Afstyla 1.000 UI polvere e solvente per soluzione iniettabile
Spagna
Indice
Foglio illustrativo: Informazioni per l'utente
Introduzione
Foglio illustrativo: informazioni per l'utente
AFSTYLA 250 UI, polvere e solvente per soluzione iniettabile
AFSTYLA 500 UI, polvere e solvente per soluzione iniettabile
AFSTYLA 1.000 UI, polvere e solvente per soluzione iniettabile
AFSTYLA 1.500 UI, polvere e solvente per soluzione iniettabile
AFSTYLA 2.000 UI, polvere e solvente per soluzione iniettabile
AFSTYLA 2.500 UI, polvere e solvente per soluzione iniettabile
AFSTYLA 3.000 UI, polvere e solvente per soluzione iniettabile
lonoctocog alfa (fattore VIII della coagulazione ricombinante a catena singola)
Legga attentamente questo foglio illustrativo prima che lei o suo figlio iniziate a usare questo medicinale perché contiene informazioni importanti per lei.
- Conservi questo foglio illustrativo: potrebbe essere necessario rileggerlo.
- Se ha domande, consulti il medico, il farmacista o l'infermiere.
- Questo medicinale è stato prescritto esclusivamente per lei o per suo figlio e non deve darlo ad altre persone, anche se presentano gli stessi sintomi, poiché potrebbe nuocere loro.
- Se manifesta effetti indesiderati, informi il medico, il farmacista o l'infermiere, anche qualora si tratti di effetti indesiderati non indicati nel presente foglio illustrativo. Vedere sezione 4.
Contenuto del foglio illustrativo :
- Che cos'è AFSTYLA e a che cosa serve
- Cosa deve sapere prima che lei o suo figlio iniziate a usare AFSTYLA
- Come usare AFSTYLA
- Possibili effetti indesiderati
- Come conservare AFSTYLA
- Contenuto della confezione e altre informazioni
1. Che cos'è AFSTYLA e a cosa serve
AFSTYLA è un prodotto contenente fattore VIII della coagulazione umano prodotto mediante tecnologia del DNA ricombinante. Il principio attivo di AFSTYLA è la lonoctocog alfa.
AFSTYLA viene utilizzato per trattare e prevenire gli episodi emorragici in pazienti con emofilia A (deficit congenito del fattore VIII). Il fattore VIII è una proteina necessaria per la coagulazione del sangue. Ai pazienti con emofilia A manca questo fattore, pertanto il sangue non si coagula con la rapidità necessaria e si ha una maggiore tendenza a sanguinare. AFSTYLA agisce sostituendo il fattore VIII carente nei pazienti con emofilia A, consentendo al sangue di coagulare normalmente.
AFSTYLA può essere utilizzato in tutti i gruppi di età.
2. Cosa deve sapere prima di iniziare a usare AFSTYLA
Non usi AFSTYLA
- Se il paziente ha avuto una reazione allergica potenzialmente letale ad AFSTYLA o a uno qualsiasi dei suoi componenti (elencati nella sezione 6).
- Se il paziente è allergico alle proteine del criceto.
Avvertenze e precauzioni
Tracciabilità
È importante registrare il numero di lotto di AFSTYLA.
Pertanto, ogni volta che utilizza un nuovo confezione di AFSTYLA, annoti la data e il numero di lotto (riportato sulla confezione dopo “Lotto”) e conservi queste informazioni in un luogo sicuro.
Consulti il suo medico, il farmacista o l’infermiere prima di iniziare a usare AFSTYLA.
- Potrebbero verificarsi reazioni allergiche (ipersensibilità). Il prodotto contiene residui di proteine del criceto (vedere anche "Non usi AFSTYLA"). Se compaiono sintomi di allergia, interrompa immediatamente il trattamento e contatti il medico. Il medico dovrà informarla sui primi segni di reazioni allergiche. Questi includono orticaria, eruzione cutanea diffusa, sensazione di oppressione al petto, difficoltà respiratorie, calo della pressione arteriosa e anafilassi (una reazione allergica grave che provoca gravi difficoltà respiratorie e capogiri).
- La formazione di inibitori (anticorpi) è una complicanza nota che può verificarsi durante il trattamento con tutti i medicinali contenenti fattore VIII. Questi inibitori, specialmente se presenti in grandi quantità, possono impedire che il trattamento sia efficace. Lei o suo figlio verranno attentamente monitorati per verificare l’eventuale sviluppo di inibitori. Se il sanguinamento suo o di suo figlio non dovesse essere controllato con AFSTYLA, consulti immediatamente il medico.
