Uman Complex 500 MO/20 ml
UcraniaContenido
INSTRUCCIONES PARA USO MÉDICO DEL MEDICAMENTO UMAN COMPLEX 500 UI/20 ml
Composición:
1 frasco contiene:
Principios activos: complejo protrombínico humano:
factor de coagulación sanguínea humana IX – 500 UI;
factor de coagulación sanguínea humana II – 500 UI;
factor de coagulación sanguínea humana X – 400 UI;
Excipientes: cloruro de sodio, citrato de sodio, glicina, heparina, antitrombina III;
Disolvente: agua para inyección – 20 ml.
El factor IX se titula según el estándar internacional.
El contenido de proteína total en el frasco es de ≤ 300 mg. La actividad específica del medicamento es superior a 0,6 UI/mg, expresada como actividad del factor IX.
Excipientes con efecto conocido: el medicamento contiene hasta 92 mg de sodio por frasco (20 ml).
Forma farmacéutica. Polvo y disolvente para solución para perfusión.
Principales características físico-químicas: el medicamento es un polvo blanco o ligeramente coloreado, muy higroscópico, o una sustancia sólida frágil.
Grupo farmacoterapéutico. Agentes antihemorrágicos. Combinación de factores de coagulación sanguínea IX, II, VII y X. Código ATC B02BD01.
Propiedades farmacológicas.
Farmacodinamia.
Los factores de la coagulación II, VII, IX y X, que se sintetizan en el hígado mediante la vitamina K, normalmente se denominan complejo de protrombina.
El factor VII es un zimógeno de la serina proteasa activa factor VIIa, que inicia la coagulación sanguínea por la vía extrínseca. El complejo formado por el factor tisular y el factor VIIa activa los factores de coagulación X e IX, dando lugar a la formación de los factores IXa y Xa. Como consecuencia de la posterior activación de la cascada de coagulación, se activa la protrombina (factor II) y se transforma en trombina. Bajo la acción de la trombina, el fibrinógeno se convierte en fibrina, formándose así un coágulo. La formación normal de trombina también es muy importante para la función plaquetaria como parte del hemostasis primario.
La deficiencia aislada y grave del factor VII conduce a una disminución en la formación de trombina y a una predisposición a hemorragias debido a una función deficiente en la formación de fibrina y un hemostasis primario debilitado. La deficiencia aislada del factor IX es una de las hemofilias clásicas (hemofilia B). La deficiencia aislada del factor II o del factor X es muy rara, pero en su forma grave provoca hemorragias similares a las de la hemofilia clásica.
La deficiencia adquirida de los factores de coagulación dependientes de la vitamina K se observa durante el tratamiento con antagonistas de la vitamina K. Si la deficiencia se vuelve grave, esto conduce a una predisposición a hemorragias severas, principalmente hemorragias retroperitoneales o cerebrales, más que hemorragias musculares o articulares. La insuficiencia hepática grave también provoca niveles notablemente reducidos de factores de coagulación dependientes de la vitamina K y una predisposición a hemorragia clínica, que sin embargo suele ser compleja debido a una coagulación intravascular continua y leve, recuentos bajos de plaquetas, deficiencia de inhibidores de la coagulación y alteraciones en la fibrinólisis.
La administración del complejo de protrombina humana proporciona un aumento en los niveles plasmáticos de los factores de coagulación dependientes de la vitamina K y puede, además, corregir durante cierto tiempo los trastornos de la coagulación en pacientes con deficiencia de uno o varios factores.
Farmacocinética.
| Factor de coagulación |
Período de semivida |
| Factor II |
40–60 horas |
| Factor IX |
16–30 horas |
| Factor X |
30–60 horas |
Datos preclínicos de seguridad
El concentrado de factores del complejo protrombínico es un componente natural del plasma humano y actúa de forma similar a los factores endógenos.
Los estudios de toxicidad de dosis única no son significativos, ya que dosis elevadas provocan hipervolemia.
Los estudios de toxicidad con administraciones repetidas en animales no son factibles debido a la interferencia de los anticuerpos que se forman frente a la proteína heteróloga.
Incluso las dosis considerablemente superiores a las recomendadas para el ser humano por kg de peso corporal no muestran ningún efecto tóxico en los animales de estudio.
Dado que la experiencia clínica con el uso de factores del complejo protrombínico humano no ha confirmado efectos carcinogénicos ni mutagénicos, no se considera necesario realizar estudios experimentales, especialmente aquellos que involucran especies heterólogas.
Características clínicas.
Indicaciones.
