesi, seguito da un periodo di osservazione di sette mesi. Il tempo medio per il ripristino del testosterone (> 0,5 ng/ml, superiore al livello post-castrazione) dopo l'interruzione del trattamento è stato di 112 giorni (calcolato dall'inizio del periodo di osservazione, cioè 28 giorni dopo l'ultima iniezione). Il tempo medio per raggiungere un livello di testosterone > 1,5 ng/ml (superiore al limite inferiore del range normale) è stato di 168 giorni.
Effetto a lungo termine
La risposta positiva durante lo studio è stata definita come il raggiungimento della castrazione farmacologica al giorno 28 e il suo mantenimento fino al giorno 364, senza alcuna singola concentrazione di testosterone superiore a 0,5 ng/ml.
Tabella 2
Probabilità cumulativa di raggiungere un livello di testosterone ≤ 0,5 ng/ml dal giorno 28 al giorno 364
| Nome del medicinale Numero di pazienti |
Degarelix 240/80 mg N=207 |
Leuprorelina 7,5 mg N=201 |
| Numero di pazienti con risposta clinica |
202 |
194 |
| Frequenza di risposta (intervalli di confidenza)* |
97,2 % (93,5; 98,8 %) |
96,4 % (92,5; 98,2 %) |
* Stime di Kaplan-Meier all'interno del gruppo
Diminuzione del livello di PSA
Durante lo studio clinico non è stata effettuata una misurazione diretta delle dimensioni tumorali, tuttavia è stata osservata una risposta indiretta ai tumori benigni, evidenziata da una riduzione del livello medio di PSA del 95% dopo 12 mesi di trattamento con degarelix.
Il livello medio di PSA allo studio di base era:
- per il gruppo trattato con degarelix 240/80 mg – 19,8 ng/ml (intervallo interquartile: P25 9,4 ng/ml, P75 46,4 ng/ml);
- per il gruppo trattato con leuprorelina 7,5 mg – 17,4 ng/ml (intervallo interquartile: P25 8,4 ng/ml, P75 56,5 ng/ml).
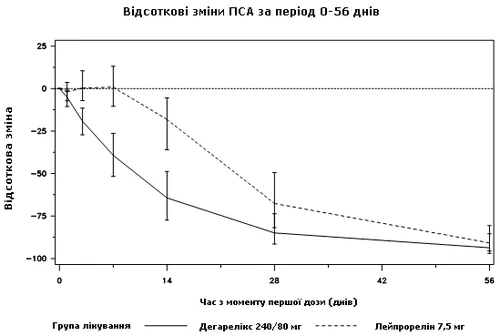
Diagramma 2. Variazione percentuale del PSA rispetto al valore basale nei gruppi di trattamento fino al giorno 56 (mediana con intervalli interquartili)
Questa differenza è risultata statisticamente significativa (p<0,001) nell'analisi per variabili prestabilite nel protocollo, al giorno 14 e al giorno 28.
I livelli di PSA si riducono del 64% dopo due settimane dall'amministrazione di degarelix, del 85% dopo un mese, del 95% dopo tre mesi e rimangono soppressi (circa il 97%) per un anno di trattamento.
Dal giorno 56 al giorno 364 non si sono osservate differenze significative tra degarelix e il farmaco di confronto riguardo alla variazione percentuale rispetto ai livelli basali.
Effetto sul volume della prostata
Dopo un trattamento di tre mesi con degarelix (regime di dosaggio 240/80 mg), si è osservata una riduzione del volume prostatico del 37%, misurato mediante ecografia transrettale (TRUS), in pazienti che richiedevano terapia ormonale prima della radioterapia e in pazienti candidati alla castrazione farmacologica. La riduzione del volume prostatico è risultata simile a quella ottenuta con goserelina e protezione antiandrogenica.
Effetto sugli intervalli QT/QTc
Negli studi di conferma di confronto tra Firmagon e leuprorelina sono stati eseguiti periodicamente ECG. Entrambe le terapie hanno mostrato intervalli QT/QTc superiori a 450 ms in circa il 20% dei pazienti. Rispetto ai valori iniziali, alla fine dello studio la variazione media di QTc è risultata pari a 12,0 ms per Firmagon e a 16,7 ms per leuprorelina.
