Fersinol
Ukraine
Table of Contents
INSTRUCTION FOR MEDICAL USE OF THE MEDICINAL PRODUCT FERSINOL (FERSINOL)
Composition:
Active substance: iron(III) hydroxide polymaltose complex;
1 ampoule (2 mL) contains iron(III) hydroxide polymaltose complex equivalent to 100 mg of elemental iron(III);
Excipient: water for injections.
Pharmaceutical form. Solution for injection.
Main physicochemical characteristics: dark red or brown solution.
Pharmacotherapeutic group.
Antianemic agents. Parenteral iron preparations. ATC code B03AC.
Pharmacological properties.
Pharmacodynamics.
After intramuscular administration, almost all iron from the iron(III) hydroxide polymaltose complex, as the active ingredient, enters the reticuloendothelial system of the liver and is also taken up by transferrin, apoferritin, spleen, and bone marrow. There, it binds to hemoglobin, myoglobin, and iron-containing enzymes, and is also stored in the body in the form of ferritin. Changes in blood parameters following parenteral iron administration occur no faster than with oral administration of iron salts. Like other iron preparations, the drug does not affect erythropoiesis and is ineffective in anemias unrelated to iron deficiency.
Pharmacokinetics.
After intramuscular injection, the complex enters the bloodstream. Maximum iron concentration is reached approximately 24 hours after injection. In the blood, iron binds to transferrin. From plasma, the macromolecular complex enters the reticuloendothelial system, where it is degraded into iron hydroxide and polymaltose. In tissues, iron is stored as part of ferritin; in the bone marrow, it binds to hemoglobin and is used in the process of erythropoiesis.
Only small amounts of iron are excreted from the body. Polymaltose is metabolized via oxidation and excreted.
In small amounts, the unchanged complex may cross the placental barrier; insignificant quantities penetrate into breast milk. Iron bound to ferritin or transferrin may cross the placental barrier, and in the form of lactoferrin, is excreted in small amounts into breast milk.
Incorporation of iron into protoporphyrin depends on the degree of iron-deficiency anemia. It is more intense when hemoglobin levels are low and decreases as hemoglobin levels normalize. The extent of iron utilization cannot exceed the iron-binding capacity of transport proteins.
Clinical characteristics.
Indications.
Treatment of iron deficiency anemia in the following cases:
- when oral therapy is contraindicated;
- in the presence of impaired intestinal iron absorption;
- in cases of non-compliance with oral iron therapy regimen or persistent gastrointestinal intolerance to oral iron preparations.
Contraindications.
- Hypersensitivity to iron hydroxide polymaltose complex;
- anemias not related to iron deficiency (e.g., hemolytic anemia, megaloblastic anemia, disorders of erythropoiesis, bone marrow hypoplasia);
- iron overload (hemosiderosis, hemochromatosis);
- Osler–Rendu–Weber syndrome;
- chronic polyarthritis;
- bronchial asthma;
- infectious kidney diseases in the acute stage;
- uncontrolled hyperparathyroidism;
- decompensated liver cirrhosis;
- infectious hepatitis;
- first trimester of pregnancy;
- severe inflammation or infection of the kidneys or liver (due to the ability of elemental iron to accumulate in inflamed tissues).
Interaction with other medicinal products and other forms of interaction.
Like other parenteral iron preparations, the medicinal product should not be used concomitantly with oral iron preparations, as this may lead to reduced absorption of orally administered iron. Therefore, treatment with oral iron preparations should not be initiated earlier than 7 days after the last administration of the parenteral iron preparation.
Concomitant use of ACE inhibitors (e.g., enalapril) may potentiate systemic effects of parenteral iron preparations, such as erythema, abdominal cramps, nausea, vomiting, and hypotension.
Interaction with laboratory test results.
High intravenous doses of iron preparations (250 mg of iron or more) may cause brown discoloration of plasma in blood samples taken within 4 hours after administration.
Iron preparations may cause falsely elevated plasma bilirubin levels and falsely decreased plasma calcium levels. Measurement of plasma iron levels (especially by colorimetric methods) may not be reliable for up to 3 weeks after administration of iron preparations. Results of plasma iron measurements obtained within 1–2 weeks after administration of high-dose iron preparations should be interpreted with caution.