- Para el tratamiento de hemorragias y la profilaxis peroperatoria de hemorragias en deficiencias adquiridas de factores de coagulación del complejo protrombínico, por ejemplo, deficiencia provocada por tratamiento con antagonistas de la vitamina K o sobredosis de antagonistas de la vitamina K, cuando se requiere una corrección rápida de la deficiencia.
- Para el tratamiento de hemorragias y la profilaxis peroperatoria en deficiencias congénitas de cualquier factor de coagulación dependiente de la vitamina K, cuando no está disponible un preparado purificado del factor de coagulación específico.
Contraindicaciones.
Hipersensibilidad al principio activo o a cualquiera de los excipientes enumerados en la sección «Composición».
Alergia conocida a la heparina o trombocitopenia inducida por heparina en la historia clínica.
Interacción con otros medicamentos y otras formas de interacción.
Los preparados del complejo protrombínico humano neutralizan el efecto de los antagonistas de la vitamina K.
No se conoce interacción con otros medicamentos.
Efecto sobre las pruebas biológicas
Al realizar pruebas de coagulación sensibles a la heparina en pacientes que reciben altas dosis de complejo protrombínico humano, debe tenerse en cuenta la heparina como componente del medicamento administrado.
Pediatría
No existen datos específicos en niños.
Características de aplicación.
Rastreabilidad
Se recomienda encarecidamente registrar el nombre y número de lote del medicamento UMANN COMPLEX 500 UOI/20 ml cada vez que se administre al paciente, con el fin de garantizar la rastreabilidad y la relación entre el paciente y el lote específico del medicamento.
El tratamiento de los trastornos de la coagulación debe ser realizado por especialistas con experiencia adecuada.
En pacientes con deficiencia adquirida de factores de coagulación dependientes de la vitamina K (por ejemplo, provocada por el tratamiento con antagonistas de la vitamina K), el medicamento UMANN COMPLEX 500 UOI/20 ml debe administrarse únicamente cuando sea necesario corregir rápidamente los niveles del complejo protrombínico, por ejemplo, en casos de hemorragias significativas o intervenciones quirúrgicas de urgencia. En otros casos, generalmente es suficiente reducir la dosis del antagonista de la vitamina K y/o administrar vitamina K.
Los pacientes que reciben antagonistas de la vitamina K pueden presentar un estado de hipercogulabilidad oculto, y la administración del complejo protrombínico humano puede agravarlo.
En caso de deficiencia congénita de cualquiera de los factores dependientes de la vitamina K, se recomienda, cuando sea posible, utilizar el factor de coagulación específico correspondiente.
Si se desarrolla una reacción alérgica o una reacción de tipo anafiláctico, se debe interrumpir inmediatamente la inyección o infusión del medicamento.
En caso de aparición de shock, deben aplicarse las medidas médicas estándar para el tratamiento del shock.
Información importante sobre los excipientes del medicamento UMANN COMPLEX 500 UOI/20 ml
Este medicamento contiene hasta 92 mg de sodio por frasco (20 ml), lo que representa el 4,6 % de la dosis diaria máxima recomendada de sodio por la OMS para adultos, que es de 2 g.
Seguridad viral
Las medidas estándar para prevenir infecciones derivadas del uso de medicamentos elaborados a partir de sangre o plasma humano incluyen la selección de donantes, el cribado (análisis) de lotes individuales de plasma donado y de pools de plasma para detectar marcadores específicos de infección, así como la implementación de procedimientos eficaces de inactivación/eliminación viral durante el proceso de fabricación.
A pesar de estas medidas, no puede excluirse completamente la posibilidad de transmisión de agentes infecciosos al administrar medicamentos derivados de sangre o plasma humano. Esto incluye también virus desconocidos o nuevos y otros patógenos.
Se considera que las medidas adoptadas son eficaces frente a virus envueltos, como el virus de la inmunodeficiencia humana (VIH), el virus de la hepatitis B (VHB) y el virus de la hepatitis C (VHC), así como frente a virus no envueltos, como el virus de la hepatitis A (VHA). Sin embargo, su eficacia puede ser limitada frente a virus no envueltos como el parvovirus B19. La infección por parvovirus B19 puede ser grave en mujeres embarazadas (infección fetal) y en personas con inmunodeficiencia o con eritropoyesis aumentada (por ejemplo, en anemia hemolítica).
Debe considerarse la posibilidad de aplicar la vacunación adecuada (contra hepatitis A y B) en pacientes que reciben regularmente complejo protrombínico humano.
La administración del complejo protrombínico humano se asocia con un mayor riesgo de coagulación intravascular diseminada, complicaciones tromboembólicas e infarto de miocardio. Los pacientes que reciben complejo protrombínico humano deben ser examinados cuidadosamente en busca de signos de coagulación intravascular diseminada o trombosis.