Anticorpi contro degarelix
Lo sviluppo di anticorpi contro degarelix è stato osservato nel 10% dei pazienti dopo un anno di trattamento con Firmagon e nel 29% dei pazienti dopo un trattamento con Firmagon della durata fino a 5,5 anni. Non ci sono evidenze che la formazione di anticorpi influisca sull'efficacia o sulla sicurezza del trattamento con Firmagon dopo un trattamento della durata fino a 5,5 anni.
Farmacocinetica.
Assorbimento
Dopo somministrazione sottocutanea di 240 mg di degarelix alla concentrazione di 40 mg/ml, in pazienti con carcinoma prostatico, l'AUC0-28 giorni è risultata pari a 635 (602–668) giorno*ng/ml, la concentrazione massima (Cmax) è stata di 66 (61–71) ng/ml, raggiunta al tempo tmax di 40 (37–42) ore. I valori minimi medi erano compresi tra 11–12 ng/ml dopo la prima dose e tra 11–16 ng/ml dopo le dosi di mantenimento di 80 mg alla concentrazione di 20 mg/ml. La Cmax di degarelix nel plasma sanguigno diminuisce in modo bifasico, con un'emivita terminale media (t1/2) di circa 29 giorni per la dose di mantenimento. Il lungo periodo di emivita dopo somministrazione sottocutanea è dovuto al rilascio molto lento di degarelix dal deposito formato nei siti di iniezione. I parametri farmacocinetici del farmaco dipendono dalla sua concentrazione nella soluzione iniettabile. Pertanto, all'aumentare della concentrazione, la Cmax e la biodisponibilità tendono a diminuire, mentre l'emivita aumenta. Per questo motivo non devono essere utilizzate concentrazioni diverse da quelle previste.
Distribuzione
Il volume di distribuzione in volontari sani anziani è di circa 1 l/kg. Il legame con le proteine plasmatiche è di circa il 90%.
Metabolismo
Degarelix è soggetto alla normale degradazione dei peptidi durante il passaggio attraverso il sistema epatobiliare, con la maggior parte dell'eliminazione che avviene sotto forma di frammenti peptidici nelle feci. Dopo somministrazione sottocutanea, non sono stati rilevati metaboliti farmacologicamente attivi nel plasma. Studi in vitro hanno dimostrato che degarelix non è un substrato del sistema del citocromo CYP450 umano.
Eliminazione
In uomini sani, circa il 20–30% di una dose endovenosa di degarelix viene eliminato attraverso i reni. Si ritiene che il 70–80% venga eliminato attraverso il sistema epatobiliare. La clearance di degarelix dopo somministrazione endovenosa singola di 0,864–49,4 µg/kg in uomini sani anziani è risultata pari a 35–50 ml/ora/kg di peso corporeo.
Gruppi specifici di pazienti
Pazienti con compromissione renale
Studi farmacocinetici non sono stati condotti su pazienti con compromissione renale. I reni eliminano in forma invariata solo il 20–30% della dose somministrata di degarelix. Un'analisi farmacocinetica di popolazione basata sui dati dello studio di fase III ha mostrato che la clearance di degarelix nei pazienti con insufficienza renale da lieve a moderata è ridotta del 23%; pertanto, non è necessario alcun aggiustamento della dose nei pazienti con insufficienza renale da lieve a moderata. I dati relativi ai pazienti con insufficienza renale grave sono limitati, pertanto si raccomanda particolare cautela in questi pazienti.
Pazienti con compromissione epatica
Degarelix è stato studiato in uno studio farmacocinetico su pazienti con compromissione epatica da lieve a moderata. Non sono state osservate evidenze di un effetto accentuato nei pazienti con compromissione epatica rispetto ai volontari sani. Non è necessario alcun aggiustamento della dose nei pazienti con insufficienza epatica da lieve a moderata. L'uso in pazienti con insufficienza epatica grave non è stato studiato; pertanto, si raccomandano misure precauzionali per questa categoria di pazienti.
