Fersinol
Ucraina
Indice
ISTRUZIONI PER L'USO MEDICINALE DEL MEDICINALE FERSINOL (FERSINOL)
Composizione:
Principio attivo: complesso polimaltosato di idrossido di ferro (III);
1 fiala (2 ml) contiene complesso polimaltosato di idrossido di ferro (III), corrispondente a 100 mg di ferro (III) elementare;
Eccipiente: acqua per preparazioni iniettabili.
Forma farmaceutica. Soluzione iniettabile.
Proprietà fisico-chimiche principali: soluzione di colore rosso scuro o marrone.
Gruppo farmacoterapeutico.
Agenti antianemici. Preparati di ferro per uso parenterale. Codice ATC B03AC.
Proprietà farmacologiche.
Farmacodinamica.
Dopo somministrazione intramuscolare, quasi tutto il ferro del complesso di idrossido di ferro (III) polimaltosato, come principio attivo, raggiunge il sistema reticolo-endoteliale del fegato ed è inoltre captato da transferrina, apoferritina, milza e midollo osseo. Lì si lega all'emoglobina, mioglobina ed enzimi contenenti ferro, e viene inoltre conservato nell'organismo sotto forma di ferritina. I cambiamenti nei parametri ematici dopo somministrazione parenterale di ferro avvengono non più rapidamente rispetto all'assunzione orale di sali di ferro. Come altri farmaci a base di ferro, il medicinale non influenza l'eritropoiesi ed è inefficace nelle anemie non correlate a carenza di ferro.
Farmacocinetica.
Dopo somministrazione intramuscolare, il complesso entra in circolo. La concentrazione massima di ferro viene raggiunta approssimativamente dopo 24 ore dall'iniezione. Nel sangue, il ferro si lega alla transferrina. Dal plasma, il complesso macromolecolare passa nel sistema reticolo-endoteliale, dove si scinde in idrossido di ferro e polimaltosio. Nei tessuti viene conservato sotto forma di ferritina; nel midollo osseo si lega all'emoglobina ed è utilizzato nel processo di eritropoiesi.
Vengono eliminate solo piccole quantità di ferro. La polimaltosio viene metabolizzata attraverso ossidazione ed eliminata.
Piccole quantità invariate del complesso possono attraversare la barriera placentare; quantità trascurabili penetrano nel latte materno. Il ferro legato a ferritina o transferrina può attraversare la barriera placentare, mentre, in forma di lattoferrina, viene escreto nel latte materno in piccole quantità.
L'inserimento del ferro nel protoporfirina dipende dal grado di anemia da carenza di ferro. È più intenso in caso di bassi livelli di emoglobina e diminuisce con la normalizzazione dei livelli di emoglobina. Il grado di utilizzazione del ferro non può superare la capacità legante del ferro dei proteine trasportatrici.
Caratteristiche cliniche.
Indicazioni.
Trattamento dell'anemia da carenza di ferro nei seguenti casi:
- quando è controindicata la terapia orale;
- in caso di alterata assorbimento del ferro a livello intestinale;
- in caso di mancato rispetto da parte del paziente del regime terapeutico con farmaci orali a base di ferro o in caso di intolleranza gastrointestinale persistente ai farmaci orali a base di ferro.
Controindicazioni.
- Ipersensibilità al complesso di idrossido di ferro con polimaltosio;
- anemie non correlate alla carenza di ferro (ad esempio anemia emolitica, anemia megaloblastica, disturbi dell'eritropoiesi, ipoplasia del midollo osseo);
- eccesso di ferro nell'organismo (emidosiderosi, emocromatosi);
- sindrome di Rendu-Osler-Weber;
- artrite reumatoide cronica;
- asma bronchiale;
- malattie renali infettive in fase acuta;
- iperparatiroidismo non controllato;
- cirrosi epatica scompensata;
- epatite infettiva;
- I trimestre di gravidanza;
- grave infiammazione o infezione renale o epatica (a causa della capacità del ferro elementare di accumularsi nei tessuti infiammati).
Interazioni con altri medicinali e altre forme di interazione.