Debido al riesgo potencial de complicaciones tromboembólicas tras la administración de este medicamento, se debe observar cuidadosamente, mediante los estudios biológicos necesarios, el estado de los pacientes con enfermedad coronaria o antecedentes de infarto de miocardio, enfermedad hepática, pacientes en período postoperatorio, recién nacidos o pacientes con riesgo de desarrollar eventos tromboembólicos o coagulación intravascular diseminada, con el fin de detectar los primeros signos de complicaciones trombóticas y de coagulopatía de consumo. En cada uno de estos casos, el beneficio potencial del tratamiento con UMANN COMPLEX 500 UOI/20 ml debe superar claramente el riesgo de dichas complicaciones.
No existen datos sobre la utilización del medicamento UMANN COMPLEX 500 UOI/20 ml en caso de hemorragia perinatal debida a deficiencia de vitamina K en recién nacidos.
Pediatría
No hay datos específicos disponibles en población pediátrica.
Uso durante el embarazo o la lactancia.
La seguridad del uso del complejo protrombínico humano durante el embarazo no ha sido establecida en estudios clínicos controlados.
Los estudios en animales no son adecuados para evaluar la seguridad del medicamento respecto a la función reproductiva, el desarrollo embrionario/fetal, el curso del embarazo y el desarrollo peri- y posnatal humano.
Por tanto, el complejo protrombínico humano solo debe utilizarse durante el embarazo y la lactancia si las indicaciones son indudables.
Capacidad para afectar la velocidad de reacción al conducir vehículos de motor o manejar otras máquinas.
No se han realizado estudios sobre el efecto de este medicamento en la capacidad para conducir vehículos de motor o manejar otras máquinas.
Vía de administración y dosis.
Dosificación
A continuación se indican únicamente recomendaciones generales sobre la dosificación. El tratamiento debe iniciarse bajo supervisión de un especialista con experiencia en el manejo de trastornos de la coagulación. La dosis y la duración de la terapia sustitutiva dependen de la gravedad del trastorno, del lugar y grado de hemorragia, así como del estado clínico del paciente.
La cantidad y frecuencia de administración deben calcularse individualmente para cada paciente.
Los intervalos de dosificación deben adaptarse a las diferentes semividas de eliminación de los distintos factores de coagulación del complejo protrombínico (ver sección «Farmacocinética»). Las necesidades individuales de dosificación solo pueden determinarse mediante mediciones regulares de los niveles plasmáticos individuales de los factores de coagulación estudiados, o bien mediante pruebas globales de niveles del complejo protrombínico (tiempo de protrombina, relación internacional normalizada (INR)), así como un monitoreo continuo del estado clínico del paciente.
En caso de intervenciones quirúrgicas extensas, es fundamental realizar un monitoreo preciso de la terapia sustitutiva mediante análisis de hemostasia (determinación del factor de coagulación específico y/o pruebas globales de niveles del complejo protrombínico).
Hemorragias y profilaxis peroperatoria de hemorragias durante el tratamiento con antagonistas de la vitamina K
La dosis dependerá del valor inicial (previo al tratamiento) y del valor objetivo de la relación internacional normalizada (INR). La corrección de los trastornos del hemostasio inducidos por antagonistas de la vitamina K se mantiene durante aproximadamente 6–8 horas. Sin embargo, el efecto de la vitamina K administrada simultáneamente generalmente se alcanza en un plazo de 4–6 horas. Por lo tanto, tras la administración de vitamina K, generalmente no es necesario repetir el tratamiento con complejo protrombínico humano.
Dado que estas recomendaciones son empíricas y que la recuperación y la duración del efecto pueden variar, el monitoreo del INR durante el tratamiento es obligatorio.
Hemorragias y profilaxis peroperatoria en deficiencias congénitas de cualquier factor de coagulación dependiente de la vitamina K, cuando no está disponible un preparado del factor de coagulación específico
La dosis necesaria para el tratamiento se calcula según datos empíricos, según los cuales aproximadamente 1 UI de factor IX por kg de peso corporal aumenta la actividad del factor IX en plasma en 0,01 UI/ml; 1 UI de factor II o factor X por kg de peso corporal aumenta la actividad del factor II o del factor X en plasma en 0,02 y 0,017 UI/ml, respectivamente.
La dosis del factor específico administrado se expresa en unidades internacionales (UI) de acuerdo con el estándar actual de la OMS para cada factor. La actividad del factor específico de coagulación en plasma se expresa en porcentaje (respecto al plasma normal) o en unidades internacionales (respecto al estándar internacional para el factor de coagulación específico).