Caratteristiche cliniche.
Indicazioni terapeutiche. Trattamento di adulti di sesso maschile con carcinoma prostatico ormono-dipendente avanzato.
Controindicazioni. Ipersensibilità al degarelix o a uno qualsiasi degli altri componenti del medicinale.
Firmagon non è destinato all'uso in donne e bambini.
Interazioni con altri medicinali ed altre forme di interazione.
Non sono stati condotti studi di interazione con altri medicinali.
Poiché la terapia di soppressione androgenica può prolungare l'intervallo QTc, l'uso concomitante di Firmagon con farmaci che prolungano l'intervallo QTc o che possono indurre tachicardia ventricolare torsione di punta, come antiaritmici di classe IA (chinidina, disopiramide) o di classe III (amiodarone, sotalolo, dofetilide, ibutilide), nonché metadone, moxifloxacina, farmaci antipsicotici, richiede una valutazione accurata.
Il degarelix non è un substrato del sistema del citocromo CYP450 nell'uomo e non induce né inibisce in modo significativo CYP1A2, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1 o CYP3A4/5 in vitro.
Pertanto, interazioni farmacocinetiche clinicamente rilevanti con altri medicinali nel metabolismo associato a questi isoenzimi sono improbabili.
Caratteristiche d'uso.
Firmagon è indicato solo per somministrazione sottocutanea.
Effetto sull'intervallo QT/QTc
Una terapia androgenica deprivante prolungata può causare un allungamento dell'intervallo QT.
Negli studi di confronto tra Firmagon e leuprorelina, è stata eseguita un'ECG mensile. Con entrambi i regimi terapeutici, è stato osservato un intervallo QT/QTc superiore a 450 ms nel 20% dei pazienti e superiore a 500 ms rispettivamente nell'1% e nel 2% dei pazienti trattati con degarelix e leuprorelina.
L'effetto di Firmagon non è stato studiato in pazienti con intervallo QT superiore a 450 ms, in pazienti con tachicardia ventricolare torsione de pointe o con rischio di svilupparla, né in pazienti che assumono medicinali concomitanti in grado di prolungare l'intervallo QT. Pertanto, in tali pazienti, il rapporto rischio/beneficio dell'uso di Firmagon deve essere attentamente valutato.
Uno studio specifico sull'intervallo QT ha dimostrato che il degarelix non ha un effetto intrinseco sull'allungamento dell'intervallo QT/QTc.
Alterazioni epatiche
I pazienti con patologie epatiche note o sospette non sono stati inclusi negli studi clinici a lungo termine con degarelix. Sono state osservate transitorie lievi aumentate dei livelli di AST e ALT, senza aumento dei livelli di bilirubina né sintomi clinici. Si raccomanda il monitoraggio della funzionalità epatica durante il trattamento nei pazienti con alterazioni epatiche note o sospette. La farmacocinetica del degarelix è stata studiata dopo somministrazione endovenosa singola in pazienti con compromissione epatica da lieve a moderata.
Alterazioni renali
Il medicinale deve essere somministrato con cautela ai pazienti con grave compromissione della funzione renale.
Ipersensibilità
Firmagon non è stato studiato in pazienti con asma grave non trattata in anamnesi, reazioni anafilattiche, orticaria grave o angioedema ereditario.
Variazioni della densità ossea
La letteratura medica riporta una riduzione della densità minerale ossea in pazienti sottoposti ad orchifuniectomia o trattati con agonisti del GnRH, indicando l'impatto della soppressione del testosterone sulla densità ossea negli uomini. La densità minerale ossea non è stata misurata durante il trattamento con Firmagon.
Tolleranza al glucosio
In pazienti sottoposti ad orchifuniectomia o trattati con agonisti del GnRH è stata osservata una ridotta tolleranza al glucosio. È possibile lo sviluppo o il peggioramento del diabete. Pertanto, nei pazienti diabetici in trattamento con terapia deprivante, è necessario un monitoraggio più frequente della glicemia. L'effetto di Firmagon sui livelli di insulina e glucosio non è stato studiato.