Come per altri farmaci di ferro per somministrazione parenterale, il medicinale non deve essere somministrato contemporaneamente a farmaci di ferro per via orale, poiché ciò potrebbe ridurre l'assorbimento del ferro somministrato per via orale. Pertanto, il trattamento con farmaci di ferro per via orale non deve essere iniziato prima di 7 giorni dall'ultima somministrazione del farmaco parenterale di ferro.
L'uso concomitante di inibitori dell'ACE (ad esempio enalapril) può causare un potenziamento degli effetti sistemici dei farmaci di ferro parenterali, come eritema, crampi addominali, nausea, vomito e ipotensione.
Interazioni con i risultati dei test di laboratorio.
Alte dosi endovenose di farmaci a base di ferro (250 mg di ferro o più) possono causare una colorazione marrone del plasma nei campioni di sangue prelevati entro 4 ore dalla somministrazione.
I farmaci a base di ferro possono causare un aumento falso del livello di bilirubina nel plasma sanguigno e una riduzione falsa del livello di calcio nel plasma sanguigno. La determinazione del livello di ferro nel plasma (in particolare con metodo colorimetrico) può non essere attendibile per un periodo di 3 settimane dopo la somministrazione dei farmaci di ferro. I risultati delle misurazioni del ferro nel plasma sanguigno ottenuti entro 1-2 settimane dalla somministrazione di alte dosi di farmaci di ferro devono essere interpretati con cautela.
Lo studio delle riserve di ferro nel midollo osseo può risultare non informativo per un lungo periodo dopo il trattamento, poiché il medicinale può permanere nelle cellule reticoloendoteliali.
La scansione ossea con difosfonato marcato con Tc-99m, eseguita da 1 a 6 giorni dopo un'iniezione intramuscolare di un farmaco a base di ferro, può mostrare aree dense di attività nel femore, che riproducono il contorno del margine iliaco. La scansione ossea con agenti di imaging marcati con Tc-99m, in presenza di alte concentrazioni di ferritina nel plasma sanguigno o dopo infusioni endovenose di farmaci a base di ferro, può mostrare ridotto assorbimento osseo, marcata attività renale, iperperfusione e accumulo nei tessuti molli.
I farmaci a base di ferro possono ridurre l'assorbimento del citrato di 67Ga durante l'imaging di tumori e/o ascessi, a causa della competizione per gli stessi siti di legame.
La presenza di ferro può portare a risultati falsamente positivi nel test ortotolidina.
Caratteristiche di impiego.
Il medicinale deve essere utilizzato solo in caso di condizione da carenza di ferro confermata da opportuni esami (ad esempio, determinazione del livello di ferritina plasmatica, emoglobina, ematocrito, numero di eritrociti e dei relativi parametri: volume corpuscolare medio, concentrazione media di emoglobina eritrocitaria).
L'uso non giustificato di preparati parenterali di ferro in pazienti la cui anemia non è correlata alla carenza di ferro (ad esempio, pazienti con emoglobinopatie) può portare ad un eccessivo accumulo di ferro e allo sviluppo di una sindrome simile all'ematosiderosi.
Poiché l'uso parenterale di preparati di ferro ha causato reazioni anafilattoidi con esito letale, il medicinale deve essere somministrato solo a pazienti con indicazione ben definita e dopo conferma dello stato del paziente tramite esami di laboratorio. In caso di reazione allergica lieve, si devono utilizzare antistaminici.
Con l'uso parenterale di preparati di ferro esiste il rischio di sviluppare reazioni di ipersensibilità e reazioni anafilattoidi con ogni dose. Le reazioni anafilattoidi si verificano più frequentemente nei primi minuti dopo l'infusione e sono generalmente caratterizzate da difficoltà respiratorie improvvisa, sviluppo di tachicardia e ipotensione. Non è necessaria una dose di prova, poiché l'assenza di reazione con la dose di prova non garantisce l'assenza di reazione con dosi successive.
Il medicinale può essere somministrato solo se personale medico esperto nella valutazione e nel trattamento delle reazioni anafilattiche è disponibile per un intervento immediato e se il luogo è adeguatamente attrezzato con strumenti per le misure di rianimazione. Ogni paziente deve essere monitorato per segni di reazioni avverse almeno per 30 minuti dopo ogni somministrazione endovenosa di preparati contenenti ferro. Se si verificano reazioni allergiche o segni di intolleranza durante la somministrazione del medicinale, il trattamento deve essere interrotto immediatamente.