- Se le è stato diagnosticato un problema cardiaco o se è a rischio di svilupparne uno, informi il medico o il farmacista.
- Se viene utilizzato un dispositivo di accesso venoso centrale (DAVC) per l’iniezione di AFSTYLA, il medico dovrà valutare e discutere con lei i rischi di complicanze, come infezioni locali, batteri nel sangue (batteriemia) e formazione di coaguli (trombosi) nei vasi sanguigni nel sito di inserzione.
Altri medicinali e AFSTYLA
Informi il medico o il farmacista se sta assumendo, ha recentemente assunto o potrebbe dover assumere altri medicinali.
Gravidanza e allattamento
- Se è in gravidanza o in allattamento, se pensa di essere in gravidanza o se intende diventare incinta, consulti il medico o il farmacista prima di usare questo medicinale.
- Durante la gravidanza e l’allattamento, AFSTYLA deve essere somministrato solo se chiaramente necessario.
Guida di veicoli e uso di macchinari
AFSTYLA non altera la capacità di guidare veicoli o di usare macchinari.
AFSTYLA contiene sodio
Questo medicinale contiene 35 mg di sodio (componente principale del sale da cucina) in ogni flaconcino. Ciò corrisponde all’1,8% dell’assunzione giornaliera massima raccomandata di sodio per un adulto.
3. Come usare AFSTYLA
Il suo trattamento deve essere supervisionato da un medico esperto nel trattamento dei disturbi della coagulazione del sangue.
Segua esattamente le istruzioni del medico per l'assunzione di questo medicamento. In caso di dubbi, consulti nuovamente il medico.
Dosi
La quantità di AFSTYLA che lei o suo figlio necessitate e la durata del trattamento dipendono da:
- la gravità della sua malattia
- il sito e l'intensità dell'emorragia
- il suo stato clinico e la risposta clinica
- il suo peso corporeo
Segua le istruzioni indicate dal medico.
Ricostituzione e somministrazione
Istruzioni generali
- La polvere deve essere mescolata con il solvente (liquido) ed estratta dalla fiala in condizioni asettiche.
- AFSTYLA non deve essere mescolato con altri medicinali o solventi, eccetto quelli menzionati nella sezione 6.
- La soluzione deve essere trasparente o leggermente opalescente, da gialla a incolore, ovvero può brillare quando esposta alla luce, ma non deve contenere particelle visibili. Dopo il filtraggio o l'estrazione della soluzione (vedere più avanti), deve essere nuovamente controllata prima dell'uso. Non utilizzi la soluzione se appare torbida o se contiene fiocchi o particelle.
- L'eliminazione del prodotto non utilizzato e di tutti i materiali residui deve avvenire in conformità con le normative locali e le indicazioni del medico.
Ricostituzione e somministrazione
Senza aprire nessuna delle fiale, assicurarsi che la polvere di AFSTYLA e il liquido siano alla temperatura ambiente o corporea. Ciò può essere ottenuto lasciando le fiale a temperatura ambiente per circa un'ora oppure tenendole in mano per alcuni minuti. Non esponga le fiale a calore diretto. Le fiale non devono essere riscaldate oltre la temperatura corporea (37 °C).
Rimuova con attenzione i tappi protettivi dalle fiale e quindi pulisca la parte esposta dei tappi di gomma con un panno imbevuto di alcol. Lasci asciugare le fiale prima di aprire l'involucro del Mix2Vial (che contiene il trasferitore con filtro) e quindi segua le istruzioni riportate di seguito.
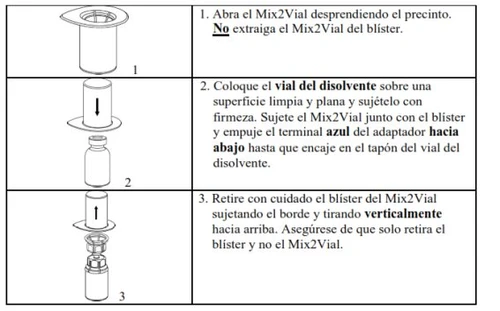

Utilizzi il kit per puntura venosa fornito con il prodotto e inserisca l'ago in una vena. Lasci scorrere il sangue fino all'estremità del tubo. Collegui la siringa all'estremità con blocco a vite del kit per puntura venosa. Inietti lentamente la soluzione ricostituita (a una velocità che le risulti comoda, fino a un massimo di 10 ml/min) nella vena, seguendo le istruzioni ricevute dal medico. Cerchi di evitare che il sangue entri nella siringa contenente il prodotto.