Patologie cardiovascolari
La letteratura medica riporta patologie cardiovascolari come ictus e infarto del miocardio in pazienti sottoposti a terapia androgenica deprivante. Pertanto, tutti i fattori di rischio cardiovascolare devono essere attentamente considerati.
Fecondità
Firmagon può inibire la fecondità maschile finché persiste l'inibizione della secrezione di testosterone.
Uso durante la gravidanza o l'allattamento.
Il medicinale non è indicato per le donne.
Capacità di guidare veicoli o utilizzare macchinari.
Firmagon non ha alcun effetto oppure un effetto trascurabile sulla capacità di guidare veicoli o di utilizzare macchinari. La stanchezza e le vertigini sono le reazioni avverse più comuni che potrebbero influire sulla capacità di guidare veicoli o utilizzare macchinari.
Modalità e dosi di somministrazione.
Dosaggio
| Dosaggio iniziale |
Dosaggio di mantenimento una volta al mese |
| 240 mg sotto forma di due iniezioni sottocutanee consecutive da 120 mg ciascuna |
80 mg sotto forma di iniezione sottocutanea |
La prima dose di mantenimento deve essere somministrata un mese dopo l’assunzione della dose iniziale.
L’effetto terapeutico di Firmagon deve essere monitorato mediante parametri clinici e misurazione del livello di PSA nel siero. Gli studi clinici hanno dimostrato che la soppressione del testosterone (T) avviene immediatamente dopo la somministrazione della dose iniziale: nei pazienti il livello di testosterone nel siero corrisponde al livello post-castrazione medica (T ≤ 0,5 ng/ml) entro tre giorni nel 96% dei casi e entro un mese nel 100% dei casi. Il trattamento a lungo termine fino a un anno con dosi di mantenimento dimostra che il 97% dei pazienti mantiene un livello di testosterone stabilmente soppresso (T ≤ 0,5 ng/ml).
Se l’effetto clinico non è sufficiente, si deve verificare che il livello sierico di testosterone sia adeguatamente ridotto.
Poiché Firmagon non induce un aumento del livello di testosterone, non è necessario somministrare farmaci antiandrogeni per proteggere dall’effetto di rilascio del testosterone all’inizio del trattamento.
Somministrazione
Firmagon è indicato esclusivamente per somministrazione sottocutanea e non deve essere somministrato per via endovenosa. La somministrazione intramuscolare non è stata studiata e pertanto non è raccomandata.
Il medicinale deve essere somministrato mediante iniezioni sottocutanee nell’area addominale. Il sito di iniezione deve essere cambiato periodicamente. Le iniezioni devono essere effettuate in aree non soggette a compressione da indumenti (in particolare non nella zona della cintura in vita o della cintura dei pantaloni) e non troppo vicino alle costole.
Adattamento del dosaggio per gruppi specifici di pazienti
Pazienti anziani e pazienti con compromissione epatica o renale
Non è necessario alcun adattamento del dosaggio nei pazienti anziani o in caso di compromissione epatica o renale lieve o moderata. L’uso del medicinale in caso di insufficienza renale o epatica grave non è stato studiato; pertanto, in tali casi, è necessario adottare particolare cautela.
Linee guida per la somministrazione del medicinale
Non è raccomandata la somministrazione di concentrazioni diverse da quelle indicate, poiché la concentrazione influenza la formazione del deposito.
La soluzione ricostituita deve essere limpida e priva di particelle non disciolte.
Avvertenze
I flaconcini non devono essere agitati.