I pazienti con asma bronchiale, ridotta capacità di legame del ferro e/o carenza di acido folico appartengono al gruppo ad alto rischio di sviluppare reazioni allergiche o anafilattiche.
Il medicinale deve essere utilizzato con cautela in pazienti con allergie, insufficienza epatica o renale o malattie cardiovascolari.
I pazienti con artrite reumatoide e, possibilmente, altre malattie infiammatorie (ad esempio, spondilite anchilosante, lupus eritematoso sistemico) possono avere un rischio aumentato di reazioni ritardate, compresi febbre e peggioramento o riattivazione del dolore articolare.
Nel periodo post-commercializzazione sono stati riportati casi di ipofosfatemia sintomatica che ha causato osteomalacia e fratture, richiedendo intervento clinico, compresa chirurgia. Ai pazienti deve essere raccomandato di consultare il medico in caso di affaticamento persistente associato a mialgia o dolore osseo. Nel caso di trattamento prolungato o di somministrazione ad alte dosi del medicinale, o in presenza di fattori di rischio per ipofosfatemia, si raccomanda il monitoraggio del livello di fosfati nel plasma. In caso di ipofosfatemia prolungata, l'uso del medicinale deve essere rivalutato.
Uso durante la gravidanza o l'allattamento.
Gravidanza.
Il medicinale è controindicato durante il I trimestre di gravidanza. Non sono disponibili dati di studi su animali o su donne in gravidanza. Negli animali trattati con preparati di ferro per via endovenosa si è osservata tossicità embrio-fetale.
Durante il II e III trimestre di gravidanza, il medicinale può essere utilizzato solo quando il beneficio atteso per la madre supera il potenziale rischio per il feto.
Periodo di allattamento.
Non sono disponibili dati di studi su donne che allattano. Si deve considerare la possibilità che il ferro passi nel latte materno. Nel caso di somministrazione del medicinale durante l'allattamento, si devono considerare il beneficio atteso per la madre e il potenziale rischio per il neonato.
Capacità di influenzare la velocità di reazione nella guida di veicoli a motore o nell'uso di macchinari.
Non sono stati condotti studi specifici.
Modalità e dosi di somministrazione
Il medicinale è indicato per l'amministrazione intramuscolare profonda. L'amministrazione endovenosa è controindicata.
Modalità di somministrazione
Prima dell'iniezione, fiale e contenuto devono essere attentamente ispezionati. Devono essere utilizzate solo fiale che non presentano sedimenti né danni. Il medicinale deve essere somministrato immediatamente dopo l'apertura della fiala.
La tecnica di iniezione è di fondamentale importanza. È vietato somministrare la soluzione nella spalla o in aree danneggiate. In caso di somministrazione errata, possono insorgere dolore e discolorazione della cute nel sito di iniezione.
Si raccomanda di somministrare il medicinale nella regione ventroglutea secondo Hochstetter, invece della tradizionale somministrazione nel quadrante superiore esterno del muscolo gluteo.
La lunghezza dell'ago deve essere di almeno 5–6 cm. Il lume dell'ago non deve essere eccessivamente ampio.
Tecnica di somministrazione
- Identificare il sito di iniezione come segue (fig. 1): individuare il punto A corrispondente al margine anteriore della cresta iliaca. Ad esempio, se il paziente è disteso sul fianco destro, il dito medio della mano sinistra deve essere posizionato sul punto A. Allontanare l'indice dal dito medio in modo che si trovi al di sotto della linea della cresta iliaca nel punto B. Il triangolo compreso tra le falangi prossimali del dito medio e dell'indice rappresenta il sito di iniezione (fig. 2). La disinfezione deve essere effettuata con metodo abituale.
- Prima di inserire l'ago, spostare la cute lateralmente rispetto al sito di iniezione di circa 2 cm (fig. 3), in modo da conferire al canale di penetrazione una forma ad S. Questo accorgimento previene il reflusso della soluzione somministrata nei tessuti sottocutanei e la discolorazione della cute.