Una unidad internacional (UI) de actividad de un factor de coagulación equivale a la cantidad de factor presente en 1 ml de plasma normal humano.
Por ejemplo, el cálculo de la dosis necesaria del factor X se basa en datos empíricos que indican que 1 unidad internacional (UI) de factor X por kg de peso corporal aumenta la actividad del factor X en plasma en 0,017 UI/ml.
La dosis necesaria se calcula mediante la siguiente fórmula:
Cantidad necesaria de unidades (UI) = peso corporal (kg) × aumento deseado del factor X (UI/ml) × 60,
donde
60 (ml/kg) es el valor recíproco del volumen de distribución previsto (normalización).
Si se conoce la normalización individual, este valor debe utilizarse para el cálculo.
Vía de administración
Disolver el medicamento como se describe a continuación. El medicamento UMAN COMPLEJO 500 UI/20 ml debe administrarse por vía intravenosa mediante inyección o infusión lenta.
Se recomienda no administrar más de 100 UI/kg de peso corporal al día.
Reconstitución del polvo con el disolvente
- Llevar el frasco con el polvo y el frasco con el disolvente a temperatura ambiente.
- Mantener la temperatura ambiente durante todo el proceso de reconstitución (disolución) (máximo 10 minutos).
- Retirar las tapas protectoras del frasco con el polvo y del frasco con el disolvente.
- Desinfectar con alcohol etílico las superficies de los tapones de ambos frascos.
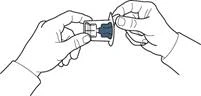
- Abrir el envase del dispositivo según se indica en la figura A, sin tocar la parte interna del envase (figura A).
- No extraer el dispositivo del envase.
- Dar la vuelta al envase con el dispositivo y perforar con la espiga plástica del dispositivo el tapón del frasco con el disolvente, de modo que la parte azul del dispositivo quede unida al frasco con el disolvente (figura B).
- Sujetando el borde del envase del dispositivo, retirarlo sin tocar el dispositivo en sí (figura C).
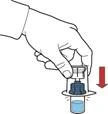
- Asegurarse de que el frasco con el polvo esté sobre una superficie estable; dar la vuelta al frasco con el disolvente conectado al dispositivo, de modo que el frasco con el disolvente quede sobre el dispositivo; presionar el adaptador transparente sobre el tapón del frasco con el polvo para que la espiga plástica atraviese el tapón del frasco con el polvo; el disolvente comenzará automáticamente a transferirse al frasco con el polvo (figura D).
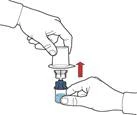
- Tras la transferencia del disolvente, desenroscar la parte azul del sistema, a la que está conectado el frasco del disolvente, y retirarla (figura E).
- Agitar suavemente el frasco hasta lograr la completa disolución del polvo. No agitar vigorosamente para evitar la formación de espuma (figura F).
Para evitar una disminución de la actividad del medicamento, asegurarse de que el polvo se haya disuelto completamente en el frasco.
| Fig. A |
Fig. B |
|
|
|
| Fig. C |
Fig. D |
|
|
|
| Fig. E |
Fig. F |
|
|
|
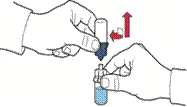
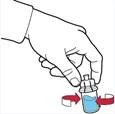
Administración de la solución
La solución preparada debe ser transparente o ligeramente opalescente.
Antes de la administración, la solución debe inspeccionarse visualmente para detectar la presencia de partículas o cambios de color. No utilizar la solución si está turbia o si contiene sedimento.
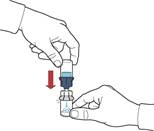
- Aspirar aire en la jeringa tirando del émbolo hacia atrás; conectar la jeringa al dispositivo e introducir el aire desde la jeringa en el frasco con la solución reconstituida (fig. E).
- Manteniendo el émbolo, invertir el sistema de forma que el frasco con la solución reconstituida quede por encima del dispositivo. Tirando lentamente del émbolo, aspirar el concentrado en la jeringa (fig. F).
- Desconectar la jeringa girándola en sentido antihorario.
- Inspeccionar visualmente la solución en la jeringa, que debe ser transparente o ligeramente opalescente y no debe contener partículas.
- Conectar una aguja-butterfly a la jeringa y administrar el medicamento por vía intravenosa.
| Fig. E |
Fig. F |
|
|
|
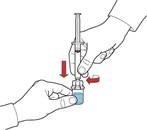
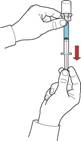
Después de la apertura, el contenido del frasco debe utilizarse inmediatamente.