Firmagon 120 mg
Poiché la confezione contiene 2 flaconcini con polvere e 2 siringhe precaricate con solvente, la procedura di ricostituzione deve essere ripetuta due volte.
|
|
|
|
|
|
Non agitare il flacone per evitare la formazione di schiuma. È ammessa la presenza di un anello con piccole bolle d'aria sulla superficie del liquido. Il processo di dissoluzione richiede di solito alcuni minuti, ma in alcuni casi può durare fino a 15 minuti. |
|
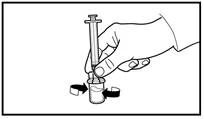
Verificare sempre con attenzione il volume aspirato e l'assenza di bolle d'aria. |
|
|
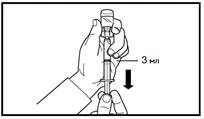
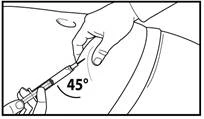
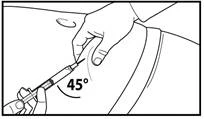
A tal fine, raccogliere la pelle dell'addome in una piega, sollevare leggermente il tessuto sottocutaneo e inserire profondamente l'ago con un angolo non inferiore a 45 gradi. Iniettare lentamente 3 ml di Firmagon 120 mg immediatamente dopo la ricostituzione. |
Non iniettare direttamente in vena. Tirare leggermente lo stantuffo verso di sé per verificare l'assenza di sangue nella siringa. Se compare sangue, il medicinale non deve essere utilizzato. Interrompere la procedura, smaltire siringa, ago e medicinale (ricostituire una nuova dose per il paziente). |
|
Firmagon 80 mg
La confezione contiene 1 flaconcino con polvere e 1 siringa preriempita con solvente per la preparazione di una soluzione per iniezione sottocutanea.
|
|
|
|
|
|
Non agitare il flacone per evitare la formazione di schiuma. È accettabile la presenza di un anello di piccole bolle d’aria sulla superficie del liquido. Il processo di dissoluzione richiede di solito alcuni minuti, ma in alcuni casi può impiegare fino a 15 minuti. |
|
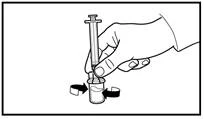
Verificare sempre con attenzione il volume aspirato e l’assenza di bolle d’aria. |
|
|
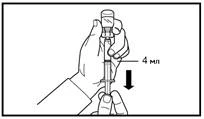
A tal fine, afferrare la pelle dell’addome formando una piega, sollevare delicatamente il tessuto sottocutaneo e inserire profondamente l’ago con un angolo di almeno 45 gradi. Iniettare lentamente 4 ml di Firmagon 80 mg immediatamente dopo la ricostituzione. |
Non iniettare direttamente in vena. Tirare leggermente lo stantuffo verso di sé per verificare l’assenza di sangue nella siringa. Se compare sangue, il medicinale non deve essere utilizzato. Interrompere la procedura, smaltire la siringa con ago e medicinale (ricostituire una nuova dose per il paziente). |
Dopo la ricostituzione, il medicinale deve essere somministrato immediatamente. La stabilità chimica e fisica della soluzione diluita è mantenuta per 2 ore dopo la ricostituzione.
Neonati e bambini. Il medicinale non deve essere utilizzato nei bambini.
Sovradosaggio.
Non esiste esperienza clinica riguardo alle conseguenze di un sovradosaggio acuto di Firmagon.
In caso di sovradosaggio, il paziente deve essere tenuto sotto osservazione e, se necessario, deve essere istituita una terapia di supporto.
Effetti indesiderati.
Gli effetti indesiderati più comunemente osservati durante la terapia con degarelix nello studio di fase III confermativo (N = 409) erano o associati agli effetti fisiologici previsti dell'inibizione della secrezione di testosterone, inclusi vampate di calore e aumento di peso (osservati rispettivamente nel 25% e nel 7% dei pazienti trattati per un anno), oppure erano effetti indesiderati a livello del sito di iniezione. Brividi transitori, febbre o sintomi simil-influenzali sono stati registrati nelle ore successive alla somministrazione del farmaco (rispettivamente nel 3%, 2% e 1% dei pazienti).