Controlli se manifesta effetti indesiderati subito dopo l'iniezione. Se dovesse manifestare effetti indesiderati che potrebbero essere correlati alla somministrazione di AFSTYLA, l'iniezione deve essere interrotta (vedere anche la sezione 2).
Uso in bambini e adolescenti
AFSTYLA può essere utilizzato in bambini e adolescenti di tutte le età. Nei bambini di età inferiore a 12 anni, potrebbero essere necessarie dosi più elevate o iniezioni più frequenti. Nei bambini di età superiore a 12 anni, può essere utilizzata la stessa dose degli adulti.
Se usa una quantità di AFSTYLA superiore a quella indicata
Se ha iniettato una quantità di AFSTYLA superiore a quella indicata, informi immediatamente il medico.
Se dimentica di usare AFSTYLA
Non inietti una dose doppia per compensare la dose dimenticata. Inietti immediatamente la dose successiva e segua le istruzioni del medico.
Se interrompe il trattamento con AFSTYLA
Se interrompe l'uso di AFSTYLA, potrebbe non essere più protetto dall'emorragia o potrebbe non smettere di sanguinare se ha un'emorragia in corso. Non interrompa l'uso di AFSTYLA senza aver prima consultato il medico.
Se ha ulteriori dubbi sull'uso di questo medicinale, chieda al medico, al farmacista o all'infermiere.
4. Possibili effetti indesiderati
Come tutti i medicinali, AFSTYLA può causare effetti indesiderati, anche se non tutte le persone li manifestano.
Sospenda immediatamente il medicinale e contatti il suo medico:
- se nota sintomi di reazioni allergiche
Possono verificarsi reazioni allergiche che includono i seguenti sintomi: orticaria localizzata, eruzione diffusa con prurito (orticaria), senso di costrizione toracica, sibilo respiratorio, pressione sanguigna bassa e anafilassi (reazione grave che causa difficoltà respiratoria severa o capogiri). In tal caso, deve interrompere immediatamente l’assunzione del medicinale e contattare il medico. - se nota che il medicinale non sta più funzionando correttamente (il sanguinamento non si arresta)
Nei bambini mai trattati in precedenza con farmaci a base di fattore VIII, possono formarsi con frequenza elevata anticorpi inibitori (vedere sezione 2) (più di 1 paziente su 10); tuttavia, nei pazienti già trattati in precedenza con fattore VIII (più di 150 giorni di trattamento) il rischio è raro (meno di 1 paziente su 100). Se lei o suo figlio avete sviluppato un inibitore a causa del medicinale, potreste manifestare un sanguinamento persistente. In tal caso, deve contattare immediatamente il medico.
Effetti indesiderati comuni (possono interessare fino a 1 persona su 10)
- Formicolio o intorpidimento (parestesia).
- Eruzioni cutanee.
- Febbre.
Effetti indesiderati non comuni (possono interessare fino a 1 persona su 100)
- Prurito.
- Arrossamento della pelle.
- Dolore nel sito di iniezione.
- Brividi.
- Sensazione di calore.
Effetti indesiderati nei bambini e negli adolescenti
Non sono state osservate differenze specifiche legate all'età nelle reazioni avverse tra bambini, adolescenti e adulti.
Segnalazione degli effetti indesiderati
Se manifesta qualsiasi tipo di effetto indesiderato, informi il medico, il farmacista o l’infermiere, anche se si tratta di possibili effetti indesiderati non indicati in questo foglio illustrativo. Può inoltre segnalarli direttamente attraverso il sistema nazionale di notifica riportato nell’Appendice V. Segnalando gli effetti indesiderati, può contribuire a fornire ulteriori informazioni sulla sicurezza di questo medicinale.
5. Conservazione di AFSTYLA
-
Tenere questo medicinale fuori dalla vista e dalla portata dei bambini.
-
Non usi questo medicinale dopo la data di scadenza indicata sull'etichetta e sulla confezione.
-
Conservare in frigorifero (tra 2 °C e 8 °C).