Bone marrow iron stores assessment may remain uninformative for a prolonged period after treatment, as the medicinal product may persist in reticuloendothelial cells.
Bone scintigraphy using Tc-99m diphosphonate, performed 1–6 days after intramuscular injection of the iron preparation, may show dense areas of activity in the femur, mirroring the contours of the iliac crest. Bone scintigraphy using Tc-99m-labeled imaging agents, in the presence of high plasma ferritin concentrations or after intravenous iron infusions, may demonstrate reduced bone uptake, increased renal activity, pronounced blood pool activity, and soft tissue deposition.
Iron preparations may reduce 67Ga-citrate uptake during tumor and/or abscess imaging due to competition for the same binding sites.
The presence of iron may lead to false-positive results in the orthotolidine test.
Special precautions for use
The medicinal product should be used only in cases of confirmed iron deficiency, as established by appropriate diagnostic tests (e.g., plasma ferritin, hemoglobin, hematocrit, erythrocyte count, and erythrocyte parameters such as mean corpuscular volume and mean corpuscular hemoglobin concentration).
Unjustified use of parenteral iron preparations in patients whose anemia is not related to iron deficiency (e.g., patients with hemoglobinopathies) may lead to excessive iron accumulation and development of a syndrome resembling hemochromatosis.
Since parenteral administration of iron preparations has led to anaphylactoid reactions with fatal outcomes, the medicinal product should be administered only to patients with a clearly established indication and after confirmation of the patient's condition by laboratory test results. In the event of a mild allergic reaction, antihistamines should be used.
Parenteral administration of iron preparations carries a risk of hypersensitivity and anaphylactoid reactions with each dose administered. Anaphylactoid reactions most commonly occur within the first few minutes after administration and are typically characterized by sudden onset of respiratory distress, tachycardia, and hypotension. A test dose is not required, as the absence of a reaction following a test dose does not guarantee the absence of a reaction with subsequent doses.
The medicinal product should be administered only if medical personnel trained in the assessment and management of anaphylactic reactions are present and immediately available, and if the facility is properly equipped with resuscitation equipment. Each patient should be monitored for adverse reactions for at least 30 minutes after each intravenous administration of iron-containing medicinal products. If any allergic reactions or signs of intolerance occur during administration, treatment must be discontinued immediately.
Patients with bronchial asthma, low iron-binding capacity, and/or folic acid deficiency are at increased risk of developing allergic or anaphylactic reactions.
The medicinal product should be used with caution in patients with allergies, hepatic or renal impairment, or cardiovascular disease.
Patients with rheumatoid arthritis and possibly other inflammatory conditions (e.g., ankylosing spondylitis, systemic lupus erythematosus) may have an increased risk of delayed reactions, including fever and exacerbation or reactivation of joint pain.
During the post-marketing period, symptomatic hypophosphatemia has been reported, leading to osteomalacia and fractures requiring clinical intervention, including surgical treatment. Patients should be advised to consult a physician if they experience increasing fatigue accompanied by myalgia or bone pain. In cases of prolonged treatment or high-dose administration, as well as in the presence of risk factors for hypophosphatemia, plasma phosphate levels should be monitored. If persistent hypophosphatemia occurs, the use of the medicinal product should be re-evaluated.
Use during pregnancy or breastfeeding
Pregnancy
The medicinal product is contraindicated during the first trimester of pregnancy. Data from animal studies or studies involving pregnant women are lacking. Embryofetal toxicity has been observed in animals receiving intravenous iron preparations.
During the second and third trimesters of pregnancy, the medicinal product may be used only if the expected benefit to the mother outweighs the potential risk to the fetus.
Breastfeeding period
Data from studies in breastfeeding women are lacking. The possibility of iron passing into breast milk should be considered. When using the medicinal product during breastfeeding, the expected benefit to the mother and the potential risk to the infant should be taken into account.
Ability to affect reaction speed when driving or operating machinery
Appropriate studies have not been conducted.
Method of Administration and Dosage
The medicinal product is intended for deep intramuscular injection. Intravenous administration is contraindicated.