Gli effetti indesiderati a livello del sito di iniezione, manifestati principalmente come dolore ed eritema, sono stati osservati rispettivamente nel 28% e nel 17% dei pazienti; meno comuni sono stati edema (6%), indurimento (4%) e formazione di noduli (3%). Tali eventi si sono verificati più frequentemente dopo la dose iniziale, mentre durante la terapia di mantenimento con 80 mg la frequenza di questi eventi ogni 100 iniezioni era pari a 3 per il dolore e < 1 per eritema, edema, indurimento e formazione di noduli. Tali manifestazioni erano in genere transitorie, di intensità da lieve a moderata e molto raramente hanno portato all'interruzione del trattamento (< 1%). Reazioni gravi a livello del sito di iniezione, come infezioni, ascessi o necrosi del sito di iniezione che richiedono trattamento chirurgico o drenaggio, si sono verificate molto raramente.
Gli effetti indesiderati sono classificati per sistemi e organi e per frequenza: molto comuni (≥ 1/10); comuni (≥ 1/100 a < 1/10); non comuni (≥ 1/1.000 a < 1/100); rari (≥ 1/10.000 a < 1/1.000). All'interno di ogni gruppo per frequenza, gli effetti indesiderati sono elencati in ordine decrescente di gravità.
Frequenza degli effetti indesiderati riportati in 1259 pazienti trattati per un totale di 1781 paziente-anni negli studi di fase II e III e nel periodo post-registrazione.
Disturbi del sistema emolinfopoietico. Comuni: anemia*. Rari: febbre neutropenica.
Disturbi del sistema immunitario. Non comuni: ipersensibilità. Rari: reazioni anafilattiche.
Disturbi del metabolismo. Comuni: aumento di peso*. Non comuni: iperglicemia/diabete mellito, aumento dei livelli di colesterolo, perdita di peso, riduzione dell'appetito, alterazioni dei livelli di calcio nel sangue.
Disturbi psichici. Comuni: insonnia. Non comuni: depressione, riduzione del libido*.
Disturbi del sistema nervoso. Comuni: capogiri, cefalea. Non comuni: riduzione delle funzioni cognitive, ipoestesia.
Disturbi della vista. Non comuni: offuscamento della vista.
Disturbi cardiaci. Non comuni: aritmie cardiache (inclusa fibrillazione atriale), palpitazioni, allungamento dell'intervallo QT*. Rari: infarto miocardico, insufficienza cardiaca.
Disturbi vascolari. Molto comuni: vampate*. Non comuni: ipertensione arteriosa, reazione vasovagale (inclusa ipotensione).
Disturbi del sistema respiratorio, toracico e mediastinico. Non comuni: dispnea.
Disturbi gastrointestinali. Comuni: diarrea, nausea. Non comuni: stitichezza, vomito, dolore addominale, fastidio addominale, secchezza della bocca.
Disturbi epatobiliari. Comuni: aumento dei livelli delle transaminasi epatiche. Non comuni: aumento dei livelli di bilirubina, aumento dei livelli di fosfatasi alcalina.
Disturbi della cute e del tessuto sottocutaneo. Comuni: iperidrosi (inclusa sudorazione notturna)*, eruzioni cutanee. Non comuni: orticaria, eruzioni nodulari, alopecia, prurito, eritema.
Disturbi del sistema muscoloscheletrico e del tessuto connettivo. Comuni: dolore e fastidio muscoloscheletrico. Non comuni: osteoporosi/osteopenia, artralgia, debolezza muscolare, crampi muscolari, gonfiore/rigidità articolare.
Disturbi del sistema urinario. Non comuni: poliuria, urgenza minzionale, disuria, nicturia, insufficienza renale, incontinenza urinaria.
Disturbi del sistema riproduttivo e delle ghiandole mammarie. Comuni: ginecomastia*, atrofia testicolare*, disfunzione erettile*. Non comuni: dolore testicolare, dolore al torace, dolore pelvico, irritazione genitale, disturbi dell'eiaculazione.
Disturbi generali e condizioni in relazione al sito di somministrazione. Molto comuni: reazioni al sito di iniezione. Comuni: brividi, febbre, affaticamento aumentato*, sintomi simil-influenzali. Non comuni: malessere, edema periferico.
* Conseguenze fisiologiche dell'inibizione della secrezione di testosterone.