-
Prima della ricostituzione della polvere di AFSTYLA, può essere conservato a temperatura ambiente (inferiore a 25 °C) per un periodo unico non superiore a 3 mesi, entro la data di scadenza riportata sulle confezioni e sui flaconcini. Riporti la data in cui inizia la conservazione di AFSTYLA a temperatura ambiente sulla confezione del medicinale.
-
Una volta tolto il medicinale dal frigorifero, non deve essere rimesso al suo interno.
-
Non congelare.
-
Conservare il flaconcino all'interno della sua confezione per proteggerlo dalla luce.
-
Dopo la ricostituzione, il medicinale deve essere utilizzato preferibilmente immediatamente.
-
Se il prodotto ricostituito non viene somministrato immediatamente, i tempi e le condizioni di conservazione prima dell'uso sono di responsabilità dell'utilizzatore.
6. Contenuto della confezione e informazioni aggiuntive
Composizione di AFSTYLA
Il principio attivo è:
250 UI per flaconcino; dopo la ricostituzione con 2,5 ml di acqua per preparazioni iniettabili, la soluzione contiene 100 UI/ml di lonoctocog alfa.
500 UI per flaconcino; dopo la ricostituzione con 2,5 ml di acqua per preparazioni iniettabili, la soluzione contiene 200 UI/ml di lonoctocog alfa.
1.000 UI per flaconcino; dopo la ricostituzione con 2,5 ml di acqua per preparazioni iniettabili, la soluzione contiene 400 UI/ml di lonoctocog alfa.
1.500 UI per flaconcino; dopo la ricostituzione con 5 ml di acqua per preparazioni iniettabili, la soluzione contiene 300 UI/ml di lonoctocog alfa.
2.000 UI per flaconcino; dopo la ricostituzione con 5 ml di acqua per preparazioni iniettabili, la soluzione contiene 400 UI/ml di lonoctocog alfa.
2.500 UI per flaconcino; dopo la ricostituzione con 5 ml di acqua per preparazioni iniettabili, la soluzione contiene 500 UI/ml di lonoctocog alfa.
3.000 UI per flaconcino; dopo la ricostituzione con 5 ml di acqua per preparazioni iniettabili, la soluzione contiene 600 UI/ml di lonoctocog alfa.
Gli altri componenti sono:
L-istidina, polisorbato 80, cloruro di calcio diidrato, cloruro sodico (vedere l'ultimo paragrafo della sezione 2), saccarosio.
Solvente: acqua per preparazioni iniettabili.
Aspetto di AFSTYLA e contenuto della confezione
AFSTYLA si presenta sotto forma di polvere o massa friabile di colore bianco o leggermente giallastro e solvente per soluzione iniettabile trasparente e incolore.
La soluzione ricostituita deve essere trasparente o leggermente opalescente, gialla o incolore, cioè può brillare quando esposta alla luce, ma non deve contenere particelle visibili.
Confezioni
Una confezione da 250, 500 o 1.000 UI contenente:
1 flaconcino con polvere
1 flaconcino con 2,5 ml di acqua per preparazioni iniettabili
1 trasferitore con filtro 20/20
Una confezione interna contenente:
1 siringa monouso da 5 ml
1 dispositivo per venopuntura
2 tamponi imbevuti di alcol
1 cerotto non sterile
Una confezione da 1.500, 2.000, 2.500 o 3.000 UI contenente:
1 flaconcino con polvere
1 flaconcino con 5 ml di acqua per preparazioni iniettabili
1 trasferitore con filtro 20/20
Una confezione interna contenente:
1 siringa monouso da 10 ml
1 dispositivo per venopuntura
2 tamponi imbevuti di alcol
1 cerotto non sterile
Possono essere commercializzate solo alcune dimensioni della confezione.