Method of Administration
Before injection, inspect the ampoules and their contents carefully. Only ampoules free from sediment and without any damage should be used. The medicinal product should be administered immediately after opening the ampoule.
The injection technique is crucial. The solution must not be injected into the shoulder or into damaged areas. Incorrect administration may result in pain and skin discoloration at the injection site.
The medicinal product is recommended to be administered into the ventrogluteal site according to Hochstetter, instead of the conventional injection into the upper outer quadrant of the gluteus muscle.
The needle length should be at least 5–6 cm. The needle lumen should not be excessively wide.
Injection Technique
- Locate the injection site as follows (Fig. 1): identify point A corresponding to the anterior surface of the iliac crest. For example, if the patient is lying on the right side, the middle finger of the left hand should be placed at point A. The index finger should be moved away from the middle finger so that it lies below the iliac crest line at point B. The triangle formed between the proximal phalanges of the middle and index fingers indicates the injection site (Fig. 2). Disinfection should be performed by the standard method.
- Before inserting the needle, displace the skin at the injection site approximately 2 cm (Fig. 3) to create an S-shaped needle track. This prevents backflow of the injected solution into subcutaneous tissues and subsequent skin discoloration.
- Insert the needle almost vertically relative to the skin surface, angled more toward the anterior superior iliac spine than toward the hip joint (Fig. 4).
- After injection, slowly withdraw the needle and apply finger pressure to the skin area adjacent to the injection site. Maintain pressure for approximately 1 minute.
- After injection, the patient should move.
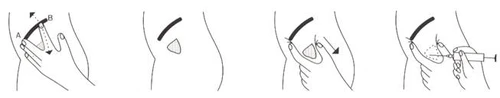
Fig. 1 Fig. 2 Fig. 3 Fig. 4
Dosage
Dose Calculation
The dose of the medicinal product is individually calculated according to the total iron deficit using the following formula:
| Dose of iron (mg) |
= |
deficit of Hb-iron (mg) + iron stores (mg) |
| Iron-deficient Hb |
= |
body weight (kg) × (normal Hb level - patient's Hb level) (g/L) × 0.24* |
* Factor 0.24 = 0.0034 × 0.07 × 1000 (iron content in hemoglobin – 0.34%, blood volume – 7% of body weight, factor 1000 – conversion from grams to milligrams).
The above formula can also be used to calculate the total iron deficit.
For body weight less than 35 kg: normal Hb level = 130 g/L, corresponding to iron stores of 15 mg/kg body weight.
For body weight over 35 kg: normal Hb level = 150 g/L, corresponding to iron stores of 500 mg.
Dosing table
Dosing table for determining the required total amount of the medicinal product
| Body weight, kg |
Hb 60 g/l |
Hb 75 g/l |
Hb 90 g/l |
Hb 105 g/l |
||||
| ml |
vials |
ml |
vials |
ml |
vials |
ml |
vials |
|
| 5 |
3 |
1.5 |
3 |
1.5 |
3 |
1.5 |
2 |
1 |
| 10 |
6 |
3 |
6 |
3 |
5 |
2.5 |
4 |
2 |
| 15 |
10 |
5 |
9 |
4.5 |
7 |
3.5 |
6 |
3 |
| 20 |
13 |
6.5 |
11 |
5.5 |
10 |
5 |
8 |
4 |
| 25 |
16 |
8 |
14 |
7 |
12 |
6 |
11 |
5.5 |
| 30 |
19 |
9.5 |
17 |
8.5 |
15 |
7.5 |
13 |
6.5 |
| 35 |
25 |
12.5 |
23 |
11.5 |
20 |
10 |
18 |
9 |
| 40 |
27 |
13.5 |
24 |
12 |
22 |
11 |
19 |
9.5 |
| 45 |
30 |
15 |
26 |
13 |
23 |
11.5 |
20 |
10 |
| 50 |
32 |
16 |
28 |
14 |
24 |
12 |
21 |
10.5 |
| 55 |
34 |
17 |
30 |
15 |
26 |
13 |
22 |
11 |
| 60 |
36 |
18 |
32 |
16 |
27 |
13.5 |
23 |
11.5 |
| 65 |
38 |
19 |
33 |
16.5 |
29 |
14.5 |
24 |
12 |
| 70 |
40 |
20 |
35 |
17.5 |
30 |
15 |
25 |
12.5 |
| 75 |
42 |
21 |
37 |
18.5 |
32 |
16 |
26 |
13 |
| 80 |
45 |
22.5 |
39 |
19.5 |
33 |
16.5 |
27 |
13.5 |
| 85 |
47 |
23.5 |
41 |
20.5 |
34 |
17 |
28 |
14 |
| 90 |
49 |
24.5 |
43 |
21.5 |
36 |
18 |
29 |
14.5 |
The drug should be administered intramuscularly at a dose of 2 ml every other day until the total cumulative dose is achieved, or 4 ml administered at longer intervals.