Descrizione di specifici effetti indesiderati
Modifiche degli esami di laboratorio.
Le modifiche degli esami di laboratorio sono state valutate durante un anno di trattamento nello studio di fase III confermativo (N = 409) e sono state simili sia per degarelix che per l'agonista del GnRH leuprorelina, utilizzato come trattamento di confronto. Un aumento significativo (3 volte superiore al limite superiore della norma) delle transaminasi epatiche (ALT, AST e GGT) durante il trattamento con entrambi i farmaci è stato osservato nel 2-6% dei pazienti con valori normali prima del trattamento. Una riduzione significativa dei parametri ematologici – ematocrito (≤ 0,37) e emoglobina (≤ 115 g/l) – è stata osservata rispettivamente nel 40% e nel 13-15% dei pazienti con valori iniziali normali. Non è ancora chiaro in quale misura questa riduzione dei parametri ematologici sia dovuta alla presenza di cancro alla prostata e in quale misura sia conseguenza del blocco androgenico. Deviazioni dai valori normali di potassio (≥ 5,8 mmol/l), creatinina (≥ 177 µmol/l) e azotemia (≥ 10,7 mmol/l) in pazienti con valori normali prima dell'inizio del trattamento sono state osservate rispettivamente nel 6%, 2% e 15% dei pazienti trattati con degarelix e nel 3%, 2% e 14% di quelli trattati con leuprorelina.
Modifiche dell'ECG.
Le modifiche dell'ECG osservate durante un anno di trattamento nello studio di fase III confermativo (N = 409) sono state simili sia per degarelix che per l'agonista del GnRH leuprorelina, utilizzato come trattamento di confronto. In tre (< 1%) su 409 pazienti trattati con degarelix e in quattro (2%) su 201 pazienti trattati con leuprorelina 7,5 mg è stato osservato un allungamento del QTcF ≥ 500 ms. Rispetto ai valori iniziali, l'aumento medio della durata del QTcF alla fine dello studio è stato di 12 ms per degarelix e di 16,7 ms per leuprorelina.
L'assenza di effetto diretto di degarelix sulla repolarizzazione cardiaca (QTcF), sulla frequenza cardiaca, sulla conduzione AV, sulla depolarizzazione cardiaca o sulla morfologia delle onde T o U è stata confermata in uno studio QT approfondito su volontari sani (N=80) che hanno ricevuto un'infusione endovenosa di degarelix per 60 minuti, raggiungendo una concentrazione media massima (Cmax) di 222 ng/ml, circa 3-4 volte superiore ai livelli di Cmax ottenuti nel trattamento del carcinoma prostatico.
Periodo di validità. 3 anni.
Non utilizzare dopo la data di scadenza indicata sull’imballaggio.
Condizioni di conservazione. Conservare nel contenitore originale a una temperatura non superiore a 25 °C. Non congelare. Tenere fuori dalla portata dei bambini.
Incompatibilità.
A causa della mancanza di studi di compatibilità, questo medicinale non deve essere miscelato con altri medicinali.
Confezione. 1 flaconcino con polvere da 80 mg in combinazione con 1 siringa preriempita con solvente (acqua per preparazioni iniettabili) da 5 ml (con segni di misura a 4,0 ml e volume di riempimento pari a 4,2 ml), 1 adattatore per flaconcino, 1 ago per iniezione e 1 stantuffo in un imballaggio cartonato.
2 flaconcini con polvere da 120 mg ciascuno in combinazione con 2 siringhe preriempite con solvente (acqua per preparazioni iniettabili) da 5 ml ciascuna (con segni di misura a 3,0 ml e volume di riempimento pari a 3,0 ml), 2 adattatori per flaconcino, 2 aghi per iniezione e 2 stantuffi in un imballaggio cartonato.
Categoria di prescrizione. Su prescrizione medica.
Produttore.
Ferring GmbH, Germania / Ferring GmbH, Germany.
Indirizzo del produttore e sede operativa.
Wittland 11, 24109 Kiel, Germania / Wittland 11, 24109 Kiel, Germany.