- Inserire l'ago quasi verticalmente rispetto alla superficie cutanea, con un angolo maggiore verso il punto dell'articolazione iliaca che verso il punto dell'articolazione dell'anca (fig. 4).
- Dopo l'iniezione, rimuovere lentamente l'ago e premere con un dito l'area cutanea adiacente al sito di iniezione. Mantenere la pressione per circa 1 minuto.
- Dopo l'iniezione, il paziente deve muoversi.
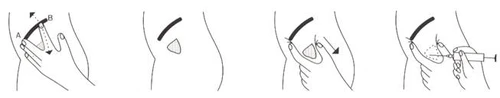
Fig.1 Fig. 2 Fig. 3 Fig. 4
Dosaggio
Calcolo della dose
La dose del medicinale deve essere calcolata individualmente in base al deficit totale di ferro, secondo la seguente formula:
| Dosaggio di ferro (mg) |
= |
deficit di ferro-Hb (mg) + ferro di riserva (mg) |
| Carenza di Hb-ferro |
= |
massa corporea (kg) × (livello normale di Hb - livello di Hb del paziente) (g/l) × 0,24* |
* Fattore 0,24 = 0,0034 × 0,07 × 1000 (contenuto di ferro nell'emoglobina - 0,34 %/volume ematico - 7 % della massa corporea/fattore 1000 - conversione da grammi a milligrammi).
La formula sopra riportata può essere utilizzata anche per calcolare il deficit totale di ferro.
Per peso corporeo inferiore a 35 kg: livello normale di Hb = 130 g/l, corrispondente a una riserva di ferro di 15 mg/kg di peso corporeo.
Per peso corporeo superiore a 35 kg: livello normale di Hb = 150 g/l, corrispondente a una riserva di ferro di 500 mg.
Tabella dei dosaggi
Tabella dei dosaggi per determinare la quantità totale necessaria del medicinale
| Massa corporea, kg |
Hb 60 g/l |
Hb 75 g/l |
Hb 90 g/l |
Hb 105 g/l |
||||
| ml |
fiale |
ml |
fiale |
ml |
fiale |
ml |
fiale |
|
| 5 |
3 |
1,5 |
3 |
1,5 |
3 |
1,5 |
2 |
1 |
| 10 |
6 |
3 |
6 |
3 |
5 |
2,5 |
4 |
2 |
| 15 |
10 |
5 |
9 |
4,5 |
7 |
3,5 |
6 |
3 |
| 20 |
13 |
6,5 |
11 |
5,5 |
10 |
5 |
8 |
4 |
| 25 |
16 |
8 |
14 |
7 |
12 |
6 |
11 |
5,5 |
| 30 |
19 |
9,5 |
17 |
8,5 |
15 |
7,5 |
13 |
6,5 |
| 35 |
25 |
12,5 |
23 |
11,5 |
20 |
10 |
18 |
9 |
| 40 |
27 |
13,5 |
24 |
12 |
22 |
11 |
19 |
9,5 |
| 45 |
30 |
15 |
26 |
13 |
23 |
11,5 |
20 |
10 |
| 50 |
32 |
16 |
28 |
14 |
24 |
12 |
21 |
10,5 |
| 55 |
34 |
17 |
30 |
15 |
26 |
13 |
22 |
11 |
| 60 |
36 |
18 |
32 |
16 |
27 |
13,5 |
23 |
11,5 |
| 65 |
38 |
19 |
33 |
16,5 |
29 |
14,5 |
24 |
12 |
| 70 |
40 |
20 |
35 |
17,5 |
30 |
15 |
25 |
12,5 |
| 75 |
42 |
21 |
37 |
18,5 |
32 |
16 |
26 |
13 |
| 80 |
45 |
22,5 |
39 |
19,5 |
33 |
16,5 |
27 |
13,5 |
| 85 |
47 |
23,5 |
41 |
20,5 |
34 |
17 |
28 |
14 |
| 90 |
49 |
24,5 |
43 |
21,5 |
36 |
18 |
29 |
14,5 |
Il medicinale deve essere somministrato per via intramuscolare alla dose di 2 ml ogni due giorni fino al raggiungimento della dose totale oppure 4 ml a intervalli più lunghi.