Confezionamenti primari
250 UI | Flacone di vetro con tappo di gomma, disco di plastica arancione e capsula di alluminio verde a strisce |
500 UI | Flacone di vetro con tappo di gomma, disco di plastica blu e capsula di alluminio verde a strisce |
1.000 UI | Flacone di vetro con tappo di gomma, disco di plastica verde e capsula di alluminio verde a strisce |
1.500 UI | Flacone di vetro con tappo di gomma, disco di plastica turchese e capsula di alluminio verde a strisce |
2.000 UI | Flacone di vetro con tappo di gomma, disco di plastica porpora e capsula di alluminio verde a strisce |
2.500 UI | Flacone di vetro con tappo di gomma, disco di plastica grigio chiaro e capsula di alluminio verde a strisce |
3.000 UI | Flacone di vetro con tappo di gomma, disco di plastica giallo e capsula di alluminio verde a strisce |
Titolare dell'autorizzazione all'immissione in commercio e responsabile della produzione
CSL Behring GmbH
Emil-von-Behring-Straße 76
35041 Marburg
Germania
È possibile richiedere ulteriori informazioni su questo medicinale rivolgendosi al rappresentante locale del titolare dell'autorizzazione all'immissione in commercio:
België/Belgique/Belgien CSL Behring NV Tél/Tel: +32 15 28 89 20 | Lietuva CentralPharma Communications UAB Tel: +370 5 243 0444 |
| Luxembourg/Luxemburg CSL Behring NV Tél/Tel: +32 15 28 89 20 |
Česká republika CSL Behring s.r.o. Tel: + 420 702 137 233 | Magyarország CSL Behring Kft. Tel.: +36 1 213 4290 |
Danmark CSL Behring AB Tlf: +46 8 544 966 70 | Malta AM Mangion Ltd. Tel: +356 2397 6333 |
Deutschland CSL Behring GmbH Tel: +49 6190 75 84810 | Nederland CSL Behring BV Tel: + 31 85 111 96 00 |
Eesti CentralPharma Communications OÜ Tel: +3726015540 | Norge CSL Behring AB Tlf: +46 8 544 966 70 |
Ελλάδα CSL Behring ΕΠΕ Τηλ: +30 210 7255 660 | Österreich CSL Behring GmbH Tel: +43 1 80101 1040 |
España CSL Behring S.A. Tel: +34 933 67 1870 | Polska CSL Behring Sp.z o.o. Tel: +48 22 213 22 65 |
France CSL Behring S.A. Tél: + 33 –(0)-1 53 58 54 00 | Portugal CSL Behring Lda Tel: +351 21 782 62 30 |
Hrvatska Marti Farm d.o.o. Tel: +385 1 5588297 | România Prisum Healthcare S.R.L. Tel: +40 21 322 0171 |
Ireland CSL Behring GmbH Tel: +49 6190 75 84700 Ísland CSL Behring AB Sími: +46 8 544 966 70 | Slovenija Emmes Biopharma Global s.r.o. podružnica v Sloveniji Tel:+ 386 41 42 0002 Slovenská republika CSL Behring Slovakia s.r.o. Tel: +421 911 653 862 |
Italia CSL Behring S.p.A. Tel: +39 02 34964 200 | Suomi/Finland CSL Behring AB Puh/Tel: +46 8 544 966 70 |
Κύπρος CSL Behring ΕΠΕ Τηλ: +30 210 7255 660 | Sverige CSL Behring AB Tel: +46 8 544 966 70 |
Latvija CentralPharma Communications SIA Tel: +371 6 7450497 | |

Data dell'ultima revisione di questo foglio illustrativo: {MM/AAAA}
Informazioni dettagliate su questo medicinale sono disponibili sul sito web dell’Agenzia Europea per i Medicinali: http://www.ema.europa.eu.
Questa informazione è destinata esclusivamente ai professionisti del settore sanitario:
Monitoraggio del trattamento
Durante il trattamento, si raccomanda di controllare adeguatamente i livelli di fattore VIII al fine di determinare la dose da somministrare e la frequenza delle iniezioni. La risposta dei pazienti al fattore VIII può variare, dimostrando una diversa emivita e recupero. La dose basata sul peso corporeo potrebbe dover essere aggiustata nei pazienti con peso insufficiente o in sovrappeso. Nel caso particolare di interventi chirurgici maggiori, è indispensabile monitorare con precisione la terapia sostitutiva mediante analisi della coagulazione (attività del fattore VIII plasmatico).
Quando si utilizza un saggio di coagulazione a stadio unico basato sul tempo di tromboplastina parziale attivata (TTPa) in vitro per determinare l'attività del fattore VIII nei campioni di sangue dei pazienti, i risultati dell'attività del fattore VIII plasmatico possono essere significativamente influenzati sia dal tipo di reagente TTPa sia dallo standard di riferimento utilizzato nel saggio. Possono inoltre verificarsi discrepanze significative tra i risultati ottenuti con il saggio di coagulazione a stadio unico basato sul TTPa e quelli ottenuti con il saggio cromogenico secondo la Farmacopea Europea. Ciò risulta particolarmente importante quando si cambia laboratorio o i reagenti utilizzati nel saggio.