During treatment with the drug, regular monitoring of Hb levels is recommended.
Maximum dosage.
Children with body weight up to 5 kg: 0.5 ml = 25 mg of iron (¼ of an ampoule).
Children with body weight from 5 to 10 kg: 1 ml = 50 mg of iron (½ of an ampoule).
Patients with body weight from 10 to 45 kg: 2 ml = 100 mg of iron (1 ampoule) per day.
Adults with body weight from 45 kg: 4 ml = 200 mg of iron (2 ampoules).
Children.
The drug can be used in children aged 4 months and older.
Overdose.
There have been no reports of overdose.
Overdose may cause acute iron overload, manifesting as symptoms of haemosiderosis. Treatment is symptomatic.
Chronic iron overload may present as haemochromatosis. This may occur when anemia unresponsive to treatment has been incorrectly diagnosed as iron-deficiency anemia.
Periodic monitoring of plasma ferritin levels may help in timely detection of progressive iron accumulation.
If necessary, iron-binding agents (chelators) such as intravenous deferoxamine should be administered.
When the drug is administered in very high doses, the complex cannot be removed from the body by haemodialysis due to the high molecular weight of the active substance.
Adverse Reactions
Adverse reactions occur infrequently. The following adverse reactions may occur after administration of parenteral iron preparations.
Blood and lymphatic system disorders:
Generalized lymphadenopathy.
Immune system disorders:
Anaphylaxis.
Gastrointestinal disorders:
Nausea, vomiting.
Nervous system disorders:
Headache, dizziness.
Respiratory, thoracic and mediastinal disorders:
Bronchospasm with dyspnea.
Cardiovascular system disorders:
Loss of consciousness, syncope, tachycardia, hypotension, circulatory collapse.
Skin and subcutaneous tissue disorders:
Rash, urticaria, angioneurotic edema.
Musculoskeletal and connective tissue disorders:
Joint and muscle pain, arthralgia, feeling of stiffness in arms, legs or facial muscles, hypophosphatemic osteomalacia.
General disorders and administration site conditions:
Facial flushing, substernal and back pain, injection site pain, local inflammation with inguinal lymphadenopathy, lower abdominal quadrant pain.
Adverse reactions may occur with a delay of 1–2 days after treatment.
Reporting of suspected adverse reactions
Reporting of suspected adverse reactions after drug registration is highly important. It enables continuous monitoring of the benefit-risk balance of the medicinal product. Healthcare and pharmaceutical professionals, as well as patients or their legal representatives, should report all suspected adverse reactions and lack of efficacy through the Automated Pharmacovigilance Information System at the following link: https://aisf.dec.gov.ua.
Shelf life
3 years.
Storage conditions
Store at temperatures not exceeding 25 °C in the original packaging and in a place inaccessible to children.
Incompatibilities
The injection solution should not be mixed with other medicinal products.
Packaging
2 ml in a vial, 5 vials in a blister pack; 1 blister pack in a cardboard box.
Prescription status
Prescription only.
Manufacturer
UORL D MEDICINE ILAC SAN. VE TIC. A.S.
WORLD MEDICINE ILAC SAN. VE TIC. A.S.
Manufacturer's address and location of operations
COSB G.O.Pasa Mah. 6. Cad. No:30, Cerkezkoy/Tekirdag, Turkey.