Durante il trattamento con il medicinale si raccomanda di effettuare regolari determinazioni del livello di Hb.
Dosaggio massimo.
Bambini con peso corporeo fino a 5 kg: 0,5 ml = 25 mg di ferro (¼ di fiala).
Bambini con peso corporeo da 5 a 10 kg: 1 ml = 50 mg di ferro (½ fiala).
Pazienti con peso corporeo da 10 a 45 kg: 2 ml = 100 mg di ferro (1 fiala) al giorno.
Adulti con peso corporeo da 45 kg: 4 ml = 200 mg di ferro (2 fiale).
Bambini.
Il medicinale può essere utilizzato nei bambini a partire dai 4 mesi di età.
Sovradosaggio.
Non sono stati segnalati casi di sovradosaggio.
Il sovradosaggio può causare un sovraccarico acuto di ferro, manifestato da sintomi di emosiderosi. Il trattamento è sintomatico.
Un sovraccarico cronico di ferro può manifestarsi come emocromatosi. Ciò può verificarsi quando un'anemia refrattaria al trattamento è stata diagnosticata erroneamente come anemia da carenza di ferro.
Un controllo periodico del livello di ferritina plasmatica può aiutare a riconoscere tempestivamente un accumulo progressivo di ferro.
Se necessario, si possono somministrare sostanze chelanti del ferro, ad esempio deferoxamina per via endovenosa.
Quando il medicinale viene somministrato in dosi molto elevate, il complesso non può essere rimosso dall'organismo mediante emodialisi a causa dell'elevata massa molecolare della sostanza attiva.
Effetti indesiderati.
Gli effetti indesiderati si verificano raramente. Dopo l'uso di farmaci parenterali contenenti ferro possono manifestarsi le seguenti reazioni avverse.
Apparato emolinfopoietico:
linfadenopatia generalizzata.
Sistema immunitario:
anafilassi.
Apparato gastrointestinale:
nausea, vomito.
Sistema nervoso:
cefalea, capogiri.
Apparato respiratorio, torace e mediastino:
broncospasmo con dispnea.
Sistema cardiocircolatorio:
perdita di coscienza, sincope, tachicardia, ipotensione, collasso circolatorio.
Pelle e tessuto sottocutaneo:
eruzioni cutanee, orticaria, angioedema.
Apparato muscoloscheletrico e tessuto connettivo:
dolore articolare e muscolare, artralgia, sensazione di rigidità alle braccia, alle gambe o ai muscoli del viso, osteomalacia da ipofosfatemia.
Disturbi generali e reazioni nel sito di somministrazione:
arrossamento del viso, dolore retrosternale e alla schiena, dolore nel sito di iniezione, infiammazione locale con linfadenopatia inguinale, dolore nel quadrante inferiore dell'addome.
Gli effetti indesiderati possono manifestarsi con un ritardo di 1-2 giorni dopo il trattamento.
Segnalazione di sospette reazioni avverse
La segnalazione di effetti indesiderati sospetti dopo la registrazione del medicinale è di grande importanza. Permette di monitorare continuamente il rapporto beneficio/rischio dell'uso di questo medicinale. Personale medico e farmaceutico, nonché pazienti o loro rappresentanti legali, devono segnalare tutti i casi sospetti di reazioni avverse e l'assenza di efficacia del medicinale attraverso il Sistema Informativo Automatico di Farmacovigilanza al seguente indirizzo: https://aisf.dec.gov.ua.
Durata della conservazione.
3 anni.
Condizioni di conservazione.
Conservare a una temperatura non superiore a 25 °C, nell'imballaggio originale e in un luogo inaccessibile ai bambini.
Incompatibilità.
La soluzione iniettabile non deve essere mescolata con altri farmaci.
Confezione.
2 ml in una fiala, 5 fiale in una confezione blister; 1 confezione blister in una scatola di cartone.
Categoria di vendita.
Sotto prescrizione medica.
Produttore.
WORLD MEDICINE ILAC SAN. VE TIC. A.S.
Indirizzo del produttore e sede operativa.
COSB G.O.Pasa Mah. 6. Cad. No:30, Cerkezkoy/Tekirdag, Turkey.