La solución reconstituida, precargada en la jeringa, debe utilizarse inmediatamente.
Los medicamentos no utilizados o los residuos derivados de su uso deben eliminarse de acuerdo con los requisitos locales.
Niños.
La seguridad y eficacia del medicamento UMAN COMPLEX 500 MO/20 ml en niños no han sido establecidas.
Sobredosificación.
La administración de dosis elevadas de complejos protrombínicos humanos se ha asociado con casos de infarto de miocardio, coagulación intravascular diseminada, trombosis venosas y embolismo pulmonar. Por lo tanto, en caso de sobredosificación, aumenta el riesgo de complicaciones tromboembólicas o de coagulación intravascular diseminada.
Niños
No existen datos específicos en niños.
Reacciones adversas.
Descripción breve del perfil de seguridad
Se han observado raramente reacciones alérgicas o reacciones de tipo anafiláctico.
La terapia sustitutiva con el complejo protrombínico humano puede provocar raramente la formación de anticuerpos circulantes que inhiben uno o varios factores del complejo protrombínico humano. Si aparecen tales inhibidores, se manifestarán mediante una respuesta clínica deficiente.
En casos raros se ha observado fiebre.
Existe un riesgo potencial de reacciones tromboembólicas tras la administración del complejo protrombínico humano, tales como embolía y trombosis, coagulación intravascular diseminada e infarto de miocardio (ver la sección «Precauciones de uso»).
Para información sobre la seguridad respecto a agentes transmisibles, ver la sección «Precauciones de uso».
Lista de reacciones adversas en forma de tabla
Los efectos adversos que pueden presentarse durante el uso del complejo protrombínico humano se muestran en la siguiente tabla, clasificados según el sistema de órganos del MedDRA [Diccionario Médico para Actividades Regulatorias] y los términos de preferencia.
La frecuencia se ha evaluado según las siguientes categorías convencionales: muy frecuente (≥ 1/10); frecuente (≥ 1/100 a < 1/10); poco frecuente (≥ 1/1 000 a < 1/100); rara (≥ 1/10 000 a < 1/1 000); muy rara (< 1/10 000) y frecuencia desconocida (no puede estimarse a partir de los datos disponibles).
| Clase de sistema de órganos según MedDRA |
Reacciones adversas(MedDRA, término de uso preferente) |
Frecuencia |
| Trastornos del sistema inmunitario |
Hipersensibilidad |
No conocida |
| Reacciones anafilácticas |
No conocida |
|
| Trastornos vasculares |
Embolia |
No conocida |
| Trombosis |
No conocida |
|
| Coagulación intravascular diseminada |
No conocida |
|
| Infarto de miocardio |
No conocida |
|
| Trastornos generales y alteraciones en el lugar de administración |
Pirexia |
No conocida |
| Pruebas diagnósticas |
Anticuerpos inhibidores |
No conocida |
Niños
No existen datos específicos en niños.
Comunicación de reacciones adversas sospechosas
La notificación de reacciones adversas tras la autorización del medicamento es importante. Permite continuar con el monitoreo de la relación beneficio/riesgo del medicamento. Los profesionales médicos y farmacéuticos, así como los pacientes o sus representantes legales, deben notificar cualquier caso sospechoso de reacción adversa o falta de eficacia del medicamento a través del Sistema Automatizado de Información para la Farmacovigilancia en el siguiente enlace: https://aisf.dec.gov.ua.
Período de validez.
3 años.
Condiciones de conservación.
Conservar en nevera a una temperatura comprendida entre 2 y 8 ºC.
Conservar el frasco en su caja exterior de cartón para protegerlo de la luz.
No congelar.
Incompatibilidades.
El concentrado del complejo protrombínico humano no debe mezclarse con otros medicamentos.
Debe utilizarse únicamente el conjunto para inyección/infusión suministrado, ya que un tratamiento inadecuado podría ser consecuencia de la adsorción de los factores de coagulación en las superficies internas de ciertos conjuntos para inyección/infusión.
Envase.
1 frasco con polvo (500 UI), junto con 1 frasco con disolvente (agua para inyección, 20 ml) y un conjunto para la reconstitución y administración, en caja de cartón.
Categoría de dispensación.
Medicamento sujeto a prescripción médica.
Fabricante.
KEDRION S.P.A.
Domicilio del fabricante y dirección del lugar de ejercicio de su actividad.
VIA PROVINCIALE (loc. BOLOGNANA) – 55027 GALLICANO (LU), Italia.