L'attività del fattore VIII plasmatico nei pazienti che ricevono AFSTYLA deve essere monitorata mediante un saggio cromogenico o un saggio di coagulazione a stadio unico per guidare la dose somministrata e la frequenza delle iniezioni ripetute. Il risultato del saggio cromogenico riflette in modo più accurato il potenziale emostatico clinico di AFSTYLA ed è pertanto il metodo preferito. Il risultato del saggio di coagulazione a stadio unico sottostima il livello di attività del fattore VIII rispetto al risultato del saggio cromogenico di circa il 45%. Se si utilizza un saggio di coagulazione a stadio unico, il risultato deve essere moltiplicato per un fattore di conversione pari a 2 per determinare il livello di attività del fattore VIII del paziente.
Posologia
La dose e la durata della terapia sostitutiva dipendono dalla gravità della carenza di fattore VIII, dalla localizzazione e dall'estensione dell'emorragia e dallo stato clinico del paziente.
Il numero di unità di fattore VIII somministrate viene espresso in Unità Internazionali (UI), corrispondenti allo standard concentrato OMS attuale per i medicinali contenenti fattore VIII. L'attività del fattore VIII nel plasma viene espressa come percentuale (rispetto al plasma umano normale) oppure, preferibilmente, in Unità Internazionali (rispetto a uno standard internazionale per il fattore VIII nel plasma).
Un'Unità Internazionale (UI) di attività del fattore VIII corrisponde alla quantità di fattore VIII presente in 1 ml di plasma umano normale.
L'assegnazione della potenza viene determinata mediante un saggio con substrato cromogenico.
I livelli plasmatici di fattore VIII possono essere monitorati mediante un saggio con substrato cromogenico o un saggio di coagulazione a stadio unico.
Trattamento su richiesta
Il calcolo della dose necessaria di fattore VIII si basa sull'osservazione empirica che 1 Unità Internazionale (UI) di fattore VIII per kg di peso corporeo aumenta l'attività plasmatica del fattore VIII di 2 UI/dl.
La dose richiesta viene determinata utilizzando la seguente formula: Dose (UI) = peso corporeo (kg) × aumento desiderato del fattore VIII (UI/dl o % del livello normale) × 0,5 (UI/kg per UI/dl)
La dose e la frequenza di somministrazione devono essere stabilite in base all'efficacia clinica osservata in ciascun caso.
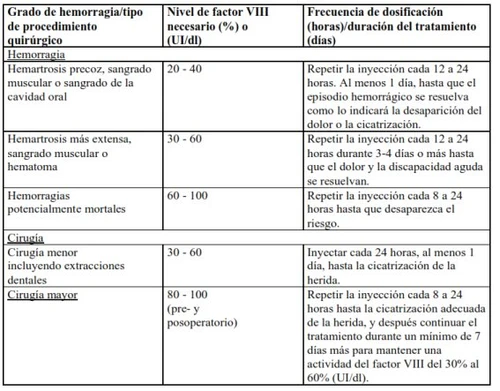
Nei seguenti eventi emorragici, l'attività del fattore VIII non deve essere inferiore al livello di attività plasmatica stabilito (in % del livello normale o UI/dl) durante il periodo corrispondente. La seguente tabella può essere utilizzata come guida posologica negli episodi emorragici e durante interventi chirurgici:
Trattamento profilattico
La posologia iniziale raccomandata è di 20-50 UI/kg di AFSTYLA somministrati 2 o 3 volte alla settimana. La posologia può essere aggiustata in base alla risposta del paziente.
Popolazione pediatrica
La posologia iniziale raccomandata nei bambini (da 0 a < 12 anni di età) è di 30-50 UI per kg di AFSTYLA somministrati 2 o 3 volte alla settimana. Nei bambini di età inferiore ai 12 anni potrebbero essere necessarie dosi più frequenti o più elevate a causa del maggiore clearance presente in questo gruppo di età.
Negli adolescenti di età pari o superiore a 12 anni, le dosi raccomandate sono le stesse previste per gli adulti.
Popolazione anziana
Negli studi clinici con AFSTYLA non sono stati inclusi soggetti di età superiore ai 65 anni.